Tu 1,*, , Sweden
|
|
|
- Amberlynn Jennings
- 5 years ago
- Views:
Transcription
























1 Supplementary Material Computational studiess of the binding profile of phosphoinositide PtdIns(,4,5)P with the pleckstrin homology domain d of an oomycetee cellulose synthase Guanglin Kuang 1, Vincent Bulone 2, and Yaoquan T Tu 1, 1 Division of Theoretical Chemistry and Biology, School of Biotechnology, Royal Institute of Technology (KTH), AlbaNova University y Center, Stockholm, 10691, Sweden 2 Division of Glycoscience, School of Biotechnology, Royal Institute of Technology (KTH), AlbaNovaa University Center, Stockholm,, , Sweden Supplementary Figure S1. Ramachandrann plot of the homologyy model of SmCesA2-PH.





















2 Supplementary Figure S2. Dynamic behaviors of SmCesA2-PH in two independent MD simulations with the Amber99SB force field. (a, c) RMSD plots of the backbone atoms of the -sandwichh and -helix core structure (black), VL1 (red) and VL2 (green) off SmCesA2-PH. (b, d) The RMSF plot of SmCesA2-PH. Supplementary Figure S. Binding mode of Ins(,4,5)P with the template structure TAPP1-PH.





")






PP")



3 Supplementary Figure S4. Some MD plots of the head groupss used to monitor their binding processes with SmCesA2-PH in i the MD simulations with different initial velocities (second run). COM distance is the distance betweenn the centers of mass (COMs) of the head group and the protein. RMSD is the root mean square deviation of the head group conformation in the MD simulations after the alignment of the protein. Minimumm distance is the minimum distance between the head group and protein heavy atoms. Supplementary Table S1. Schrödinger XP docking scores off Ins(,4,5)PP with the mutated forms of the hot-spot residues identified by molecular docking and molecular dynamics simulations. wild type K85A K88A K100A R102A Glide XP Docking Score (kcal/mol)




















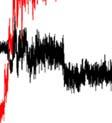






















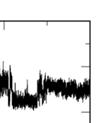

4 Supplementary Figure S5. (a) Binding mode of Ins(,4,5)P in i a meta-stable state.(b) Some parameters to monitor the binding profilee of the meta-stable state. COM distance is the t distance between the centers of mass (COMs) of thee inositol head group and the protein. RMSD is the root mean square deviation of the head group conformationn in the MD simulation n after the alignment of the protein. Minimumm distance iss the minimum distance between the t head group and protein heavy atoms. Dihedral is the same as that t definedd in the Method section.








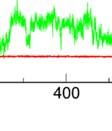



















5 Supplementary Figure S6. (a) Bindingg mode of SmCesA2-PH with h the PtdIns(,4,5)P molecule in solution. (b) Some parameters used to monitor the binding profile in i the MD simulations. COM distance is the distance between the centers of mass m (COMs) of the inositol head group and the protein. Head RMSD is the RMSD of the inositol head group. Tail RMSD is the RMSD of the two hydrophobic tails. Minimum distance is the minimum distance between the lipid molecule and protein heavy atoms.















































6 Supplementary Figure S7. Binding profile of SmCesA2-PHH on the PtdIns(,4,5)P /POPC membranee in two independent MD simulations. The figures show the plotss of the RMSDs of the backbone atoms of the -sandwich and -helix core structure (RMSD1, black), VL1 (RMSD2, blue) and VL2 (RMSD, cyan) as well as thee minimumm distance between SmCesA2-PH and the PtdIns(,4,,5)P /POPC membranee (green). a b








































7 c d Supplementary Figure S8. Binding profilee of SmCesA2-PH with PtdIns(,4)P 2. (a) The binding mode of SmCesA2-PH with the head groupp Ins(,4)P 2. (b) Somee parameters used to monitor the binding profile of the head group in MD simulations. COM distance is thee distance between the centers of mass (COMs) of the inositol headd group and the protein. RMSD iss the root mean square deviation of the head group conformation inn the MD simulation after a the alignment of the t protein. Minimum distance is the minimum distancee between the head group and protein heavy atoms. (c) The binding profile of SmCesA2-PH with the PtdIns( (,4)P 2 /POPC membrane which includes the contact (within Å) probability plots of the amino acids of SmCesA2-PH with the PtdIns(,4) )P 2 /POPC membrane, and the binding mode of SmCesA2-PH with the membrane. (d)




 and")


P 2")
.")





, VL1")







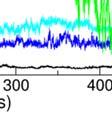





8 The figures showing the plots of the RMSDs of the backbone atoms of the -sandwich and -helixx core structure (RMSD1, black),, VL1 (RMSD2, blue) and VL2 (RMSD, cyan) as well as the minimum distance between SmCesA2-PH and the PtdIns(,4)P 2 /POPC membrane (green). Supplementary Figure S9. Binding profile of SmCesA2-PH on the POPC membrane in two independent MD simulations. The figures show the plots of the RMSDs R of the backbone atoms of the -sandwich and -helix core structure (RMSD1, black), VL1 (RMSD2, blue) and VL2 (RMSD, cyan) as well as the minimum distance between SmCesA2-PH andd the POPC membrane (green). Supplementary Figure S10. Binding profile of SmCesA2-PH on o the PtdIns/POPC membranee in two independent MD simulations. The figures show the plots of the RMSDs of the backbone atoms of the -sandwich and -helix core structure (RMSD1, black), VL1 (RMSD2, blue) and VL2 (RMSD, cyan) as well as the minimum distance between SmCesA2-PH and the PtdIns/POPC membrane.






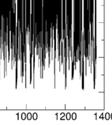
























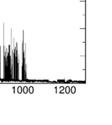
9 Supplementary Figure S11. metadynamics simulations of Illustration of the collective variables selected for the the SmCesA2-Ins complex. CV1 is the distance d between the center of mass (COM) of the ligand and thatt of SmCesA2-PH. CV2 is the dihedral angle (torsion ) ) defined by the two end points of the -helix,, the COM of the protein, and the COM of the ligand. Supplementary Figure S12. Evolutions off COM distance (CV1) and Gaussian HILLS height in the metadynamics simulations. Several recrossing events of the distance CV and smalll Gaussian HILLS height (close to zero) are two common indicators of the convergence of the









P")









P")
10 metadynamics simulations. Thesee plots indicate that the three systems have converged. Supplementary Figure S1. Evolutions of the ALPHARMSD in the three metadynamics simulations, which can be used to monitorr the conten of -helix. Supplementary Figure S14. Initial setup of the SmCesA2-PH are shown in the thick stick SmCesA2-PH and PtdIns(,4,5)P /POPC membranee system. The PtdIns(,4,5)P molecule and three critical residuess in the binding site of mode and the POPC molecules m in the thin stick s mode. The protein is shown in the cartoon mode. Supplementary Videos Supplementary Video S1. Trajectory of SmCesA2-PH in a 500 ns MD simulation. Supplementary Video S2. Binding profilee of SmCesA2-PH with the PtdIns(,4,5)P moleculee in solution in a 500 ns MD simulation.
11 Supplementary Video S. Binding profile of SmCesA2-PH on the PtdIns(,4,5)P /POPC membrane in a 500 ns MD simulation. Supplementary Video S4. Binding profile of SmCesA2-PH on the PtdIns(,4)P 2 /POPC membrane in a 500 ns MD simulation. Supplementary Video S5. Binding profile of SmCesA2-PH on the POPC membrane in a 500 ns MD simulation. Supplementary Video S6. Binding profile of SmCesA2-PH on the PtdIns/POPC membrane in a 500 ns MD simulation. Appendix S1 Force field parameters of the head groups of phosphoinositides, obtained from CHARMM ParamChem web server Ins Toppar stream file generated by CHARMM General Force Field (CGenFF) program version beta For use with CGenFF version 2b8 read rtf card append Topologies generated by CHARMM General Force Field (CGenFF) program version beta 6 1! "penalty" is the highest penalty score of the associated parameters.! Penalties lower than 10 indicate the analogy is fair; penalties between 10! and 50 mean some basic validation is recommended; penalties higher than! 50 indicate poor analogy and mandate extensive validation/optimization. RESI INS GROUP! param penalty= ; charge penalty=! CHARGE CH_PENALTY ATOM O2 ATOM O ATOM O4 ATOM O5 ATOM O6 ATOM C ! ATOM C ! ATOM O12 ATOM C ! ATOM C ! ATOM C15 ATOM C ! 0.146!
12 ATOM H1 ATOM H2 ATOM H ATOM H4 ATOM H5 ATOM H6 ATOM H7 ATOM H8 ATOM H9 ATOM H10 ATOM H11 ATOM H12 BOND O2 BOND O BOND O4 BOND O5 BOND O6 BOND C1 BOND O2 BOND O BOND O4 BOND O5 BOND O6 BOND O12 BOND C1 BOND C16 C12 C1 C14 C15 C16 C16 C12 O12 C1 C14 C15 C16 H1 H2 H H4 H5 H6 H7 H8 H9 H10 H11 H12 END read param card flex append Parameters generated by analogy by CHARMM General Force Field (CGenFF) program version beta! Penalties lower than 10 indicate the analogy is fair; penalties between 10! and 50 mean some basic validation is recommended; penalties higher than! 50 indicate poor analogy and mandate extensive validation/optimization. BONDS ANGLES DIHEDRALS ! INS, from CG21, penalty= ! INS, from CG21, penalty= 4 IMPROPERS END RETURN Ins(1)P (PIP) Toppar stream file generated by CHARMM General Force Field (CGenFF) program version beta For use with CGenFF version 2b8
13 read rtf card append Topologies generated by CHARMM General Force Field (CGenFF) program version beta 6 1! "penalty" is the highest penalty score of the associated parameters.! Penalties lower than 10 indicate the analogy is fair; penalties between 10! and 50 mean some basic validation is recommended; penalties higher than! 50 indicate poor analogy and mandate extensive validation/optimization. RESI PIP 2.000! param penalty= ; charge penalty= 2.12 GROUP! CHARGE CH_PENALTY ATOM C11 ATOM C ! 0.146! ATOM C ! ATOM C14 ATOM C ! 0.098! ATOM C ! 2.12 ATOM O5 ATOM P5 0.99! 1.099! ATOM OP ! ATOM OP5 ATOM OP ! 0.900! ATOM O ATOM O6 ATOM O 0.00 ATOM O12 ATOM O2 ATOM H1 ATOM H2 ATOM H ATOM H ATOM H5 ATOM H6 ATOM H ATOM H8 ATOM H9 ATOM H10 ATOM H11 BOND C1 BOND C1 BOND C16 BOND O5 BOND P5 BOND P5 BOND P5 BOND C1 BOND C16 BOND O4 BOND O6 BOND O BOND O12 BOND O2 C16 O12 C12 O2 C1 C14 O O4 C15 O5 C16 O6 P5 OP4 OP6 OP5 H1 H2 H H4 H5 H6 H7 H8 H9 H10 H11 END read param card flex append
14 Parameters generated by analogy by CHARMM General Force Field (CGenFF) program version beta! Penalties lower than 10 indicate the analogy is fair; penalties between 10! and 50 mean some basic validation is recommended; penalties higher than! 50 indicate poor analogy and mandate extensive validation/optimization. BONDS ANGLES ! PIP, from CG1, penalty= 1.5 DIHEDRALS ! PIP, from CG21, penalty= ! PIP, from CG21 CG21, penalty= ! PIP, from CG21, penalty= ! PIP, from CG21, penalty= ! PIP, from CG21, penalty= ! PIP, from CG1, penalty= ! PIP, from CG1, penalty= ! PIP, from CG1, penalty= 1.5 IMPROPERS END RETURN Ins(,4)P 2 (PIP2) Toppar stream file generated by CHARMM General Force Field (CGenFF) program version beta For use with CGenFF version 2b8 read rtf card append Topologies generated by CHARMM General Force Field (CGenFF) program version beta 6 1! "penalty" is the highest penalty score of the associated parameters.! Penalties lower than 10 indicate the analogy is fair; penalties between 10! and 50 mean some basic validation is recommended; penalties higher than! 50 indicate poor analogy and mandate extensive validation/optimization. RESI PIP2 GROUP.000! param penalty= ; charge penalty=! CHARGE CH_PENALTY ATOM C ! ATOM C12 ATOM C ! 0.094! ATOM C ! ATOM C15 ATOM C ! 0.146! ATOM O 0.99! ATOM P ATOM O 1.099! 0.900! ATOM O ! ATOM O1 ATOM O ! 0.00 ATOM O ! ATOM P4 ATOM O ! 0.670! ATOM O ! 0.00 ATOM O4 ATOM O ! 0.00 ATOM O ATOM O6 ATOM H11
15 ATOM H ATOM H1 ATOM H ATOM H ATOM H16 ATOM HO2 ATOM HO ! ATOM HO12 ATOM HO5 ATOM HO6 BOND C1 BOND C1 BOND C1 BOND C16 BOND C16 BOND O BOND P BOND P BOND P BOND O2 BOND O4 BOND P4 BOND P4 BOND P4 BOND O42 BOND O12 BOND O5 BOND O6 C12 C16 O12 H11 C1 O2 H12 C14 O H1 C15 O4 H14 C16 O5 H15 O6 H16 P O O2 O1 HO2 P4 O42 O41 O4 HO42 HO12 HO5 HO6 END read param card flex append Parameters generated by analogy by CHARMM General Force Field (CGenFF) program version beta! Penalties lower than 10 indicate the analogy is fair; penalties between 10! and 50 mean some basic validation is recommended; penalties higher than! 50 indicate poor analogy and mandate extensive validation/optimization. BONDS ANGLES ! PIP2, from CG1, penalty= ! PIP2, from CG21, penalty= 0.6 DIHEDRALS ! PIP2, from CG21, penalty= ! PIP2, from CG21 CG21, penalty= ! PIP2, from OG02 CG21, penalty= ! PIP2, from CG21, penalty= ! PIP2, from CG21, penalty= ! PIP2, from CG21, penalty= ! PIP2, from CG1, penalty= ! PIP2, from CG1, penalty= ! PIP2, from CG1, penalty= ! PIP2, from CG1, penalty= ! PIP2, from CG1, penalty= 1.5
16 ! PIP2, from CG1, penalty= ! PIP2, from, penalty= ! PIP2, from CG21, penalty= ! PIP2, from CG21, penalty= ! PIP2, from CG21, penalty= 0.6 IMPROPERS END RETURN Ins(,4,5)P (PIP) Toppar stream file generated by CHARMM General Force Field (CGenFF) program version beta For use with CGenFF version 2b8 read rtf card append Topologies generated by CHARMM General Force Field (CGenFF) program version beta 6 1! "penalty" is the highest penalty score of the associated parameters.! Penalties lower than 10 indicate the analogy is fair; penalties between 10! and 50 mean some basic validation is recommended; penalties higher than! 50 indicate poor analogy and mandate extensive validation/optimization. RESI PIP 5.000! param penalty= ; charge penalty=.268 GROUP! CHARGE CH_PENALTY ATOM P ATOM O ! 0.82! ATOM O2 0.82! 0.00 ATOM O ATOM C ! 0.00! ATOM C ! ATOM C11 ATOM C ! 0.150! ATOM C !.22 ATOM C14 ATOM O ATOM O12 ATOM O6 ATOM O5 0.99! ATOM P ! ATOM O51 ATOM O ! 0.900! ATOM O ! ATOM O4 ATOM P4 0.99! 1.099! ATOM O ! ATOM O4 ATOM O ! 0.900! ATOM O 0.670! ATOM H1 ATOM H ATOM H11 ATOM H16 ATOM H ATOM H ATOM HO2 ATOM HO12 ATOM HO6 ATOM HO 0.41! BOND P BOND P BOND P O1 O2 O
17 BOND P BOND O BOND C1 BOND C1 BOND C1 BOND C16 BOND C16 BOND C16 BOND O2 BOND O12 BOND O6 BOND O5 BOND P5 BOND P5 BOND P5 BOND O4 BOND P4 BOND P4 BOND P4 BOND O O C1 C12 C14 H1 C11 O2 H12 C16 O12 H11 C15 O6 H16 C14 O5 H15 O4 H14 HO2 HO12 HO6 P5 O51 O5 O52 P4 O41 O4 O42 HO END read param card flex append Parameters generated by analogy by CHARMM General Force Field (CGenFF) program version beta! Penalties lower than 10 indicate the analogy is fair; penalties between 10! and 50 mean some basic validation is recommended; penalties higher than! 50 indicate poor analogy and mandate extensive validation/optimization. BONDS ANGLES ! PIP, from CG1, penalty= ! PIP, from CG21, penalty= 0.6 DIHEDRALS ! PIP, from CG21, penalty= ! PIP, from CG21 CG21, penalty= ! PIP, from OG02 CG21, penalty= ! PIP, from CG21, penalty= ! PIP, from CG21, penalty= ! PIP, from CG21, penalty= ! PIP, from CG1, penalty= ! PIP, from CG1, penalty= ! PIP, from CG1, penalty= ! PIP, from CG1, penalty= ! PIP, from CG1, penalty= ! PIP, from CG1, penalty= ! PIP, from, penalty= ! PIP, from CG21, penalty= ! PIP, from CG21, penalty= ! PIP, from CG21, penalty= 0.6 IMPROPERS END RETURN
read rtf card append * Initial topology guesses generated by * CHARMM General Force Field (CGenFF) program version 0.9.
 program version 0.9.") Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2014 * Stream file for dehydro-amino residues * If using these parameters please cite: * Turpin,
Electronic Supplementary Material (ESI) for RSC Advances. This journal is The Royal Society of Chemistry 2014 * Stream file for dehydro-amino residues * If using these parameters please cite: * Turpin,
Computational engineering of cellulase Cel9A-68 functional motions through mutations in its linker region. WT 1TF4 (crystal) -90 ERRAT PROVE VERIFY3D
 -90 ERRAT PROVE VERIFY3D") Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 218 Supplementary Material: Computational engineering of cellulase Cel9-68 functional
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 218 Supplementary Material: Computational engineering of cellulase Cel9-68 functional
SUPPLEMENTAL MATERIAL
 SUPPLEMENTAL MATERIAL Systematic Coarse-Grained Modeling of Complexation between Small Interfering RNA and Polycations Zonghui Wei 1 and Erik Luijten 1,2,3,4,a) 1 Graduate Program in Applied Physics, Northwestern
SUPPLEMENTAL MATERIAL Systematic Coarse-Grained Modeling of Complexation between Small Interfering RNA and Polycations Zonghui Wei 1 and Erik Luijten 1,2,3,4,a) 1 Graduate Program in Applied Physics, Northwestern
Hands-on Course in Computational Structural Biology and Molecular Simulation BIOP590C/MCB590C. Course Details
 Hands-on Course in Computational Structural Biology and Molecular Simulation BIOP590C/MCB590C Emad Tajkhorshid Center for Computational Biology and Biophysics Email: emad@life.uiuc.edu or tajkhors@uiuc.edu
Hands-on Course in Computational Structural Biology and Molecular Simulation BIOP590C/MCB590C Emad Tajkhorshid Center for Computational Biology and Biophysics Email: emad@life.uiuc.edu or tajkhors@uiuc.edu
Supporting Information
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2016 Supporting Information Lipid molecules can induce an opening of membrane-facing
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2016 Supporting Information Lipid molecules can induce an opening of membrane-facing
Equilibrated atomic models of outward-facing P-glycoprotein and effect of ATP binding on structural dynamics (Supplementary Information)
") Equilibrated atomic models of outward-facing P-glycoprotein and effect of ATP binding on structural dynamics (Supplementary Information) Lurong Pan 1 and Stephen G. Aller 2 * 1,2 Department of Pharmacology
Equilibrated atomic models of outward-facing P-glycoprotein and effect of ATP binding on structural dynamics (Supplementary Information) Lurong Pan 1 and Stephen G. Aller 2 * 1,2 Department of Pharmacology
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Structural Insights from Molecular Dynamics. Simulations of Tryptophan 7-Halogenase and
 Supporting Information Structural Insights from Molecular Dynamics Simulations of Tryptophan 7-Halogenase and Tryptophan 5-halogenase Jon Ainsley 1, Adrian J. Mulholland 2, Gary W. Black 1, Olivier Sparagano
Supporting Information Structural Insights from Molecular Dynamics Simulations of Tryptophan 7-Halogenase and Tryptophan 5-halogenase Jon Ainsley 1, Adrian J. Mulholland 2, Gary W. Black 1, Olivier Sparagano
Supplementary Information
 Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
What makes a good graphene-binding peptide? Adsorption of amino acids and peptides at aqueous graphene interfaces: Electronic Supplementary
 Electronic Supplementary Material (ESI) for Journal of Materials Chemistry B. This journal is The Royal Society of Chemistry 21 What makes a good graphene-binding peptide? Adsorption of amino acids and
Electronic Supplementary Material (ESI) for Journal of Materials Chemistry B. This journal is The Royal Society of Chemistry 21 What makes a good graphene-binding peptide? Adsorption of amino acids and
Computational Structural Biology and Molecular Simulation. Introduction to VMD Molecular Visualization and Analysis
 Computational Structural Biology and Molecular Simulation Introduction to VMD Molecular Visualization and Analysis Emad Tajkhorshid Department of Biochemistry, Beckman Institute, Center for Computational
Computational Structural Biology and Molecular Simulation Introduction to VMD Molecular Visualization and Analysis Emad Tajkhorshid Department of Biochemistry, Beckman Institute, Center for Computational
Supplementary Figure S1. Urea-mediated buffering mechanism of H. pylori. Gastric urea is funneled to a cytoplasmic urease that is presumably attached
 Supplementary Figure S1. Urea-mediated buffering mechanism of H. pylori. Gastric urea is funneled to a cytoplasmic urease that is presumably attached to HpUreI. Urea hydrolysis products 2NH 3 and 1CO 2
Supplementary Figure S1. Urea-mediated buffering mechanism of H. pylori. Gastric urea is funneled to a cytoplasmic urease that is presumably attached to HpUreI. Urea hydrolysis products 2NH 3 and 1CO 2
Rho1 binding site PtdIns(4,5)P2 binding site Both sites
P2 binding site Both sites") localization Mutation site DMSO LatB WT F77A I115A I131A K134A Rho1 binding site PtdIns(4,5)P2 binding site Both sites E186A E199A N201A R84A-E186A-E199A L131A-K136A-E186A L131A-E186A-E199A K136A-E186A-E199A
localization Mutation site DMSO LatB WT F77A I115A I131A K134A Rho1 binding site PtdIns(4,5)P2 binding site Both sites E186A E199A N201A R84A-E186A-E199A L131A-K136A-E186A L131A-E186A-E199A K136A-E186A-E199A
T H E J O U R N A L O F G E N E R A L P H Y S I O L O G Y. jgp
 S u p p l e m e n ta l m at e r i a l jgp Lee et al., http://www.jgp.org/cgi/content/full/jgp.201411219/dc1 T H E J O U R N A L O F G E N E R A L P H Y S I O L O G Y S u p p l e m e n ta l D I S C U S
S u p p l e m e n ta l m at e r i a l jgp Lee et al., http://www.jgp.org/cgi/content/full/jgp.201411219/dc1 T H E J O U R N A L O F G E N E R A L P H Y S I O L O G Y S u p p l e m e n ta l D I S C U S
A conserved P-loop anchor limits the structural dynamics that mediate. nucleotide dissociation in EF-Tu.
 Supplemental Material for A conserved P-loop anchor limits the structural dynamics that mediate nucleotide dissociation in EF-Tu. Evan Mercier 1,2, Dylan Girodat 1, and Hans-Joachim Wieden 1 * 1 Alberta
Supplemental Material for A conserved P-loop anchor limits the structural dynamics that mediate nucleotide dissociation in EF-Tu. Evan Mercier 1,2, Dylan Girodat 1, and Hans-Joachim Wieden 1 * 1 Alberta
Figure 1. Molecules geometries of 5021 and Each neutral group in CHARMM topology was grouped in dash circle.
 Project I Chemistry 8021, Spring 2005/2/23 This document was turned in by a student as a homework paper. 1. Methods First, the cartesian coordinates of 5021 and 8021 molecules (Fig. 1) are generated, in
Project I Chemistry 8021, Spring 2005/2/23 This document was turned in by a student as a homework paper. 1. Methods First, the cartesian coordinates of 5021 and 8021 molecules (Fig. 1) are generated, in
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Focus on PNA Flexibility and RNA Binding using Molecular Dynamics and Metadynamics
 SUPPLEMENTARY INFORMATION Focus on PNA Flexibility and RNA Binding using Molecular Dynamics and Metadynamics Massimiliano Donato Verona 1, Vincenzo Verdolino 2,3,*, Ferruccio Palazzesi 2,3, and Roberto
SUPPLEMENTARY INFORMATION Focus on PNA Flexibility and RNA Binding using Molecular Dynamics and Metadynamics Massimiliano Donato Verona 1, Vincenzo Verdolino 2,3,*, Ferruccio Palazzesi 2,3, and Roberto
Type II Kinase Inhibitors Show an Unexpected Inhibition Mode against Parkinson s Disease-Linked LRRK2 Mutant G2019S.
 Type II Kinase Inhibitors Show an Unexpected Inhibition Mode against Parkinson s Disease-Linked LRRK2 Mutant G219S. Min Liu@&*, Samantha A. Bender%*, Gregory D Cuny@, Woody Sherman, Marcie Glicksman@ Soumya
Type II Kinase Inhibitors Show an Unexpected Inhibition Mode against Parkinson s Disease-Linked LRRK2 Mutant G219S. Min Liu@&*, Samantha A. Bender%*, Gregory D Cuny@, Woody Sherman, Marcie Glicksman@ Soumya
Insights into the Biotransformation of 2,4,6- Trinitrotoluene by the Old Yellow Enzyme Family of Flavoproteins. A Computational Study
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2019 Supporting Information for Insights into the Biotransformation of 2,4,6- Trinitrotoluene
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2019 Supporting Information for Insights into the Biotransformation of 2,4,6- Trinitrotoluene
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Enhancing Specificity in the Janus Kinases: A Study on the Thienopyridine. JAK2 Selective Mechanism Combined Molecular Dynamics Simulation
 Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is The Royal Society of Chemistry 2015 Supporting Information Enhancing Specificity in the Janus Kinases: A Study on the Thienopyridine
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is The Royal Society of Chemistry 2015 Supporting Information Enhancing Specificity in the Janus Kinases: A Study on the Thienopyridine
Molecular Dynamics Simulations of the Mammalian Glutamate Transporter EAAT3
 Molecular Dynamics Simulations of the Mammalian Glutamate Transporter EAAT3 Germano Heinzelmann, Serdar Kuyucak* School of Physics, University of Sydney, NSW, Australia Abstract Excitatory amino acid transporters
Molecular Dynamics Simulations of the Mammalian Glutamate Transporter EAAT3 Germano Heinzelmann, Serdar Kuyucak* School of Physics, University of Sydney, NSW, Australia Abstract Excitatory amino acid transporters
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
A: Up regulated proteins B: Down regulated proteins. Susceptible Resistant Susceptible Resistant Resistant Susceptible
 Supplementary Materials: Identification of Biomarkers for Resistance to Fusarium oxysporum f. sp. cubense Infection and in Silico Studies in Musa paradisiaca Cultivar Puttabale through Proteomic Approach
Supplementary Materials: Identification of Biomarkers for Resistance to Fusarium oxysporum f. sp. cubense Infection and in Silico Studies in Musa paradisiaca Cultivar Puttabale through Proteomic Approach
Electro-Mechanical Conductance Modulation of a Nanopore Using a Removable Gate
 Electro-Mechanical Conductance Modulation of a Nanopore Using a Removable Gate Shidi Zhao a, Laura Restrepo-Pérez b, Misha Soskine c, Giovanni Maglia c, Chirlmin Joo b, Cees Dekker b and Aleksei Aksimentiev
Electro-Mechanical Conductance Modulation of a Nanopore Using a Removable Gate Shidi Zhao a, Laura Restrepo-Pérez b, Misha Soskine c, Giovanni Maglia c, Chirlmin Joo b, Cees Dekker b and Aleksei Aksimentiev
Molecular Modeling Lecture 7. Homology modeling insertions/deletions manual realignment
 Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Supplementary Figure 1 Irregular arrangement of E,E-8-mer on TMA. STM height images formed when
 Supplementary Figure 1 Irregular arrangement of E,E-8-mer on TMA. STM height images formed when a 5 µl heptanoic acid solution of E,E-8-mer is applied on: (a) a TMA templated HOPG substrate, U s = +1.00
Supplementary Figure 1 Irregular arrangement of E,E-8-mer on TMA. STM height images formed when a 5 µl heptanoic acid solution of E,E-8-mer is applied on: (a) a TMA templated HOPG substrate, U s = +1.00
ψ j φ j+1 Motif position
 1.0 % Occupancy 0.8 0.6 0.4 0.2 0.0 ψ i-1 φ i ψ j φ j+1 ψ k-1 Motif position φ Supplementary Figure 1 Residue profiles for the positions in the β-sheet motif. Residue types as percentages for the six positions
1.0 % Occupancy 0.8 0.6 0.4 0.2 0.0 ψ i-1 φ i ψ j φ j+1 ψ k-1 Motif position φ Supplementary Figure 1 Residue profiles for the positions in the β-sheet motif. Residue types as percentages for the six positions
Nature Structural & Molecular Biology: doi: /nsmb Supplementary Figure 1
 Supplementary Figure 1 Crystallization. a, Crystallization constructs of the ET B receptor are shown, with all of the modifications to the human wild-type the ET B receptor indicated. Residues interacting
Supplementary Figure 1 Crystallization. a, Crystallization constructs of the ET B receptor are shown, with all of the modifications to the human wild-type the ET B receptor indicated. Residues interacting
Goals. Structural Analysis of the EGR Family of Transcription Factors: Templates for Predicting Protein DNA Interactions
 Structural Analysis of the EGR Family of Transcription Factors: Templates for Predicting Protein DNA Interactions Jamie Duke 1,2 and Carlos Camacho 3 1 Bioengineering and Bioinformatics Summer Institute,
Structural Analysis of the EGR Family of Transcription Factors: Templates for Predicting Protein DNA Interactions Jamie Duke 1,2 and Carlos Camacho 3 1 Bioengineering and Bioinformatics Summer Institute,
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Comparing crystal structure of M.HhaI with and without DNA1, 2 (PDBID:1hmy and PDBID:2hmy),
,") Supporting Information 1. Constructing the starting structure Comparing crystal structure of M.HhaI with and without DNA1, 2 (PDBID:1hmy and PDBID:2hmy), we find that: the RMSD of overall structure and
Supporting Information 1. Constructing the starting structure Comparing crystal structure of M.HhaI with and without DNA1, 2 (PDBID:1hmy and PDBID:2hmy), we find that: the RMSD of overall structure and
Supplementary information for cloud computing approaches for prediction of ligand binding poses and pathways
 Supplementary information for cloud computing approaches for prediction of ligand binding poses and pathways Morgan Lawrenz 1, Diwakar Shukla 1,2 & Vijay S. Pande 1,2 1 Department of Chemistry, Stanford
Supplementary information for cloud computing approaches for prediction of ligand binding poses and pathways Morgan Lawrenz 1, Diwakar Shukla 1,2 & Vijay S. Pande 1,2 1 Department of Chemistry, Stanford
Structural and mechanistic insight into the substrate. binding from the conformational dynamics in apo. and substrate-bound DapE enzyme
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 215 Structural and mechanistic insight into the substrate binding from the conformational
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 215 Structural and mechanistic insight into the substrate binding from the conformational
Carbazole Derivatives Binding to c-kit G-quadruplex DNA
 Supplementary Materials Carbazole Derivatives Binding to c-kit G-quadruplex DNA Agata Głuszyńska 1, *, Bernard Juskowiak 1, Martyna Kuta-Siejkowska 2, Marcin Hoffmann 2 and Shozeb Haider 3 1 Laboratory
Supplementary Materials Carbazole Derivatives Binding to c-kit G-quadruplex DNA Agata Głuszyńska 1, *, Bernard Juskowiak 1, Martyna Kuta-Siejkowska 2, Marcin Hoffmann 2 and Shozeb Haider 3 1 Laboratory
SUPPLEMENTARY INFORMATION
 Supplementary Results DNA binding property of the SRA domain was examined by an electrophoresis mobility shift assay (EMSA) using synthesized 12-bp oligonucleotide duplexes containing unmodified, hemi-methylated,
Supplementary Results DNA binding property of the SRA domain was examined by an electrophoresis mobility shift assay (EMSA) using synthesized 12-bp oligonucleotide duplexes containing unmodified, hemi-methylated,
Destruction of Amyloid Fibrils by Graphene through Penetration and Extraction of Peptides
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2015 Destruction of Amyloid Fibrils by Graphene through Penetration and Extraction of Peptides Zaixing
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2015 Destruction of Amyloid Fibrils by Graphene through Penetration and Extraction of Peptides Zaixing
Electronic Supplementary Information. A biologically relevant fluorescent probe to assess the binding of ceramide to the CERT transfer protein
 This journal is The Royal Society of Chemistry 20 Electronic Supplementary Information A biologically relevant fluorescent probe to assess the binding of ceramide to the CERT transfer protein Stéphanie
This journal is The Royal Society of Chemistry 20 Electronic Supplementary Information A biologically relevant fluorescent probe to assess the binding of ceramide to the CERT transfer protein Stéphanie
Structure Investigation of Fam20C, a Golgi Casein Kinase
 Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Electronic Supplementary Information
 Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2016 Electronic Supplementary Information Active Site Structure and Absorption Spectrum of Channelrhodopsin-2
Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2016 Electronic Supplementary Information Active Site Structure and Absorption Spectrum of Channelrhodopsin-2
A Brief Guide to All Atom/Coarse Grain Simulations 1.1
 A Brief Guide to All Atom/Coarse Grain Simulations 1.1 Contents 1. Introduction 2. AACG simulations 2.1. Capabilities and limitations 2.2. AACG commands 2.3. Visualization and analysis 2.4. AACG simulation
A Brief Guide to All Atom/Coarse Grain Simulations 1.1 Contents 1. Introduction 2. AACG simulations 2.1. Capabilities and limitations 2.2. AACG commands 2.3. Visualization and analysis 2.4. AACG simulation
SANDRO BOTTARO, PAVEL BANÁŠ, JIŘÍ ŠPONER, AND GIOVANNI BUSSI
 FREE ENERGY LANDSCAPE OF GAGA AND UUCG RNA TETRALOOPS. SANDRO BOTTARO, PAVEL BANÁŠ, JIŘÍ ŠPONER, AND GIOVANNI BUSSI Contents 1. Supplementary Text 1 2 Sample PLUMED input file. 2. Supplementary Figure
FREE ENERGY LANDSCAPE OF GAGA AND UUCG RNA TETRALOOPS. SANDRO BOTTARO, PAVEL BANÁŠ, JIŘÍ ŠPONER, AND GIOVANNI BUSSI Contents 1. Supplementary Text 1 2 Sample PLUMED input file. 2. Supplementary Figure
SUPPLEMENTARY INFORMATION
 SUPPLEMENTARY INFORMATION Structure of human carbamoyl phosphate synthetase: deciphering the on/off switch of human ureagenesis Sergio de Cima, Luis M. Polo, Carmen Díez-Fernández, Ana I. Martínez, Javier
SUPPLEMENTARY INFORMATION Structure of human carbamoyl phosphate synthetase: deciphering the on/off switch of human ureagenesis Sergio de Cima, Luis M. Polo, Carmen Díez-Fernández, Ana I. Martínez, Javier
Prediction and refinement of NMR structures from sparse experimental data
 Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
β1 Structure Prediction and Validation
 13 Chapter 2 β1 Structure Prediction and Validation 2.1 Overview Over several years, GPCR prediction methods in the Goddard lab have evolved to keep pace with the changing field of GPCR structure. Despite
13 Chapter 2 β1 Structure Prediction and Validation 2.1 Overview Over several years, GPCR prediction methods in the Goddard lab have evolved to keep pace with the changing field of GPCR structure. Despite
Energetics of Ion Permeation in an Open-Activated TRPV1 Channel
 Biophysical Journal, Volume 111 Supplemental Information Energetics of Ion Permeation in an Open-Activated TRPV1 Channel Christian Jorgensen, Simone Furini, and Carmen Domene Supporting Material Energetics
Biophysical Journal, Volume 111 Supplemental Information Energetics of Ion Permeation in an Open-Activated TRPV1 Channel Christian Jorgensen, Simone Furini, and Carmen Domene Supporting Material Energetics
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Medical Research, Medicinal Chemistry, University of Leuven, Leuven, Belgium.
 Supporting Information Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins Muhammad Usman Mirza
Supporting Information Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins Muhammad Usman Mirza
SUPPLEMENTARY INFORMATION
 doi:10.1038/nature17991 Supplementary Discussion Structural comparison with E. coli EmrE The DMT superfamily includes a wide variety of transporters with 4-10 TM segments 1. Since the subfamilies of the
doi:10.1038/nature17991 Supplementary Discussion Structural comparison with E. coli EmrE The DMT superfamily includes a wide variety of transporters with 4-10 TM segments 1. Since the subfamilies of the
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Time-dependence of key H-bond/electrostatic interaction distances in the sirna5-hago2 complexes... Page S14
 Supporting Information Probing the Binding Interactions between Chemically Modified sirnas and Human Argonaute 2 Using Microsecond Molecular Dynamics Simulations S. Harikrishna* and P. I. Pradeepkumar*
Supporting Information Probing the Binding Interactions between Chemically Modified sirnas and Human Argonaute 2 Using Microsecond Molecular Dynamics Simulations S. Harikrishna* and P. I. Pradeepkumar*
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Modeling Biological Systems Opportunities for Computer Scientists
 Modeling Biological Systems Opportunities for Computer Scientists Filip Jagodzinski RBO Tutorial Series 25 June 2007 Computer Science Robotics & Biology Laboratory Protein: πρώτα, "prota, of Primary Importance
Modeling Biological Systems Opportunities for Computer Scientists Filip Jagodzinski RBO Tutorial Series 25 June 2007 Computer Science Robotics & Biology Laboratory Protein: πρώτα, "prota, of Primary Importance
Supplementary Information Intrinsic Localized Modes in Proteins
 Supplementary Information Intrinsic Localized Modes in Proteins Adrien Nicolaï 1,, Patrice Delarue and Patrick Senet, 1 Department of Physics, Applied Physics and Astronomy, Rensselaer Polytechnic Institute,
Supplementary Information Intrinsic Localized Modes in Proteins Adrien Nicolaï 1,, Patrice Delarue and Patrick Senet, 1 Department of Physics, Applied Physics and Astronomy, Rensselaer Polytechnic Institute,
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Effect of intracellular loop 3 on intrinsic dynamics of human β 2 -adrenergic receptor. Ozcan et al.
 Effect of intracellular loop 3 on intrinsic dynamics of human β 2 -adrenergic receptor Ozcan et al. Ozcan et al. BMC Structural Biology 2013, 13:29 Ozcan et al. BMC Structural Biology 2013, 13:29 RESEARCH
Effect of intracellular loop 3 on intrinsic dynamics of human β 2 -adrenergic receptor Ozcan et al. Ozcan et al. BMC Structural Biology 2013, 13:29 Ozcan et al. BMC Structural Biology 2013, 13:29 RESEARCH
β 2 -microglobulin fibrillation?
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2014 Can small hydrophobic gold nanoparticles inhibit β 2 -microglobulin fibrillation? G. Brancolini,,
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2014 Can small hydrophobic gold nanoparticles inhibit β 2 -microglobulin fibrillation? G. Brancolini,,
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
of the Guanine Nucleotide Exchange Factor FARP2
 Structure, Volume 21 Supplemental Information Structural Basis for Autoinhibition of the Guanine Nucleotide Exchange Factor FARP2 Xiaojing He, Yi-Chun Kuo, Tyler J. Rosche, and Xuewu Zhang Inventory of
Structure, Volume 21 Supplemental Information Structural Basis for Autoinhibition of the Guanine Nucleotide Exchange Factor FARP2 Xiaojing He, Yi-Chun Kuo, Tyler J. Rosche, and Xuewu Zhang Inventory of
Molecular dynamics simulations of anti-aggregation effect of ibuprofen. Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov
 Biophysical Journal, Volume 98 Supporting Material Molecular dynamics simulations of anti-aggregation effect of ibuprofen Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov Supplemental
Biophysical Journal, Volume 98 Supporting Material Molecular dynamics simulations of anti-aggregation effect of ibuprofen Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov Supplemental
Molecular Modeling lecture 2
 Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
Semi Empirical Force Fields and Their Limitations. Potential Energy Surface (PES)
") Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
SUPPLEMENTARY INFORMATION
 SUPPLEMENTARY INFORMATION doi:10.1038/nature11524 Supplementary discussion Functional analysis of the sugar porter family (SP) signature motifs. As seen in Fig. 5c, single point mutation of the conserved
SUPPLEMENTARY INFORMATION doi:10.1038/nature11524 Supplementary discussion Functional analysis of the sugar porter family (SP) signature motifs. As seen in Fig. 5c, single point mutation of the conserved
Improving Protein Function Prediction with Molecular Dynamics Simulations. Dariya Glazer Russ Altman
 Improving Protein Function Prediction with Molecular Dynamics Simulations Dariya Glazer Russ Altman Motivation Sometimes the 3D structure doesn t score well for a known function. The experimental structure
Improving Protein Function Prediction with Molecular Dynamics Simulations Dariya Glazer Russ Altman Motivation Sometimes the 3D structure doesn t score well for a known function. The experimental structure
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Building 3D models of proteins
 Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
SUPPLEMENTARY INFORMATION
 SUPPLEMENTARY INFORMATION doi:10.1038/nature11539 Supplementary Figure 1 Schematic representation of plant (A) and mammalian (B) P 2B -ATPase domain organization. Actuator (A-), nucleotide binding (N-),
SUPPLEMENTARY INFORMATION doi:10.1038/nature11539 Supplementary Figure 1 Schematic representation of plant (A) and mammalian (B) P 2B -ATPase domain organization. Actuator (A-), nucleotide binding (N-),
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
L718Q mutant EGFR escapes covalent inhibition by stabilizing. a non-reactive conformation of the lung cancer drug. osimertinib
 Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2018 Electronic Supplementary Information (ESI) for L718Q mutant EGFR escapes covalent inhibition
Electronic Supplementary Material (ESI) for Chemical Science. This journal is The Royal Society of Chemistry 2018 Electronic Supplementary Information (ESI) for L718Q mutant EGFR escapes covalent inhibition
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
Novel Quinoline and Naphthalene derivatives as potent Antimycobacterial agents
 ovel Quinoline and aphthalene derivatives as potent Antimycobacterial agents Ram Shankar Upadhayaya a, Jaya Kishore Vandavasi a, Ramakant A. Kardile a, Santosh V. Lahore a, Shailesh S. Dixit a, Hemantkumar
ovel Quinoline and aphthalene derivatives as potent Antimycobacterial agents Ram Shankar Upadhayaya a, Jaya Kishore Vandavasi a, Ramakant A. Kardile a, Santosh V. Lahore a, Shailesh S. Dixit a, Hemantkumar
Substrate Binding and Formation of an Occluded State in the Leucine Transporter
 1600 Biophysical Journal Volume 94 March 2008 1600 1612 Substrate Binding and Formation of an Occluded State in the Leucine Transporter Leyla Celik,* y Birgit Schiøtt, y and Emad Tajkhorshid* *Department
1600 Biophysical Journal Volume 94 March 2008 1600 1612 Substrate Binding and Formation of an Occluded State in the Leucine Transporter Leyla Celik,* y Birgit Schiøtt, y and Emad Tajkhorshid* *Department
Supporting Information
 Supporting Information The Predicted Ensemble of Low-Energy Conformations of Human Somatostatin Receptor Subtype 5 and the Binding of Antagonists Sijia S. Dong, [a] Ravinder Abrol, [a, b] and William A.
Supporting Information The Predicted Ensemble of Low-Energy Conformations of Human Somatostatin Receptor Subtype 5 and the Binding of Antagonists Sijia S. Dong, [a] Ravinder Abrol, [a, b] and William A.
How is molecular dynamics being used in life sciences? Davide Branduardi
 How is molecular dynamics being used in life sciences? Davide Branduardi davide.branduardi@schrodinger.com Exploring molecular processes with MD Drug discovery and design Protein-protein interactions Protein-DNA
How is molecular dynamics being used in life sciences? Davide Branduardi davide.branduardi@schrodinger.com Exploring molecular processes with MD Drug discovery and design Protein-protein interactions Protein-DNA
Insights on Antibiotic Translocation : Molecular Dynamics Simulations
 I Insights on Antibiotic Translocation : Molecular Dynamics Simulations Amit Kumar, Eric Hajjar,Enrico Spiga, Francesa Collu, Paolo Ruggerone & Matteo Ceccarelli Marseille : 11 04 2008 Outline METHODS
I Insights on Antibiotic Translocation : Molecular Dynamics Simulations Amit Kumar, Eric Hajjar,Enrico Spiga, Francesa Collu, Paolo Ruggerone & Matteo Ceccarelli Marseille : 11 04 2008 Outline METHODS
Visualization of Macromolecular Structures
 Visualization of Macromolecular Structures Present by: Qihang Li orig. author: O Donoghue, et al. Structural biology is rapidly accumulating a wealth of detailed information. Over 60,000 high-resolution
Visualization of Macromolecular Structures Present by: Qihang Li orig. author: O Donoghue, et al. Structural biology is rapidly accumulating a wealth of detailed information. Over 60,000 high-resolution
Supporting Information for. Models for the Metal Transfer Complex of the N-terminal Region of CusB and. CusF
 Supporting Information for Models for the Metal Transfer Complex of the N-terminal Region of CusB and CusF Melek N. Ucisik, Dhruva K. Chakravorty, and Kenneth M. Merz Jr. * Department of Chemistry and
Supporting Information for Models for the Metal Transfer Complex of the N-terminal Region of CusB and CusF Melek N. Ucisik, Dhruva K. Chakravorty, and Kenneth M. Merz Jr. * Department of Chemistry and
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Thiamine Diphosphate Activation in. 1-deoxy-D-xylulose 5-Phosphate Synthase: Insights. into the Mechanism and Underlying Intermolecular.
 Thiamine Diphosphate Activation in 1-deoxy-D-xylulose 5-Phosphate Synthase: Insights into the Mechanism and Underlying Intermolecular Interactions Justin K. White,, Sumit Handa,,, Sai Lakshmana Vankayala,,
Thiamine Diphosphate Activation in 1-deoxy-D-xylulose 5-Phosphate Synthase: Insights into the Mechanism and Underlying Intermolecular Interactions Justin K. White,, Sumit Handa,,, Sai Lakshmana Vankayala,,
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Bulk behaviour. Alanine. FIG. 1. Chemical structure of the RKLPDA peptide. Numbers on the left mark alpha carbons.
 Bulk behaviour To characterise the conformational behaviour of the peptide, first we looked at the statistics of alpha carbons and the torsion angles. Then they were correlated with positions of positively
Bulk behaviour To characterise the conformational behaviour of the peptide, first we looked at the statistics of alpha carbons and the torsion angles. Then they were correlated with positions of positively
A dynamic interaction process between KaiA and KaiC is critical to
 A dynamic interaction process between KaiA and KaiC is critical to the cyanobacterial circadian oscillator Pei Dong 1,2, Ying Fan 2, Jianqiang Sun 3, Mengting Lv 2, Ming Yi 4, Xiao Tan 1,2, Sen Liu 1,2,*
A dynamic interaction process between KaiA and KaiC is critical to the cyanobacterial circadian oscillator Pei Dong 1,2, Ying Fan 2, Jianqiang Sun 3, Mengting Lv 2, Ming Yi 4, Xiao Tan 1,2, Sen Liu 1,2,*
Outline. The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation. Unfolded Folded. What is protein folding?
 The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation By Jun Shimada and Eugine Shaknovich Bill Hawse Dr. Bahar Elisa Sandvik and Mehrdad Safavian Outline Background on protein
The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation By Jun Shimada and Eugine Shaknovich Bill Hawse Dr. Bahar Elisa Sandvik and Mehrdad Safavian Outline Background on protein
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Bioinformatics. Macromolecular structure
 Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Supplementary Figures:
 Supplementary Figures: Supplementary Figure 1: The two strings converge to two qualitatively different pathways. A) Models of active (red) and inactive (blue) states used as end points for the string calculations
Supplementary Figures: Supplementary Figure 1: The two strings converge to two qualitatively different pathways. A) Models of active (red) and inactive (blue) states used as end points for the string calculations
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Protein structure analysis. Risto Laakso 10th January 2005
 Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Multi-Scale Hierarchical Structure Prediction of Helical Transmembrane Proteins
 Multi-Scale Hierarchical Structure Prediction of Helical Transmembrane Proteins Zhong Chen Dept. of Biochemistry and Molecular Biology University of Georgia, Athens, GA 30602 Email: zc@csbl.bmb.uga.edu
Multi-Scale Hierarchical Structure Prediction of Helical Transmembrane Proteins Zhong Chen Dept. of Biochemistry and Molecular Biology University of Georgia, Athens, GA 30602 Email: zc@csbl.bmb.uga.edu
Can protein model accuracy be. identified? NO! CBS, BioCentrum, Morten Nielsen, DTU
 Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality