The Molecular Dynamics Method
|
|
|
- Roy Powell
- 5 years ago
- Views:
Transcription



1 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport
2 Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation (x)
3 Classical Molecular Dynamics r ( t + δ t) = r( t) + v( t) δt v ( t + δ t) = v( t) + a( t) δt a ( t ) = F(t) / m d F = U (r) dr
4 Classical Molecular Dynamics U ( r) = 1 4πε 0 q q Coulomb interaction i r ij j U ( r) = ε ij &, R $ * $ % + r min, ij ij ) ' ( 12, R 2* + r van der Waals interaction min, ij ij ) ' ( 6 #!! "
5 Classical Molecular Dynamics U(r) = 1 + q i q $ j + ε ij - R min,ij 4πε 0 r ij - & % r, ij ' ) ( 12 $ 2 R min,ij & % r ij ' ) ( / % F(r) = ' 1 q i q j 2 ' 4πε 0 r & ij 12 ε ij r ij + % - - ',& R min,ij r ij ( * ) 12 % R min,ij ' & r ij ( * ) 6.( 0 * 0 * ˆ /) r ij
6 Classical Molecular Dynamics
7 Classical Molecular Dynamics Bond definitions, atom types, atom names, parameters,.
8 What is a Force Field? In molecular dynamics a molecule is described as a series of charged points (atoms) linked by springs (bonds). To describe the time evolution of bond lengths, bond angles and torsions, also the non-bonding van der Waals and elecrostatic interactions between atoms, one uses a force field. The force field is a collection of equations and associated constants designed to reproduce molecular geometry and selected properties of tested structures.
9 Energy Functions U bond = oscillations about the equilibrium bond length U angle = oscillations of 3 atoms about an equilibrium bond angle U dihedral = torsional rotation of 4 atoms about a central bond U nonbond = non-bonded energy terms (electrostatics and Lenard-Jones)





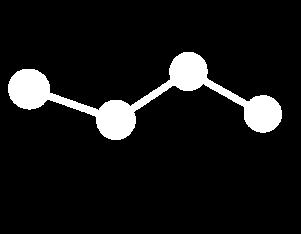
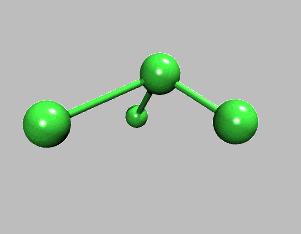
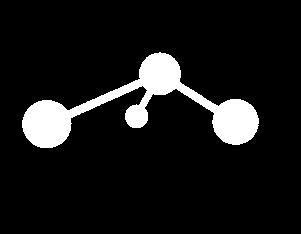
10 Energy Terms Described in the CHARMm Force Field Bond Angle Dihedral Improper
 2 V dihedral = K ( 1+ cos( nφ δ )) φ")
11 Interactions between bonded atoms V angle = K θ ( ) 2 θ θ o V bond = K b ( b b o ) 2 V dihedral = K ( 1+ cos( nφ δ )) φ
12 Classical Dynamics F=ma at 300K Energy function: used to determine the force on each atom: yields a set of 3N coupled 2 nd -order differential equations that can be propagated forward (or backward) in time. Initial coordinates obtained from crystal structure, velocities taken at random from Boltzmann distribution. Maintain appropriate temperature by adjusting velocities.
13 Langevin Dynamics Langevin dynamics deals with each atom separately, balancing a small friction term with Gaussian noise to control temperature:

 Hinge")


14 The most serious bottleneck steps Rotation of buried sidechains Local denaturations Allosteric transitions s ms µs 10 9 (year) Hinge bending Rotation of surface sidechains Elastic vibrations ns ps 10 6 (day) 10 3 SPEED LIMIT Bond stretching Molecular dynamics timestep fs 10 0 δt = 1 fs
15 Potential Energy (hyper)surface r ( t + δ t) = r( t) + v( t) δt Energy U(x) Conformation (x)

16 V bond = K b ( b b ) 2 o Chemical type K bond b o C-C 100 kcal/mole/å Å C=C 200 kcal/mole/å Å C=C 400 kcal/mole/å Å Bond Energy versus Bond length 400 Potential Energy, kcal/mol Single Bond Double Bond Triple Bond Bond length, Å Bond angles and improper terms have similar quadratic forms, but with softer spring constants. The force constants can be obtained from vibrational analysis of the molecule (experimentally or theoretically).

17 Dihedral Potential V dihedral = K ( 1+ cos( nφ δ )) φ Dihedral energy versus dihedral angle 20 Potential Energy, kcal/mol K=10, n=1 K=5, n=2 K=2.5, N= Dihedral Angle, degrees δ = 0
18 van der Waals interaction Lennard-Jones Energy versus Distance 0.9 Interaction Energy, kcal/mol e=0.2,rmin= eps,i,j Rmin,i,j Distance, Å ε ij *#,, % + $ R min,ij r ij & ( ' 12 # 2 R min,ij % $ r ij & ( ' 6 - / /. Short range
19 Electrostatic Energy versus Distance Interaction energy, kcal/mol q1=1, q2=1 q1=-1, q2= Distance, Å From MacKerell Note that the effect is long range.
20 Steps in a Typical MD Simulation 1. Prepare molecule Read in pdb and psf file 2. Minimization Reconcile the structure with force field used (T = 0) 3. Heating Raise temperature of the system 4. Equilibration Ensure system is stable 5. Dynamics Simulate under desired conditions (NVE, NpT, etc) Collect your data 6. Analysis Evaluate observables (macroscopic level properties) Or relate to single molecule experiments Solvation



21 Preparing Your System for MD Solvation Biological activity is the result of interactions between molecules and occurs at the interfaces between molecules (protein-protein, protein- DNA, protein-solvent, DNA-solvent, etc). Why model solvation? many biological processes occur in aqueous solution solvation effects play a crucial role in determining molecular conformation, electronic properties, binding energies, etc How to model solvation? explicit treatment: solvent molecules are added to the molecular system implicit treatment: solvent is modeled as a continuum dielectric
22 Maxwell Distribution of Atomic Velocities
23 CHARMM Potential Function PDB file geometry Topology PSF file parameters Parameter file
24 File Format/Structure The structure of a pdb file The structure of a psf file The topology file The parameter file Connection to potential energy terms
25 Structure of a PDB file resname index name chain resid X Y Z segname ATOM 22 N ALA B BH ATOM 23 HN ALA B BH ATOM 24 CA ALA B BH ATOM 25 HA ALA B BH ATOM 26 CB ALA B BH ATOM 27 HB1 ALA B BH ATOM 28 HB2 ALA B BH ATOM 29 HB3 ALA B BH ATOM 30 C ALA B BH ATOM 31 O ALA B BH ATOM 32 N ALA B BH ATOM 33 HN ALA B BH ATOM 34 CA ALA B BH ATOM 35 HA ALA B BH ATOM 36 CB ALA B BH ATOM 37 HB1 ALA B BH ATOM 38 HB2 ALA B BH ATOM 39 HB3 ALA B BH ATOM 40 C ALA B BH ATOM 41 O ALA B BH >>> It is an ascii, fixed-format file <<< No connectivity information
26 Checking file structures PDB file Topology file PSF file Parameter file
27 Parameter Optimization Strategies Check if it has been parameterized by somebody else Literature Google Minimal optimization By analogy (i.e. direct transfer of known parameters) Quick, starting point - dihedrals?? Maximal optimization Time-consuming Requires appropriate experimental and target data Choice based on goal of the calculations Minimal database screening NMR/X-ray structure determination Maximal free energy calculations, mechanistic studies, subtle environmental effects
28 Major Difficulties in Simulating Size problem Biological Systems Biological Systems are Complex Natural environment: Membrane, solvent, ions, Many biosystems function in assemblies Photosynthetic apparatus Nuclear receptors, GPCRs, Time scale problem Many biological events happen at µs ms time scales Signaling and other regulatory mechanisms Protein folding


29 Large Systems F 1 unit: 330,000 atoms β α β γ δ ε F 0 unit: 110,000 atoms a c 10 ~ 10 millisecond timescale ATP synthase



")
 SGI Origin")
")
")

30 Size scale can be addressed effectively by larger clusters of computers HP 735 cluster 12 processors (1993)? Blue Waters (UIUC) 200,000+ processors (2012) SGI Origin processors (1997) Anton/DESHAW/PSC 512 processors (2010) PSC LeMieux AlphaServer SC 3000 processors (2002) Ranger/Kraken ~60,000 processors (2007)
 D wat = 2.5 x 10-5 cm 2.")
 ~9 orders of magnitude slower")
31 Lipid Diffusion in a Membrane D lip = 10-8 cm 2.s -1 (50 Å in ~ 5 x 10-6 s) D wat = 2.5 x 10-5 cm 2.s -1 Modeling mixed lipid bilayers! Once in several hours! (~ 50 Å in ~ 10 4 s) ~9 orders of magnitude slower ensuring bilayer asymmetry
32 Technical difficulties in Simulations of Biological Membranes Time scale Heterogeneity of biological membranes L 60 x 60 Å Pure POPE 5 ns ~100,000 atoms

33 Coarse-grained modeling of lipids 150 particles 9 particles! Also, increasing the time step by orders of magnitude.

34 by: J. Siewert-Jan Marrink and Alan E. Mark, University of Groningen, The Netherlands
35 Steered Molecular Dynamics Single Molecule Experiments: AFM Accelerating Events Ligand Binding/Unbinding Unfolding Experiments Applying Surface Tension Torque Application Pressure induction Inducing Large Domain Conformational Changes Constant-force: f(t) = C Constant-velocity: f(t) = k [vt (x t - x 0 )]



36 displacement applied force A wide variety of events that are inaccessible to conventional molecular dynamics simulations can be probed. The system will be driven, however, away from equilibrium, resulting in problems in describing the energy landscape associated with the event of interest. Second law of thermodynamics W ΔG
37 Jarzynski s Equality Transition between two equilibrium states λ = λ i T λ = λ(t) T work W heat Q λ = λ f e -βw p(w) ΔG W W ΔG Δ G = G G f i p(w) C. Jarzynski, Phys. Rev. Lett., 78, 2690 (1997) C. Jarzynski, Phys. Rev. E, 56, 5018 (1997) e βw = e βδg In principle, it is possible to obtain free energy surfaces from repeated nonequilibrium experiments. β = 1 k T B

38 Aquaporins Membrane water channels
39
40 Water Bipolar Configuration in Aquaporins
41
42 R E M E M B E R: One of the most useful advantages of simulations over experiments is that you can modify the system as you wish: You can do modifications that are not even possible at all in reality! This is a powerful technique to test hypotheses developed during your simulations. Use it!

43 Electrostatic Stabilization of Water Bipolar Arrangement
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Don t forget to bring your MD tutorial. Potential Energy (hyper)surface
surface") Don t forget to bring your MD tutorial Lab session starts at 1pm You will have to finish an MD/SMD exercise on α-conotoxin in oxidized and reduced forms Potential Energy (hyper)surface What is Force? Energy
Don t forget to bring your MD tutorial Lab session starts at 1pm You will have to finish an MD/SMD exercise on α-conotoxin in oxidized and reduced forms Potential Energy (hyper)surface What is Force? Energy
The Molecular Dynamics Method
 H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
Principles and Applications of Molecular Dynamics Simulations with NAMD
 Principles and Applications of Molecular Dynamics Simulations with NAMD Nov. 14, 2016 Computational Microscope NCSA supercomputer JC Gumbart Assistant Professor of Physics Georgia Institute of Technology
Principles and Applications of Molecular Dynamics Simulations with NAMD Nov. 14, 2016 Computational Microscope NCSA supercomputer JC Gumbart Assistant Professor of Physics Georgia Institute of Technology
Force Fields for MD simulations
 Force Fields for MD simulations Topology/parameter files Where do the numbers an MD code uses come from? ow to make topology files for ligands, cofactors, special amino acids, ow to obtain/develop missing
Force Fields for MD simulations Topology/parameter files Where do the numbers an MD code uses come from? ow to make topology files for ligands, cofactors, special amino acids, ow to obtain/develop missing
The Molecular Dynamics Method
 H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
The Molecular Dynamics Simulation Process
 The Molecular Dynamics Simulation Process For textbooks see: M.P. Allen and D.J. Tildesley. Computer Simulation of Liquids.Oxford University Press, New York, 1987. D. Frenkel and B. Smit. Understanding
The Molecular Dynamics Simulation Process For textbooks see: M.P. Allen and D.J. Tildesley. Computer Simulation of Liquids.Oxford University Press, New York, 1987. D. Frenkel and B. Smit. Understanding
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
The Computational Microscope
 The Computational Microscope Computational microscope views at atomic resolution... Rs SER RER C E M RER N GA L... how living cells maintain health and battle disease John Stone Our Microscope is Made
The Computational Microscope Computational microscope views at atomic resolution... Rs SER RER C E M RER N GA L... how living cells maintain health and battle disease John Stone Our Microscope is Made
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
Computational Studies of the Photoreceptor Rhodopsin. Scott E. Feller Wabash College
 Computational Studies of the Photoreceptor Rhodopsin Scott E. Feller Wabash College Rhodopsin Photocycle Dark-adapted Rhodopsin hn Isomerize retinal Photorhodopsin ~200 fs Bathorhodopsin Meta-II ms timescale
Computational Studies of the Photoreceptor Rhodopsin Scott E. Feller Wabash College Rhodopsin Photocycle Dark-adapted Rhodopsin hn Isomerize retinal Photorhodopsin ~200 fs Bathorhodopsin Meta-II ms timescale
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Dynamics Simulations. Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia
 Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Computational Structural Biology and Molecular Simulation. Introduction to VMD Molecular Visualization and Analysis
 Computational Structural Biology and Molecular Simulation Introduction to VMD Molecular Visualization and Analysis Emad Tajkhorshid Department of Biochemistry, Beckman Institute, Center for Computational
Computational Structural Biology and Molecular Simulation Introduction to VMD Molecular Visualization and Analysis Emad Tajkhorshid Department of Biochemistry, Beckman Institute, Center for Computational
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Molecular Dynamics Simulation of a Nanoconfined Water Film
 Molecular Dynamics Simulation of a Nanoconfined Water Film Kyle Lindquist, Shu-Han Chao May 7, 2013 1 Introduction The behavior of water confined in nano-scale environment is of interest in many applications.
Molecular Dynamics Simulation of a Nanoconfined Water Film Kyle Lindquist, Shu-Han Chao May 7, 2013 1 Introduction The behavior of water confined in nano-scale environment is of interest in many applications.
Protein Structure Analysis
 BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Computational Modeling of Protein Dynamics. Yinghao Wu Department of Systems and Computational Biology Albert Einstein College of Medicine Fall 2014
 Computational Modeling of Protein Dynamics Yinghao Wu Department of Systems and Computational Biology Albert Einstein College of Medicine Fall 2014 Outline Background of protein dynamics Basic computational
Computational Modeling of Protein Dynamics Yinghao Wu Department of Systems and Computational Biology Albert Einstein College of Medicine Fall 2014 Outline Background of protein dynamics Basic computational
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia
 University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
Exercise 2: Solvating the Structure Before you continue, follow these steps: Setting up Periodic Boundary Conditions
 Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Water models in classical simulations
 Water models in classical simulations Maria Fyta Institut für Computerphysik, Universität Stuttgart Stuttgart, Germany Water transparent, odorless, tasteless and ubiquitous really simple: two H atoms attached
Water models in classical simulations Maria Fyta Institut für Computerphysik, Universität Stuttgart Stuttgart, Germany Water transparent, odorless, tasteless and ubiquitous really simple: two H atoms attached
Energy functions and their relationship to molecular conformation. CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror
 Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Outline Energy functions for proteins (or biomolecular systems more generally)
Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Outline Energy functions for proteins (or biomolecular systems more generally)
Folding WT villin in silico Three folding simulations reach native state within 5-8 µs
 Modeling of Cryo-EM Maps Workshop Baylor College of Medicine Klaus Schulten, U. Illinois at Urbana-Champaign Molecular Modeling Flexible Fitting 1: Introduction to Molecular Dynamics MD force field Equation
Modeling of Cryo-EM Maps Workshop Baylor College of Medicine Klaus Schulten, U. Illinois at Urbana-Champaign Molecular Modeling Flexible Fitting 1: Introduction to Molecular Dynamics MD force field Equation
Coupling the Level-Set Method with Variational Implicit Solvent Modeling of Molecular Solvation
 Coupling the Level-Set Method with Variational Implicit Solvent Modeling of Molecular Solvation Bo Li Math Dept & CTBP, UCSD Li-Tien Cheng (Math, UCSD) Zhongming Wang (Math & Biochem, UCSD) Yang Xie (MAE,
Coupling the Level-Set Method with Variational Implicit Solvent Modeling of Molecular Solvation Bo Li Math Dept & CTBP, UCSD Li-Tien Cheng (Math, UCSD) Zhongming Wang (Math & Biochem, UCSD) Yang Xie (MAE,
Energy functions and their relationship to molecular conformation. CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror
 Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Yesterday s Nobel Prize: single-particle cryoelectron microscopy 2 Outline Energy
Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Yesterday s Nobel Prize: single-particle cryoelectron microscopy 2 Outline Energy
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Semi Empirical Force Fields and Their Limitations. Potential Energy Surface (PES)
") Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro. Karel Berka, Ph.D.
 4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
Molecular Mechanics. Yohann Moreau. November 26, 2015
 Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Simulation III
 Molecular Simulation III Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Molecular Dynamics Jeffry D. Madura Department of Chemistry & Biochemistry Center
Molecular Simulation III Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Molecular Dynamics Jeffry D. Madura Department of Chemistry & Biochemistry Center
Hyeyoung Shin a, Tod A. Pascal ab, William A. Goddard III abc*, and Hyungjun Kim a* Korea
 The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
This semester. Books
 Models mostly proteins from detailed to more abstract models Some simulation methods This semester Books None necessary for my group and Prof Rarey Molecular Modelling: Principles and Applications Leach,
Models mostly proteins from detailed to more abstract models Some simulation methods This semester Books None necessary for my group and Prof Rarey Molecular Modelling: Principles and Applications Leach,
arxiv: v1 [cond-mat.soft] 22 Oct 2007
![arxiv: v1 [cond-mat.soft] 22 Oct 2007](/thumbs/78/78401876.jpg "arxiv: v1 [cond-mat.soft] 22 Oct 2007") Conformational Transitions of Heteropolymers arxiv:0710.4095v1 [cond-mat.soft] 22 Oct 2007 Michael Bachmann and Wolfhard Janke Institut für Theoretische Physik, Universität Leipzig, Augustusplatz 10/11,
Conformational Transitions of Heteropolymers arxiv:0710.4095v1 [cond-mat.soft] 22 Oct 2007 Michael Bachmann and Wolfhard Janke Institut für Theoretische Physik, Universität Leipzig, Augustusplatz 10/11,
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Free energy, electrostatics, and the hydrophobic effect
 Protein Physics 2016 Lecture 3, January 26 Free energy, electrostatics, and the hydrophobic effect Magnus Andersson magnus.andersson@scilifelab.se Theoretical & Computational Biophysics Recap Protein structure
Protein Physics 2016 Lecture 3, January 26 Free energy, electrostatics, and the hydrophobic effect Magnus Andersson magnus.andersson@scilifelab.se Theoretical & Computational Biophysics Recap Protein structure
Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability
 Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability Dr. Andrew Lee UNC School of Pharmacy (Div. Chemical Biology and Medicinal Chemistry) UNC Med
Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability Dr. Andrew Lee UNC School of Pharmacy (Div. Chemical Biology and Medicinal Chemistry) UNC Med
Computational Molecular Biophysics. Computational Biophysics, GRS Jülich SS 2013
 Computational Molecular Biophysics Computational Biophysics, GRS Jülich SS 2013 Computational Considerations Number of terms for different energy contributions / E el ~N ~N ~N ~N 2 Computational cost for
Computational Molecular Biophysics Computational Biophysics, GRS Jülich SS 2013 Computational Considerations Number of terms for different energy contributions / E el ~N ~N ~N ~N 2 Computational cost for
Molecular Dynamics. A very brief introduction
 Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Methods of Computer Simulation. Molecular Dynamics and Monte Carlo
 Molecular Dynamics Time is of the essence in biological processes therefore how do we understand time-dependent processes at the molecular level? How do we do this experimentally? How do we do this computationally?
Molecular Dynamics Time is of the essence in biological processes therefore how do we understand time-dependent processes at the molecular level? How do we do this experimentally? How do we do this computationally?
Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model. John Dood Hope College
 Water Model. John Dood Hope College") Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model John Dood Hope College What are MD simulations? Model and predict the structure and dynamics of large macromolecules.
Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model John Dood Hope College What are MD simulations? Model and predict the structure and dynamics of large macromolecules.
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Hands-on Course in Computational Structural Biology and Molecular Simulation BIOP590C/MCB590C. Course Details
 Hands-on Course in Computational Structural Biology and Molecular Simulation BIOP590C/MCB590C Emad Tajkhorshid Center for Computational Biology and Biophysics Email: emad@life.uiuc.edu or tajkhors@uiuc.edu
Hands-on Course in Computational Structural Biology and Molecular Simulation BIOP590C/MCB590C Emad Tajkhorshid Center for Computational Biology and Biophysics Email: emad@life.uiuc.edu or tajkhors@uiuc.edu
Gromacs Workshop Spring CSC
 Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
Variational Implicit Solvation of Biomolecules: From Theory to Numerical Computations
 Variational Implicit Solvation of Biomolecules: From Theory to Numerical Computations Bo Li Department of Mathematics and Center for Theoretical Biological Physics UC San Diego CECAM Workshop: New Perspectives
Variational Implicit Solvation of Biomolecules: From Theory to Numerical Computations Bo Li Department of Mathematics and Center for Theoretical Biological Physics UC San Diego CECAM Workshop: New Perspectives
Modeling Biological Systems Opportunities for Computer Scientists
 Modeling Biological Systems Opportunities for Computer Scientists Filip Jagodzinski RBO Tutorial Series 25 June 2007 Computer Science Robotics & Biology Laboratory Protein: πρώτα, "prota, of Primary Importance
Modeling Biological Systems Opportunities for Computer Scientists Filip Jagodzinski RBO Tutorial Series 25 June 2007 Computer Science Robotics & Biology Laboratory Protein: πρώτα, "prota, of Primary Importance
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D.
 3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
Protein Structures. 11/19/2002 Lecture 24 1
 Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Sunyia Hussain 06/15/2012 ChE210D final project. Hydration Dynamics at a Hydrophobic Surface. Abstract:
 Hydration Dynamics at a Hydrophobic Surface Sunyia Hussain 6/15/212 ChE21D final project Abstract: Water is the universal solvent of life, crucial to the function of all biomolecules. Proteins, membranes,
Hydration Dynamics at a Hydrophobic Surface Sunyia Hussain 6/15/212 ChE21D final project Abstract: Water is the universal solvent of life, crucial to the function of all biomolecules. Proteins, membranes,
LAMMPS Performance Benchmark on VSC-1 and VSC-2
 LAMMPS Performance Benchmark on VSC-1 and VSC-2 Daniel Tunega and Roland Šolc Institute of Soil Research, University of Natural Resources and Life Sciences VSC meeting, Neusiedl am See, February 27-28,
LAMMPS Performance Benchmark on VSC-1 and VSC-2 Daniel Tunega and Roland Šolc Institute of Soil Research, University of Natural Resources and Life Sciences VSC meeting, Neusiedl am See, February 27-28,
Applications of Molecular Dynamics
 June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation
 Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Why Proteins Fold. How Proteins Fold? e - ΔG/kT. Protein Folding, Nonbonding Forces, and Free Energy
 Why Proteins Fold Proteins are the action superheroes of the body. As enzymes, they make reactions go a million times faster. As versatile transport vehicles, they carry oxygen and antibodies to fight
Why Proteins Fold Proteins are the action superheroes of the body. As enzymes, they make reactions go a million times faster. As versatile transport vehicles, they carry oxygen and antibodies to fight
MD Thermodynamics. Lecture 12 3/26/18. Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky
 MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
Our Microscope is Made of...
 The Computational Microscope Computational microscope views at atomic resolution... Rs E M RER SER N RER C GA L... how living cells maintain health and battle disease John%Stone Our Microscope is Made
The Computational Microscope Computational microscope views at atomic resolution... Rs E M RER SER N RER C GA L... how living cells maintain health and battle disease John%Stone Our Microscope is Made
Review. CS/CME/BioE/Biophys/BMI 279 Nov. 30 and Dec. 5, 2017 Ron Dror
 Review CS/CME/BioE/Biophys/BMI 279 Nov. 30 and Dec. 5, 2017 Ron Dror 1 Information on the exam The exam will focus on high-level concepts You may use two (single-sided) pages of notes, but the emphasis
Review CS/CME/BioE/Biophys/BMI 279 Nov. 30 and Dec. 5, 2017 Ron Dror 1 Information on the exam The exam will focus on high-level concepts You may use two (single-sided) pages of notes, but the emphasis
Parallel Utility for Modeling of Molecular Aggregation
 Parallel Utility for Modeling of Molecular Aggregation Peter Spijker, Bart Markvoort and Peter Hilbers BioModeling and bioinformatics Eindhoven University of Technology Eindhoven,The Netherlands p.spijker@pumma.nl
Parallel Utility for Modeling of Molecular Aggregation Peter Spijker, Bart Markvoort and Peter Hilbers BioModeling and bioinformatics Eindhoven University of Technology Eindhoven,The Netherlands p.spijker@pumma.nl
Free energy simulations
 Free energy simulations Marcus Elstner and Tomáš Kubař January 14, 2013 Motivation a physical quantity that is of most interest in chemistry? free energies Helmholtz F or Gibbs G holy grail of computational
Free energy simulations Marcus Elstner and Tomáš Kubař January 14, 2013 Motivation a physical quantity that is of most interest in chemistry? free energies Helmholtz F or Gibbs G holy grail of computational
Introduction to Classical Molecular Dynamics. Giovanni Chillemi HPC department, CINECA
 Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
Biophysics II. Hydrophobic Bio-molecules. Key points to be covered. Molecular Interactions in Bio-molecular Structures - van der Waals Interaction
 Biophysics II Key points to be covered By A/Prof. Xiang Yang Liu Biophysics & Micro/nanostructures Lab Department of Physics, NUS 1. van der Waals Interaction 2. Hydrogen bond 3. Hydrophilic vs hydrophobic
Biophysics II Key points to be covered By A/Prof. Xiang Yang Liu Biophysics & Micro/nanostructures Lab Department of Physics, NUS 1. van der Waals Interaction 2. Hydrogen bond 3. Hydrophilic vs hydrophobic
Our Microscope is Made of...
 The Computational Microscope Computational microscope views at atomic resolution... Rs E M RER SER N RER C GA L... how living cells maintain health and battle disease John%Stone Our Microscope is Made
The Computational Microscope Computational microscope views at atomic resolution... Rs E M RER SER N RER C GA L... how living cells maintain health and battle disease John%Stone Our Microscope is Made
A generalized Born formalism for heterogeneous dielectric environments: Application to the implicit modeling of biological membranes
 THE JOURNAL OF CHEMICAL PHYSICS 122, 124706 2005 A generalized Born formalism for heterogeneous dielectric environments: Application to the implicit modeling of biological membranes Seiichiro Tanizaki
THE JOURNAL OF CHEMICAL PHYSICS 122, 124706 2005 A generalized Born formalism for heterogeneous dielectric environments: Application to the implicit modeling of biological membranes Seiichiro Tanizaki
Biomolecular modeling I
 2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
Biomolecules are dynamic no single structure is a perfect model
 Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Hands-on : Model Potential Molecular Dynamics
 Hands-on : Model Potential Molecular Dynamics OUTLINE 0. DL_POLY code introduction 0.a Input files 1. THF solvent molecule 1.a Geometry optimization 1.b NVE/NVT dynamics 2. Liquid THF 2.a Equilibration
Hands-on : Model Potential Molecular Dynamics OUTLINE 0. DL_POLY code introduction 0.a Input files 1. THF solvent molecule 1.a Geometry optimization 1.b NVE/NVT dynamics 2. Liquid THF 2.a Equilibration
INTERMOLECULAR AND SURFACE FORCES
 INTERMOLECULAR AND SURFACE FORCES SECOND EDITION JACOB N. ISRAELACHVILI Department of Chemical & Nuclear Engineering and Materials Department University of California, Santa Barbara California, USA ACADEMIC
INTERMOLECULAR AND SURFACE FORCES SECOND EDITION JACOB N. ISRAELACHVILI Department of Chemical & Nuclear Engineering and Materials Department University of California, Santa Barbara California, USA ACADEMIC
Protein Dynamics, Allostery and Function
 Protein Dynamics, Allostery and Function Lecture 3. Protein Dynamics Xiaolin Cheng UT/ORNL Center for Molecular Biophysics SJTU Summer School 2017 1 Obtaining Dynamic Information Experimental Approaches
Protein Dynamics, Allostery and Function Lecture 3. Protein Dynamics Xiaolin Cheng UT/ORNL Center for Molecular Biophysics SJTU Summer School 2017 1 Obtaining Dynamic Information Experimental Approaches
Solutions and Non-Covalent Binding Forces
 Chapter 3 Solutions and Non-Covalent Binding Forces 3.1 Solvent and solution properties Molecules stick together using the following forces: dipole-dipole, dipole-induced dipole, hydrogen bond, van der
Chapter 3 Solutions and Non-Covalent Binding Forces 3.1 Solvent and solution properties Molecules stick together using the following forces: dipole-dipole, dipole-induced dipole, hydrogen bond, van der
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water?
 Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
SECOND PUBLIC EXAMINATION. Honour School of Physics Part C: 4 Year Course. Honour School of Physics and Philosophy Part C C7: BIOLOGICAL PHYSICS
 2757 SECOND PUBLIC EXAMINATION Honour School of Physics Part C: 4 Year Course Honour School of Physics and Philosophy Part C C7: BIOLOGICAL PHYSICS TRINITY TERM 2013 Monday, 17 June, 2.30 pm 5.45 pm 15
2757 SECOND PUBLIC EXAMINATION Honour School of Physics Part C: 4 Year Course Honour School of Physics and Philosophy Part C C7: BIOLOGICAL PHYSICS TRINITY TERM 2013 Monday, 17 June, 2.30 pm 5.45 pm 15
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Van Gunsteren et al. Angew. Chem. Int. Ed. 45, 4064 (2006)
") Van Gunsteren et al. Angew. Chem. Int. Ed. 45, 4064 (2006) Martini Workshop 2015 Coarse Graining Basics Alex de Vries Every word or concept, clear as it may seem to be, has only a limited range of applicability
Van Gunsteren et al. Angew. Chem. Int. Ed. 45, 4064 (2006) Martini Workshop 2015 Coarse Graining Basics Alex de Vries Every word or concept, clear as it may seem to be, has only a limited range of applicability
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 1 David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Time/s Multi-Scale Modeling Based on SDSC Blue Horizon (SP3) 1.728 Tflops
1 CE 530 Molecular Simulation Lecture 1 David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Time/s Multi-Scale Modeling Based on SDSC Blue Horizon (SP3) 1.728 Tflops
2008 Biowerkzeug Ltd.
 2008 Biowerkzeug Ltd. 1 Contents Summary...3 1 Simulation...4 1.1 Setup...4 1.2 Output...4 2 Settings...5 3 Analysis...9 3.1 Setup...9 3.2 Input options...9 3.3 Descriptions...10 Please note that we cannot
2008 Biowerkzeug Ltd. 1 Contents Summary...3 1 Simulation...4 1.1 Setup...4 1.2 Output...4 2 Settings...5 3 Analysis...9 3.1 Setup...9 3.2 Input options...9 3.3 Descriptions...10 Please note that we cannot
A Brief Guide to All Atom/Coarse Grain Simulations 1.1
 A Brief Guide to All Atom/Coarse Grain Simulations 1.1 Contents 1. Introduction 2. AACG simulations 2.1. Capabilities and limitations 2.2. AACG commands 2.3. Visualization and analysis 2.4. AACG simulation
A Brief Guide to All Atom/Coarse Grain Simulations 1.1 Contents 1. Introduction 2. AACG simulations 2.1. Capabilities and limitations 2.2. AACG commands 2.3. Visualization and analysis 2.4. AACG simulation
T6.2 Molecular Mechanics
 T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
Biomolecular modeling I
 2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
What can be learnt from MD
 - 4.0 H-bond energy (kcal/mol) 0 What can be learnt from MD Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis ATPase, a molecular motor
- 4.0 H-bond energy (kcal/mol) 0 What can be learnt from MD Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis ATPase, a molecular motor
University of Groningen. Characterization of oil/water interfaces van Buuren, Aldert Roelf
 University of Groningen Characterization of oil/water interfaces van Buuren, Aldert Roelf IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it.
University of Groningen Characterization of oil/water interfaces van Buuren, Aldert Roelf IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it.
Scientific Computing II
 Scientific Computing II Molecular Dynamics Simulation Michael Bader SCCS Summer Term 2015 Molecular Dynamics Simulation, Summer Term 2015 1 Continuum Mechanics for Fluid Mechanics? Molecular Dynamics the
Scientific Computing II Molecular Dynamics Simulation Michael Bader SCCS Summer Term 2015 Molecular Dynamics Simulation, Summer Term 2015 1 Continuum Mechanics for Fluid Mechanics? Molecular Dynamics the
BIOC : Homework 1 Due 10/10
 Contact information: Name: Student # BIOC530 2012: Homework 1 Due 10/10 Department Email address The following problems are based on David Baker s lectures of forces and protein folding. When numerical
Contact information: Name: Student # BIOC530 2012: Homework 1 Due 10/10 Department Email address The following problems are based on David Baker s lectures of forces and protein folding. When numerical
The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii
 And the Basic Force Field Chem 4021/8021 Video II.iii") The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii Fundamental Points About Which to Be Thinking It s clear the PES is useful, so how can I construct it for an arbitrary
The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii Fundamental Points About Which to Be Thinking It s clear the PES is useful, so how can I construct it for an arbitrary
3. An Introduction to Molecular Mechanics
 3. An Introduction to Molecular Mechanics Introduction When you use Chem3D to draw molecules, the program assigns bond lengths and bond angles based on experimental data. The program does not contain real
3. An Introduction to Molecular Mechanics Introduction When you use Chem3D to draw molecules, the program assigns bond lengths and bond angles based on experimental data. The program does not contain real
Advanced in silico drug design
 Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
CHEM 498Q / CHEM 630Q: Molecular Modeling of Proteins TUTORIAL #3a: Empirical force fields
 Department of Chemistry and Biochemistry, Concordia University page 1 of 6 CHEM 498Q / CHEM 630Q: Molecular Modeling of Proteins TUTORIAL #3a: Empirical force fields INTRODUCTION The goal of this tutorial
Department of Chemistry and Biochemistry, Concordia University page 1 of 6 CHEM 498Q / CHEM 630Q: Molecular Modeling of Proteins TUTORIAL #3a: Empirical force fields INTRODUCTION The goal of this tutorial
Non equilibrium thermodynamics: foundations, scope, and extension to the meso scale. Miguel Rubi
 Non equilibrium thermodynamics: foundations, scope, and extension to the meso scale Miguel Rubi References S.R. de Groot and P. Mazur, Non equilibrium Thermodynamics, Dover, New York, 1984 J.M. Vilar and
Non equilibrium thermodynamics: foundations, scope, and extension to the meso scale Miguel Rubi References S.R. de Groot and P. Mazur, Non equilibrium Thermodynamics, Dover, New York, 1984 J.M. Vilar and
Protein Structure Analysis
 BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
Principles of Physical Biochemistry
 Principles of Physical Biochemistry Kensal E. van Hold e W. Curtis Johnso n P. Shing Ho Preface x i PART 1 MACROMOLECULAR STRUCTURE AND DYNAMICS 1 1 Biological Macromolecules 2 1.1 General Principles
Principles of Physical Biochemistry Kensal E. van Hold e W. Curtis Johnso n P. Shing Ho Preface x i PART 1 MACROMOLECULAR STRUCTURE AND DYNAMICS 1 1 Biological Macromolecules 2 1.1 General Principles
Martini Basics. Hands on: how to prepare a Martini
 Martini Basics Hands on: how to prepare a Martini gin and vermouth are combined at a ratio of 2:1, stirred in a mixing glass with ice cubes, then strained into a chilled cocktail glass and garnished with
Martini Basics Hands on: how to prepare a Martini gin and vermouth are combined at a ratio of 2:1, stirred in a mixing glass with ice cubes, then strained into a chilled cocktail glass and garnished with