Methods of Computer Simulation. Molecular Dynamics and Monte Carlo
|
|
|
- Gary Mosley
- 5 years ago
- Views:
Transcription

1 Molecular Dynamics Time is of the essence in biological processes therefore how do we understand time-dependent processes at the molecular level? How do we do this experimentally? How do we do this computationally? 1

2 Methods of Computer Simulation Molecular Dynamics and Monte Carlo 2
3 3
4 What Is Molecular Dynamics? Simulating the Brownian motion of proteins (and other macromolecules). Formally, solving Newton s equations of motion for every atom in the system. Connecting the trajectory with thermodynamic properties. Ultimately, understanding macromolecular properties from first principles. What is a molecular dynamics simulation? Simulation that shows how the atoms in the system move with time Typically on the nanosecond timescale Atoms are treated like hard balls, and their motions are described by Newton s laws. 4
5 5
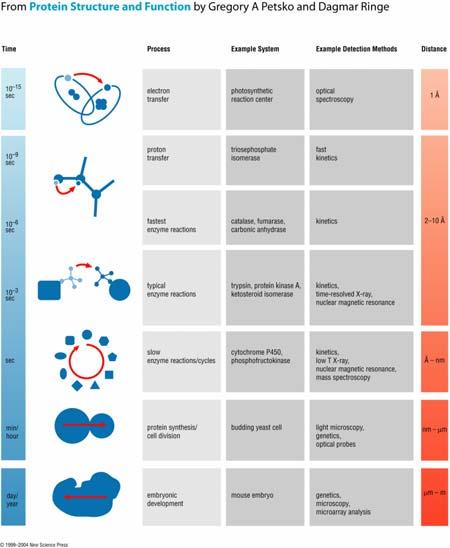
 Global protein tumbling (water tumbling) protein folding 20 ns (20 ps) ms hrs > 5 Å Random (stochastic) Energy Landscape 1 ms to 1s Barrier crossing")
6 Characteristic protein motions Type of motion Timescale Amplitude Local: bond stretching angle bending methyl rotation Medium scale loop motions SSE formation 0.01 ps 0.1 ps 1 ps ns µs < 1 Å 1-5 Å Periodic (harmonic) Global protein tumbling (water tumbling) protein folding 20 ns (20 ps) ms hrs > 5 Å Random (stochastic) Energy Landscape 1 ms to 1s Barrier crossing time ~ exp[barrier Height] 1 µs Barrier Height Unfolded State Expanded, disordered Molten Globule Compact, disordered Native State Compact, Ordered 6
7 Why MD simulations? Link physics, chemistry and biology Model phenomena that cannot be observed experimentally Understand protein folding Access to thermodynamics quantities (free energies, binding energies, ) 7
8 Why do we do computer simulations in biophysics and biochemistry? Systems with many particles behave in qualitatively different ways than systems with a small number of particles By simulating a system in thermal equilibrium, we can see qualitativelyand quantitatively what is happening. We can calculate explicit observable quantities to confirm that our model (and our understanding of the system) is correct. If we succeed, we will be able to predict behavior that we would not expect without the simulations. The goal of computation is understanding not numbers. 8
9 How do you run a MD simulation? Get the initial configuration From x-ray crystallography or NMR spectroscopy (PDB) Assign initial velocities At thermal equilibrium, the expected value of the kinetic energy of the system at temperature T is: N Ekin = m v 2 i i = (3N ) kbt 2 i= 1 2 This can be obtained by assigning the velocity components vi from a random Gaussian distribution with mean 0 and standard deviation (k B T/m i ): v = 2 i kbt m i How do you run a MD simulation? For each time step: Compute the force on each atom: E F( X ) = E( X ) = X Solve Newton s 2 nd law of motion for each atom, to get new coordinates and velocities M X = Store coordinates F(X ) X: cartesian vector of the system M diagonal mass matrix.. means second order differentiation with respect to time Stop Newton s equation cannot be solved analytically: Use stepwise numerical integration 9
10 What the integration algorithm does Advance the system by a small time step t during which forces are considered constant Recalculate forces and velocities Repeat the process If t is small enough, solution is a reasonable approximation 10
11 Molecular dynamics (MD) simulations A deterministic method based on the solution of Newton s equation of motion F i = m i a i for the ith particle; the acceleration at each step is calculated from the negative gradient of the overall potential, using F i = - grad E i -= - E i Newton s Laws r r F= ma 2r dr r r m = UE 2 ( ) dt r In general, is a 3Ndimensional vector 11
12 E i = Σ k (energies of interactions between i and all other residues k located within a cutoff distance of R c from i) E i = Gradient of potential? Derivative of E with respect to the position vector r i = (x i, y i, z i ) T at each step a xi ~ - E/ x i a yi ~ - E/ y i a zi ~ E/ z i Interaction potentials include; Non-Bonded Interaction Potentials Electrostatic interactions of the form E ik (es) = q i q k /r ik Van de Waals interactions E ij (vdw) = - a ik /r ik 6 + b ik /r ik 12 Bonded Interaction Potentials Bond stretching E i (bs) = (k bs /2) (l i l i0 ) 2 Bond angle distortion E i (bad) = (k θ /2) (θ i θ i0 ) 2 Bond torsional rotation E i (tor) = (k φ /2) f(cosφ i ) 12
13 A Potential Energy Function 1 1 V = Kb b b + K 2 2 bonds ( ) ( θ θ ) 1 + Kφ 1+ cos 2 dihedral angles θ 0 bond angles ( nφ δ) 1 A C qq nonbonded pairs r 6 r εr 13

14 14
15 Statistical Mechanics Ensemble very many copies of the system of interest evolving independently. Calculate averages of important quantities over the ensemble. Thermodynamic quantities can be derived from the appropriate averages. The Ergodic Hypothesis Averages taken over a single system for a sufficiently long time will yield the same values as ensemble averages. Molecular dynamics generates the values to be averaged. Practical issues: How long is long enough? Is the force field U(r) good enough? 15
16 16
17 17
18 Without the out-of-plane term, the bonded structures would behave differently To achieve the desired geometry (known from experiments) additional terms should be added in the force field (these help a benzene ring to be planar!) E(w)= k(1-cos2w) is one way... Cross-terms: 1, 2 and 3 force fields The presece of cross terms in a force field reflects couplings between the internal coordinates. As a bond angle is decreased, it is found that the adjacent bonds stretch to reduce the interaction between the 1,3 atoms. 18
19 Types of cross-terms Stretch-stretch Stretch-torsion Stretch-bend Bend-torsion Bend-bend 19
20 Example 1: gradient of vdw interaction with k, with respect to r i E ik (vdw) = - a ik /r ik 6 + b ik /r ik 12 r ik = r k r i x ik = x k x i y ik = y k y i z ik = z k z i r ik = [ (x k x i ) 2 + (y k y i ) 2 + (z k z i ) 2 ] 1/2 E/ x i = [- a ik /r ik 6 + b ik /r ik 12 ] / x i where r ik6 = [ (x k x i ) 2 + (y k y i ) 2 + (z k z i ) 2 ] 3 20
21 Example 2: gradient of bond stretching potential with respect to r i E i (bs) = (k bs /2) (l i l i0 ) 2 l i = r i+1 r i l ix = x i+1 x i l iy = y i+1 y i l iz = z i+1 z i l i = [ (x i+1 x i ) 2 + (y i+1 y i ) 2 + (z i+1 z i ) 2 ] 1/2 Ei(bs) = (k bs /2) (l i l i0 ) 2 E i (bs) / x i = - m i a ix (bs) (induced by deforming bond l i ) = (k bs /2) {[ (x i+1 x i ) 2 + (y i+1 y i ) 2 + (z i+1 z i ) 2 ] 1/2 l i0 } 2 / x i = k bs (l i l i0 ) {[ (x i+1 x i ) 2 + (y i+1 y i ) 2 + (z i+1 z i ) 2 ] 1/2 l i0 }/ x i = k bs (l i l i0 ) (1/2) (l i -1 ) (x i+1 x i ) 2 / x i = - k bs (1 l i0 / l i ) (x i+1 x i ) 21
22 The Verlet algorithm Perhaps the most widely used method of integrating the equations of motion is that initially adopted by Verlet [1967].The method is based on positions r(t), accelerations a (t), and the positions r(t -dt) from the previous step. The equation for advancing the positions reads as r(t+dt) = 2r(t)-r(t-dt)+ dt 2 a(t) There are several points to note about this equation. It will be seen that the velocities do not appear at all. They have been eliminated by addition of the equations obtained by Taylor expansion about r(t): You need r(t) and r(t-dt) r(t+dt) = r(t) + dt v(t) + (1/2) dt 2 a(t)+... to find r r(t+dt) r(t-dt) = r(t) - dt v(t) + (1/2) dt 2 a(t)- The velocities are not needed to compute the trajectories, but they are useful for estimating the kinetic energy (and hence the total energy). They may be obtained from the formula v(t)= [r(t+dt)-r(t-dt)]/2dt i F i Time=t+dt Time=t 22
23 Initial velocities (v i ) using the Boltzmann distribution at the given temperature v i = (m i /2πkT) 1/2 exp (- m i v i2 /2kT) How to generate MD trajectories? Known initial conformation, i.e. r i (0) for all atom i Assign v i (0), based on Boltzmann distribution at given T Calculate r i (δt) = r i (0) + δt v i (0) Using new r i (δt) evaluate the total potential V i on atom i Calculate negative gradient of V i to find a i (δt) = - V i /m i Start Verlet algorithm using r i (0), r i (δt) and a i (δt) Repeat for all atoms (including solvent, if any) Repeat the last three steps for ~ 10 6 successive times (MD steps) 23
24 A widely-used algorithm: Leap-frog Verlet Using accelerations of the current time step, compute the velocities at half-time step: v(t+ t/2) = v(t t/2) + a(t) t Then determine positions at the next time step: X(t+ t) = X(t) + v(t + t/2) t v X v t- t/2 t t+ t/2 t+ t t+3 t/2 t+2 t Choosing a time step t Not too short so that conformations are efficiently sampled Not too long to prevent wild fluctuations or system blow-up An order of magnitude less than the fastest motion is ideal Usually bond stretching is the fastest motion: C-H is ~10fs so use time step of 1fs Not interested in these motions? Constrain these bonds and double the time step 24
25 Minimization Energy minimization is very widely used in molecular modelling and is an integral part of techniques such as conformational search procedures. Min is also used to prepare a system for MD in order to relieve any unfavorable interactions in the initial configuration of the system. Especially important in complex systems such as proteins. Energy Minimization Local minima: A conformation X is a local minimum if there exists a domain D in the neighborhood of X such that for all Y X in D: U(X) <U(Y) Global minimum: A conformation X is a global minimum if U(X) <U(Y) for all conformations Y X 25
26 The minimizers Some definitions: Gradient: The gradient of a smooth function f with continuous first and second derivatives is defined as: Hessian f f f ( X ) =... f... x1 x The n x n symmetric matrix of second derivatives, H(x), is called the Hessian: 2 f 2 x f H ( x) = xi x f xn x f x1 x j... 2 f xi x j... 2 f x x N i x N j f x1 x... 2 f xi x... 2 f 2 x N N N The minimizers Minimization of a multi-variable function is usually an iterative process, in which updates of the state variable x are computed using the gradient, and in some (favorable) cases the Hessian. Iterations are stopped either when the maximum number of steps (user s input) is reached, or when the gradient norm is below a given threshold. Steepest descent (SD): The simplest iteration scheme consists of following the steepest descent direction: (α sets the minimum along the line defined xk+ 1 = xk α f ( xk ) by the gradient) Usually, SD methods leads to improvement quickly, but then exhibit slow progress toward a solution. They are commonly recommended for initial minimization iterations, when the starting function and gradient-norm values are very large. 26
27 Minimization Minimization follows gradient of potential to identify stable points on energy surface Let U(x) = a/2(x-x0)2 Begin at x, how do we find x0 if we don t know U(x) in detail? How can we move from x to x0? Steepest descent based algorithms (SD): x x = x+δ δ = -κ U(x)/ x = -κa(x- x 0) This moves us, depending on κ, toward the minimum. On a simple harmonic surface, we will reach the minimum, x0, i.e. converge, in a certain number of steps related to κ. SD methods use first derivatives only SD methods are useful for large systems with large forces Conjugate gradients (CG): The minimizers In each step of conjugate gradient methods, a search vector p k is defined by a recursive formula: pk + 1 = f ( xk ) + βk+ 1pk The corresponding new position is found by line minimization along p k : the CG methods differ in their definition of b: - Fletcher-Reeves: - Polak-Ribiere - Hestenes-Stiefel x k +1 β β β = x k + λ p k f ( xk+ 1) f ( xk 1) f ( x ) f ( x ) FR + k+ 1 = k k PR f ( xk + 1) f ( xk+ 1 k+ 1 = f ( xk ) f HS k+ 1 f ( xk+ 1 = p k k [ ) f ( x )] ( x ) ) [ f ( xk+ 1) f ( xk )] [ f ( x ) f ( x )] k + 1 k k k 27
28 Newton s methods: The minimizers Newton s method is a popular iterative method for finding the 0 of a one-dimensional function: x k+ 1 = xk g g ( xk ) '( x ) k It can be adapted to the minimization of a one dimensional function, in which case the iteration formula is: g' ( xk ) xk+ 1 = xk g' '( x ) The equivalent iterative scheme for multivariate functions is based on: x k+ 1 = x k H 1 k x 3 ( x ) f ( x ) k Several implementations of Newton s method exist, that avoid computing the full Hessian matrix: quasi-newton, truncated Newton, adopted-basis Newton-Raphson (ABNR), k k x 2 x 1 x 0 28

29 29

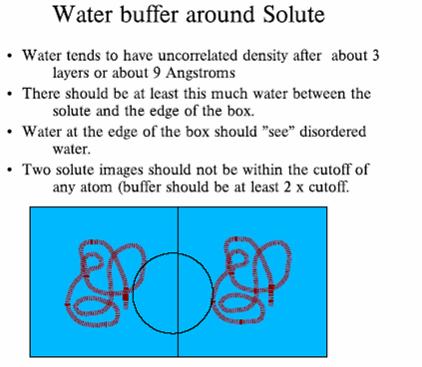
30 Periodic boundary conditions 30
31 31
32 32

33 33

34 34
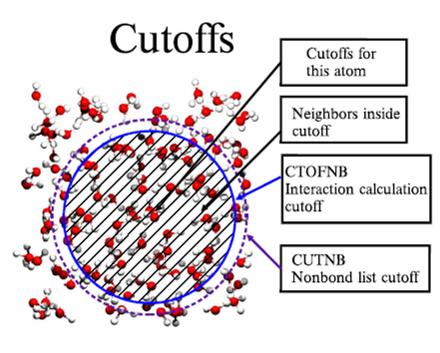
35 Notes on computing the energy Bonded interactions are local, and therefore their computation has a linear computational complexity (O(N), where N is the number of atoms in the molecule considered. The direct computation of the non bonded interactions involve all pairs of atoms and has a quadratic complexity (O(N2)). This is usually prohibitive for large molecules. U NB = 12 6 R ij Rij ε ij 2 + rij rij i, j nonbonded q q i, j nonbonded 0 i j 4πε εr ij Reducing the computing time: use of cutoff in U NB Notes on computing the energy Tamar Schlick, Molecular Modeling and Simulation, Springer 35
36 36 ( ) ( ) + = j i ij j i ij ij j i ij ij ij ij ij ij ij NB r q q r S r R r R r S U, 0, ε πε ω ε ω Cutoff schemes for faster energy computation ω ij : weights (0< ω ij <1). Can be used to exclude bonded terms, or to scale some interactions (usually 1-4) S(r) : cutoff function. Three types: 1) Truncation: < = b r b r r S 0 1 ) ( b Cutoff schemes for faster energy computation 2. Switching a b [ ] > + < = b r b r a r y r y a r r S 0 3 ) ( 2 ) ( 1 1 ) ( 2 with ) ( a b a r r y = 3. Shifting b b r b r r S = ) ( b r b r r S = ) ( or
37 Shifting function is an important concept if the non bonded interactions are concerned. In a simulation using periodic boundary condition, minimal image convention should be seriously taken in to account. The cut off distance should be half of the periodic box in order to prevent system from interacting with itself. However, this truncation causes discontinuity in potential energy function, so the potential function can not be differentiated. Therefore, a cut off length rc < Lx/2 is introduced and the potential energy functions are modified as follows: For the Lennard-Jones potential it is conventionally taken as rc = 2.5σ. Notes on computing the energy Switch function alters the non-bonded energy smoothly and gradually over the buffer region [a-b]. Shift functionavoid the sudden changes in force that occur with truncation and switch functions. Various cutoff schemes, potential switch and two types of potential shift with buffer regions of 8-12A (electrostatic), and 6-10 A (vdw) Tamar Schlick, Molecular Modeling and Simulation, Springer 37
38 Stoichastic Dynamics: Governing dynamic equations include stochastic forces that mimic solvent effects, in addition to systematic force. 38
39 An ensemble is a collection of all possible systems which have different microscopic states but have an identical macroscopic or thermodynamic state. There exist different ensembles with different characteristics. Microcanonical ensemble (NVE) : The thermodynamic state characterized by a fixed number of atoms, N, a fixed volume, V, and a fixed energy, E. This corresponds to an isolated system. Canonical Ensemble (NVT): This is a collection of all systems whose thermodynamic state is characterized by a fixed number of atoms, N, a fixed volume, V, and a fixed temperature, T. Isobaric-Isothermal Ensemble (NPT): This ensemble is characterized by a fixed number of atoms, N, a fixed pressure, P, and a fixed temperature, T. Grand canonical Ensemble (µvt): The thermodynamic state for this ensemble is characterized by a fixed chemical potential, µ, a fixed volume, V, and a fixed temperature, T. MD at Constant T and P The study of molecular properties as a function of T and P, rather than V and E is of general interest Thus microcanonical ensembles that allow energy and volume to fluctuate and require constant T and/or P are inappropriate for systems. 39
40 Molecular Dynamics ensembles The method discussed above is appropriate for the micro-canonical ensemble: constant N (# of particles) V (volume) and E T (total energy = E + E kin ) In some cases, it might be more appropriate to simulate under constant Temperature (T) or constant Pressure (P): Canonical ensemble: NVT Isothermal-isobaric: NPT Constant pressure and enthalpy: NPH How do you simulate at constant temperature and pressure? Simulating at constant T: the Berendsen scheme system Heat bath Bath supplies or removes heat from the system as appropriate Exponentially scale the velocities at each time step by the factor λ: λ = t T 1 1 τ T where τ determines how strong the bath influences the system Berendsen et al. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684 (1984) bath T: kinetic temperature 40
 where P = Ekin + xi Fi 3v i= 1 τ P where κ : isothermal compressibility τ P : coupling constant υ : volume x i : position of particle i F i : force on particle i Berendsen et al.")
41 Simulating at constant P: Berendsen scheme system pressure bath Couple the system to a pressure bath Exponentially scale the volume of the simulation box at each time step by a factor λ: N t 2 λ = 1 κ ( P Pbath ) where P = Ekin + xi Fi 3v i= 1 τ P where κ : isothermal compressibility τ P : coupling constant υ : volume x i : position of particle i F i : force on particle i Berendsen et al. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684 (1984) Energy MD as a tool for minimization State A Molecular dynamics uses thermal energy to explore the energy surface State B Energy minimization stops at local minima position 41

42 Crossing energy barriers Energy A I G Position State B State A B Position time The actual transition time from A to B is very quick (a few pico seconds). What takes time is waiting. The average waiting time for going from A to B can be expressed as: G τ = kt A B Ce R.H. Stote et al, Biochemistry, v.43, no.24, p (2004) 42

43 kı HW: ht 34<Z< >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>> >>> Baaaaaaaaaaaaaaaaaaaaaaaaaaaaaq vccbvb y 43
44 Molecular Simulations Molecular Mechanics: energy minimization Molecular Dynamics: simulation of motions Monte Carlo methods: sampling techniques 44
45 Monte Carlo: random sampling A simple example: Evaluate numerically the one-dimensional integral: I f ( x) dx = b a Instead of using classical quadrature, the integral can be rewritten as I = ( b a) f ( x) <f(x)> denotes the unweighted average of f(x) over [a,b], and can be determined by evaluating f(x) at a large number of x values randomly distributed over [a,b] Monte Carlo method! 45
46 46
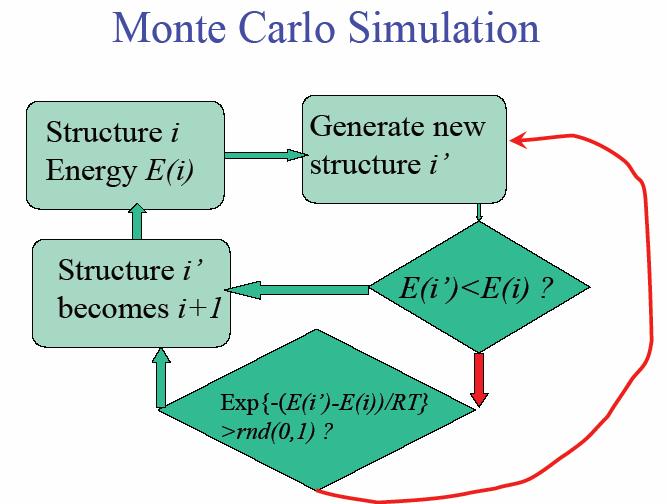
47 Brief Account of MC, MD N ~ ,000 Periodic boundaries Prescribed intermolecular potential Monte Carlo Specify N, V, T Molecular Dynamics Specify N, V, E Generate random moves Sample with P α exp(-υ /kt) Solve Newton s equations F i = m i a i Calculate r i (t), v i (t) Obtain equilibrium properties Take averages Take averages Obtain equilibrium and nonequilibrium properties Monte Carlo Simulation MC techniques applied to molecular simulation, almost always involves a Markov process move to a new configuration from an existing one according to a well-defined transition probability Simulation procedure generate a new trial configuration by making a perturbation to the present configuration accept the new configuration based on the ratio of the probabilities for the new and old configurations, according to the Metropolis algorithm if the trial is rejected, the present configuration is taken as the next one in the Markov chain repeat this many times, accumulating sums for averages taken from D. A. Kofke s lectures on Molecular Simulation, SUNY Buffalo e new βu e old βu State k State k+1 47
48 Differences Between MC and MD MD gives information about dynamical behavior, as well as equilibrium, thermodynamic properties. Thus, transport properties can be calculated. MC can only give static, equilibrium properties MC can be more easily adapted to other ensembles: - µ, V, T (grand canonical) - N, V, T (canonical) - N, P, T (isobaric-isothermic) In MC motions are artificial - in MD they are natural In MC we can use special techniques to achieve equilibrium. For example, can observe formation of micelles, slow phase transitions. 48
49 The Monte Carlo Method In MC simulations the average value is approximated by generating a large number of trial configurations x N using a random number generator, and replacing the integrals by sums over a finite number of configurations. Random sampling: Suppose we choose configurations x N randomly, so the average becomes A = j j ( ) exp U( ) A j j kt exp U ( j) kt Monte Carlo Sampling for protein structure The probability of finding a protein (biomolecule) with a total energy E(X) is: P( X ) = E( X ) exp kt E( Z) exp dz kt Partition function Estimates of any average quantity of the form: A = A( X ) P( X ) dx using uniform sampling would therefore be extremely inefficient. Metropolis and coll. developed a method for directly sampling according to the actual distribution. Metropolis et al. Equation of state calculations by fast computing machines. J. Chem. Phys. 21: (1953) 49
50 Monte Carlo for the canonical ensemble The canonical ensemble corresponds to constant NVT. The total energy (Hamiltonian) is the sum of the kinetic energy and potential energy: E=E k (p)+e p (X) If the quantity to be measured is velocity independent, it is enough to consider the potential energy: A = = E E X k ( p) p( ) A( X )exp exp dxdp kt kt E E X k ( p) p( ) exp exp dxdp kt kt E X A X p( ) ( )exp dx kt E X p( ) exp dx kt The kinetic energy depends on the momentum p; it can be factored and canceled. Monte Carlo for the canonical ensemble Let: P( X ) = E p( X ) exp kt E Z p( ) exp dz kt π ( X Y ) And let be the transition probability from state X to state Y. Let us suppose we carry out a large number of Monte Carlo simulations, such that the number of points observed in conformation X is proportional to N(X). The transition probability must satisfy one obvious condition: it should not destroy this equilibrium once it is reached. Metropolis proposed to realize this using the detailed balance condition: P( X ) π ( X Y ) = P( Y ) π ( Y X ) or π ( X Y ) P( Y ) E = = exp π ( Y X ) P( X ) ( Y ) E kt ( X p p ) 50
51 Monte Carlo for the canonical ensemble There are many choices for the transition probability that satisfy the balance condition. The choice of Metropolis is: E exp π ( X Y ) = 1 ( Y ) E ( X p kt The Metropolis Monte Carlo algorithm: p ) if if E ( Y ) > E p E ( Y ) E p p p ( X ) ( X ) 1. Select a conformation X at random. Compute its energy E(X) 2. Generate a new trial conformation Y. Compute its energy E(Y) 3. Accept the move from X to Y with probability: E p ( Y ) E p( X ) P = min(1, exp kt 4. Repeat 2 and 3. Pick a random number RN, uniform in [0,1]. If RN < P, accept the move. Monte Carlo for the canonical ensemble Notes: 1. There are many types of Metropolis Monte Carlo simulations, characterized by the generation of the trial conformation. 2. The random number generator is crucial 3. Metropolis Monte Carlo simulations are used for finding thermodynamics quantities, for optimization, 4. An extension of the Metropolis algorithm is often used for minimization: the simulated annealing technique, where the temperature is lowered as the simulation evolves, in an attempt to locate the global minimum. 51
What is Classical Molecular Dynamics?
 What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
 A Nobel Prize for Molecular Dynamics and QM/MM What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential
A Nobel Prize for Molecular Dynamics and QM/MM What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential
Molecular Dynamics Simulations. Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia
 Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Monte Carlo (MC) Simulation Methods. Elisa Fadda
 Simulation Methods. Elisa Fadda") Monte Carlo (MC) Simulation Methods Elisa Fadda 1011-CH328, Molecular Modelling & Drug Design 2011 Experimental Observables A system observable is a property of the system state. The system state i is
Monte Carlo (MC) Simulation Methods Elisa Fadda 1011-CH328, Molecular Modelling & Drug Design 2011 Experimental Observables A system observable is a property of the system state. The system state i is
MD Thermodynamics. Lecture 12 3/26/18. Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky
 MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Energy functions and their relationship to molecular conformation. CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror
 Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Outline Energy functions for proteins (or biomolecular systems more generally)
Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Outline Energy functions for proteins (or biomolecular systems more generally)
Exploring the energy landscape
 Exploring the energy landscape ChE210D Today's lecture: what are general features of the potential energy surface and how can we locate and characterize minima on it Derivatives of the potential energy
Exploring the energy landscape ChE210D Today's lecture: what are general features of the potential energy surface and how can we locate and characterize minima on it Derivatives of the potential energy
Simulations with MM Force Fields. Monte Carlo (MC) and Molecular Dynamics (MD) Video II.vi
 and Molecular Dynamics (MD) Video II.vi") Simulations with MM Force Fields Monte Carlo (MC) and Molecular Dynamics (MD) Video II.vi Some slides taken with permission from Howard R. Mayne Department of Chemistry University of New Hampshire Walking
Simulations with MM Force Fields Monte Carlo (MC) and Molecular Dynamics (MD) Video II.vi Some slides taken with permission from Howard R. Mayne Department of Chemistry University of New Hampshire Walking
Molecular Simulation II. Classical Mechanical Treatment
 Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Biomolecules are dynamic no single structure is a perfect model
 Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Temperature and Pressure Controls
 Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Biomolecular modeling I
 2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Introduction to Simulation - Lectures 17, 18. Molecular Dynamics. Nicolas Hadjiconstantinou
 Introduction to Simulation - Lectures 17, 18 Molecular Dynamics Nicolas Hadjiconstantinou Molecular Dynamics Molecular dynamics is a technique for computing the equilibrium and non-equilibrium properties
Introduction to Simulation - Lectures 17, 18 Molecular Dynamics Nicolas Hadjiconstantinou Molecular Dynamics Molecular dynamics is a technique for computing the equilibrium and non-equilibrium properties
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
FALL 2018 MATH 4211/6211 Optimization Homework 4
 FALL 2018 MATH 4211/6211 Optimization Homework 4 This homework assignment is open to textbook, reference books, slides, and online resources, excluding any direct solution to the problem (such as solution
FALL 2018 MATH 4211/6211 Optimization Homework 4 This homework assignment is open to textbook, reference books, slides, and online resources, excluding any direct solution to the problem (such as solution
Introduction to Classical Molecular Dynamics. Giovanni Chillemi HPC department, CINECA
 Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
Understanding Molecular Simulation 2009 Monte Carlo and Molecular Dynamics in different ensembles. Srikanth Sastry
 JNCASR August 20, 21 2009 Understanding Molecular Simulation 2009 Monte Carlo and Molecular Dynamics in different ensembles Srikanth Sastry Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore
JNCASR August 20, 21 2009 Understanding Molecular Simulation 2009 Monte Carlo and Molecular Dynamics in different ensembles Srikanth Sastry Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore
Unconstrained optimization
 Chapter 4 Unconstrained optimization An unconstrained optimization problem takes the form min x Rnf(x) (4.1) for a target functional (also called objective function) f : R n R. In this chapter and throughout
Chapter 4 Unconstrained optimization An unconstrained optimization problem takes the form min x Rnf(x) (4.1) for a target functional (also called objective function) f : R n R. In this chapter and throughout
Computer simulation methods (2) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Principles and Applications of Molecular Dynamics Simulations with NAMD
 Principles and Applications of Molecular Dynamics Simulations with NAMD Nov. 14, 2016 Computational Microscope NCSA supercomputer JC Gumbart Assistant Professor of Physics Georgia Institute of Technology
Principles and Applications of Molecular Dynamics Simulations with NAMD Nov. 14, 2016 Computational Microscope NCSA supercomputer JC Gumbart Assistant Professor of Physics Georgia Institute of Technology
Langevin Methods. Burkhard Dünweg Max Planck Institute for Polymer Research Ackermannweg 10 D Mainz Germany
 Langevin Methods Burkhard Dünweg Max Planck Institute for Polymer Research Ackermannweg 1 D 55128 Mainz Germany Motivation Original idea: Fast and slow degrees of freedom Example: Brownian motion Replace
Langevin Methods Burkhard Dünweg Max Planck Institute for Polymer Research Ackermannweg 1 D 55128 Mainz Germany Motivation Original idea: Fast and slow degrees of freedom Example: Brownian motion Replace
Ab initio molecular dynamics. Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy. Bangalore, 04 September 2014
 Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Introduction Statistical Thermodynamics. Monday, January 6, 14
 Introduction Statistical Thermodynamics 1 Molecular Simulations Molecular dynamics: solve equations of motion Monte Carlo: importance sampling r 1 r 2 r n MD MC r 1 r 2 2 r n 2 3 3 4 4 Questions How can
Introduction Statistical Thermodynamics 1 Molecular Simulations Molecular dynamics: solve equations of motion Monte Carlo: importance sampling r 1 r 2 r n MD MC r 1 r 2 2 r n 2 3 3 4 4 Questions How can
Wang-Landau Monte Carlo simulation. Aleš Vítek IT4I, VP3
 Wang-Landau Monte Carlo simulation Aleš Vítek IT4I, VP3 PART 1 Classical Monte Carlo Methods Outline 1. Statistical thermodynamics, ensembles 2. Numerical evaluation of integrals, crude Monte Carlo (MC)
Wang-Landau Monte Carlo simulation Aleš Vítek IT4I, VP3 PART 1 Classical Monte Carlo Methods Outline 1. Statistical thermodynamics, ensembles 2. Numerical evaluation of integrals, crude Monte Carlo (MC)
Temperature and Pressure Controls
 Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Exercise 2: Solvating the Structure Before you continue, follow these steps: Setting up Periodic Boundary Conditions
 Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Protein Dynamics, Allostery and Function
 Protein Dynamics, Allostery and Function Lecture 3. Protein Dynamics Xiaolin Cheng UT/ORNL Center for Molecular Biophysics SJTU Summer School 2017 1 Obtaining Dynamic Information Experimental Approaches
Protein Dynamics, Allostery and Function Lecture 3. Protein Dynamics Xiaolin Cheng UT/ORNL Center for Molecular Biophysics SJTU Summer School 2017 1 Obtaining Dynamic Information Experimental Approaches
Advanced sampling. fluids of strongly orientation-dependent interactions (e.g., dipoles, hydrogen bonds)
") Advanced sampling ChE210D Today's lecture: methods for facilitating equilibration and sampling in complex, frustrated, or slow-evolving systems Difficult-to-simulate systems Practically speaking, one is
Advanced sampling ChE210D Today's lecture: methods for facilitating equilibration and sampling in complex, frustrated, or slow-evolving systems Difficult-to-simulate systems Practically speaking, one is
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Introduction to Geometry Optimization. Computational Chemistry lab 2009
 Introduction to Geometry Optimization Computational Chemistry lab 9 Determination of the molecule configuration H H Diatomic molecule determine the interatomic distance H O H Triatomic molecule determine
Introduction to Geometry Optimization Computational Chemistry lab 9 Determination of the molecule configuration H H Diatomic molecule determine the interatomic distance H O H Triatomic molecule determine
Simulation of molecular systems by molecular dynamics
 Simulation of molecular systems by molecular dynamics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Dynamics, Label RFCT 2015 November 26, 2015 1 / 35 Introduction
Simulation of molecular systems by molecular dynamics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Dynamics, Label RFCT 2015 November 26, 2015 1 / 35 Introduction
Monte Carlo. Lecture 15 4/9/18. Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky
 Monte Carlo Lecture 15 4/9/18 1 Sampling with dynamics In Molecular Dynamics we simulate evolution of a system over time according to Newton s equations, conserving energy Averages (thermodynamic properties)
Monte Carlo Lecture 15 4/9/18 1 Sampling with dynamics In Molecular Dynamics we simulate evolution of a system over time according to Newton s equations, conserving energy Averages (thermodynamic properties)
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Length Scales (c) ICAMS: http://www.icams.de/cms/upload/01_home/01_research_at_icams/length_scales_1024x780.png Goals for today: Background
André Schleife Department of Materials Science and Engineering Length Scales (c) ICAMS: http://www.icams.de/cms/upload/01_home/01_research_at_icams/length_scales_1024x780.png Goals for today: Background
Advanced Molecular Dynamics
 Advanced Molecular Dynamics Introduction May 2, 2017 Who am I? I am an associate professor at Theoretical Physics Topics I work on: Algorithms for (parallel) molecular simulations including GPU acceleration
Advanced Molecular Dynamics Introduction May 2, 2017 Who am I? I am an associate professor at Theoretical Physics Topics I work on: Algorithms for (parallel) molecular simulations including GPU acceleration
Energy Barriers and Rates - Transition State Theory for Physicists
 Energy Barriers and Rates - Transition State Theory for Physicists Daniel C. Elton October 12, 2013 Useful relations 1 cal = 4.184 J 1 kcal mole 1 = 0.0434 ev per particle 1 kj mole 1 = 0.0104 ev per particle
Energy Barriers and Rates - Transition State Theory for Physicists Daniel C. Elton October 12, 2013 Useful relations 1 cal = 4.184 J 1 kcal mole 1 = 0.0434 ev per particle 1 kj mole 1 = 0.0104 ev per particle
Ab initio molecular dynamics and nuclear quantum effects
 Ab initio molecular dynamics and nuclear quantum effects Luca M. Ghiringhelli Fritz Haber Institute Hands on workshop density functional theory and beyond: First principles simulations of molecules and
Ab initio molecular dynamics and nuclear quantum effects Luca M. Ghiringhelli Fritz Haber Institute Hands on workshop density functional theory and beyond: First principles simulations of molecules and
Nonlinear Programming
 Nonlinear Programming Kees Roos e-mail: C.Roos@ewi.tudelft.nl URL: http://www.isa.ewi.tudelft.nl/ roos LNMB Course De Uithof, Utrecht February 6 - May 8, A.D. 2006 Optimization Group 1 Outline for week
Nonlinear Programming Kees Roos e-mail: C.Roos@ewi.tudelft.nl URL: http://www.isa.ewi.tudelft.nl/ roos LNMB Course De Uithof, Utrecht February 6 - May 8, A.D. 2006 Optimization Group 1 Outline for week
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 If you throw up a stone, it is Physics. If you throw up a stone, it is Physics. If it lands on your head, it is
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 If you throw up a stone, it is Physics. If you throw up a stone, it is Physics. If it lands on your head, it is
Javier Junquera. Statistical mechanics
 Javier Junquera Statistical mechanics From the microscopic to the macroscopic level: the realm of statistical mechanics Computer simulations Thermodynamic state Generates information at the microscopic
Javier Junquera Statistical mechanics From the microscopic to the macroscopic level: the realm of statistical mechanics Computer simulations Thermodynamic state Generates information at the microscopic
Set the initial conditions r i. Update neighborlist. r i. Get new forces F i
 Set the initial conditions r i t 0, v i t 0 Update neighborlist Get new forces F i r i Solve the equations of motion numerically over time step t : r i t n r i t n + v i t n v i t n + Perform T, P scaling
Set the initial conditions r i t 0, v i t 0 Update neighborlist Get new forces F i r i Solve the equations of motion numerically over time step t : r i t n r i t n + v i t n v i t n + Perform T, P scaling
Energy functions and their relationship to molecular conformation. CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror
 Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Yesterday s Nobel Prize: single-particle cryoelectron microscopy 2 Outline Energy
Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Yesterday s Nobel Prize: single-particle cryoelectron microscopy 2 Outline Energy
Monte Carlo Methods in Statistical Mechanics
 Monte Carlo Methods in Statistical Mechanics Mario G. Del Pópolo Atomistic Simulation Centre School of Mathematics and Physics Queen s University Belfast Belfast Mario G. Del Pópolo Statistical Mechanics
Monte Carlo Methods in Statistical Mechanics Mario G. Del Pópolo Atomistic Simulation Centre School of Mathematics and Physics Queen s University Belfast Belfast Mario G. Del Pópolo Statistical Mechanics
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 1 David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Time/s Multi-Scale Modeling Based on SDSC Blue Horizon (SP3) 1.728 Tflops
1 CE 530 Molecular Simulation Lecture 1 David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Time/s Multi-Scale Modeling Based on SDSC Blue Horizon (SP3) 1.728 Tflops
Ab Ini'o Molecular Dynamics (MD) Simula?ons
 Simula?ons") Ab Ini'o Molecular Dynamics (MD) Simula?ons Rick Remsing ICMS, CCDM, Temple University, Philadelphia, PA What are Molecular Dynamics (MD) Simulations? Technique to compute statistical and transport properties
Ab Ini'o Molecular Dynamics (MD) Simula?ons Rick Remsing ICMS, CCDM, Temple University, Philadelphia, PA What are Molecular Dynamics (MD) Simulations? Technique to compute statistical and transport properties
Multiscale Materials Modeling
 Multiscale Materials Modeling Lecture 02 Capabilities of Classical Molecular Simulation These notes created by David Keffer, University of Tennessee, Knoxville, 2009. Outline Capabilities of Classical
Multiscale Materials Modeling Lecture 02 Capabilities of Classical Molecular Simulation These notes created by David Keffer, University of Tennessee, Knoxville, 2009. Outline Capabilities of Classical
Introduction to model potential Molecular Dynamics A3hourcourseatICTP
 Introduction to model potential Molecular Dynamics A3hourcourseatICTP Alessandro Mattoni 1 1 Istituto Officina dei Materiali CNR-IOM Unità di Cagliari SLACS ICTP School on numerical methods for energy,
Introduction to model potential Molecular Dynamics A3hourcourseatICTP Alessandro Mattoni 1 1 Istituto Officina dei Materiali CNR-IOM Unità di Cagliari SLACS ICTP School on numerical methods for energy,
2. Thermodynamics. Introduction. Understanding Molecular Simulation
 2. Thermodynamics Introduction Molecular Simulations Molecular dynamics: solve equations of motion r 1 r 2 r n Monte Carlo: importance sampling r 1 r 2 r n How do we know our simulation is correct? Molecular
2. Thermodynamics Introduction Molecular Simulations Molecular dynamics: solve equations of motion r 1 r 2 r n Monte Carlo: importance sampling r 1 r 2 r n How do we know our simulation is correct? Molecular
Protein Structure Analysis
 BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
Statistical thermodynamics for MD and MC simulations
 Statistical thermodynamics for MD and MC simulations knowing 2 atoms and wishing to know 10 23 of them Marcus Elstner and Tomáš Kubař 22 June 2016 Introduction Thermodynamic properties of molecular systems
Statistical thermodynamics for MD and MC simulations knowing 2 atoms and wishing to know 10 23 of them Marcus Elstner and Tomáš Kubař 22 June 2016 Introduction Thermodynamic properties of molecular systems
Gear methods I + 1/18
 Gear methods I + 1/18 Predictor-corrector type: knowledge of history is used to predict an approximate solution, which is made more accurate in the following step we do not want (otherwise good) methods
Gear methods I + 1/18 Predictor-corrector type: knowledge of history is used to predict an approximate solution, which is made more accurate in the following step we do not want (otherwise good) methods
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
Computational Chemistry - MD Simulations
 Computational Chemistry - MD Simulations P. Ojeda-May pedro.ojeda-may@umu.se Department of Chemistry/HPC2N, Umeå University, 901 87, Sweden. May 2, 2017 Table of contents 1 Basics on MD simulations Accelerated
Computational Chemistry - MD Simulations P. Ojeda-May pedro.ojeda-may@umu.se Department of Chemistry/HPC2N, Umeå University, 901 87, Sweden. May 2, 2017 Table of contents 1 Basics on MD simulations Accelerated
G : Statistical Mechanics
 G25.2651: Statistical Mechanics Notes for Lecture 15 Consider Hamilton s equations in the form I. CLASSICAL LINEAR RESPONSE THEORY q i = H p i ṗ i = H q i We noted early in the course that an ensemble
G25.2651: Statistical Mechanics Notes for Lecture 15 Consider Hamilton s equations in the form I. CLASSICAL LINEAR RESPONSE THEORY q i = H p i ṗ i = H q i We noted early in the course that an ensemble
Basics of Statistical Mechanics
 Basics of Statistical Mechanics Review of ensembles Microcanonical, canonical, Maxwell-Boltzmann Constant pressure, temperature, volume, Thermodynamic limit Ergodicity (see online notes also) Reading assignment:
Basics of Statistical Mechanics Review of ensembles Microcanonical, canonical, Maxwell-Boltzmann Constant pressure, temperature, volume, Thermodynamic limit Ergodicity (see online notes also) Reading assignment:
Time-Dependent Statistical Mechanics 5. The classical atomic fluid, classical mechanics, and classical equilibrium statistical mechanics
 Time-Dependent Statistical Mechanics 5. The classical atomic fluid, classical mechanics, and classical equilibrium statistical mechanics c Hans C. Andersen October 1, 2009 While we know that in principle
Time-Dependent Statistical Mechanics 5. The classical atomic fluid, classical mechanics, and classical equilibrium statistical mechanics c Hans C. Andersen October 1, 2009 While we know that in principle
Computer simulation methods (1) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (1) Dr. Vania Calandrini Why computational methods To understand and predict the properties of complex systems (many degrees of freedom): liquids, solids, adsorption of molecules
Computer simulation methods (1) Dr. Vania Calandrini Why computational methods To understand and predict the properties of complex systems (many degrees of freedom): liquids, solids, adsorption of molecules
Organization of NAMD Tutorial Files
 Organization of NAMD Tutorial Files .1.1. RMSD for individual residues Objective: Find the average RMSD over time of each residue in the protein using VMD. Display the protein with the residues colored
Organization of NAMD Tutorial Files .1.1. RMSD for individual residues Objective: Find the average RMSD over time of each residue in the protein using VMD. Display the protein with the residues colored
Programming, numerics and optimization
 Programming, numerics and optimization Lecture C-3: Unconstrained optimization II Łukasz Jankowski ljank@ippt.pan.pl Institute of Fundamental Technological Research Room 4.32, Phone +22.8261281 ext. 428
Programming, numerics and optimization Lecture C-3: Unconstrained optimization II Łukasz Jankowski ljank@ippt.pan.pl Institute of Fundamental Technological Research Room 4.32, Phone +22.8261281 ext. 428
Unfolding CspB by means of biased molecular dynamics
 Chapter 4 Unfolding CspB by means of biased molecular dynamics 4.1 Introduction Understanding the mechanism of protein folding has been a major challenge for the last twenty years, as pointed out in the
Chapter 4 Unfolding CspB by means of biased molecular dynamics 4.1 Introduction Understanding the mechanism of protein folding has been a major challenge for the last twenty years, as pointed out in the
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
4/18/2011. Titus Beu University Babes-Bolyai Department of Theoretical and Computational Physics Cluj-Napoca, Romania
 1. Introduction Titus Beu University Babes-Bolyai Department of Theoretical and Computational Physics Cluj-Napoca, Romania Bibliography Computer experiments Ensemble averages and time averages Molecular
1. Introduction Titus Beu University Babes-Bolyai Department of Theoretical and Computational Physics Cluj-Napoca, Romania Bibliography Computer experiments Ensemble averages and time averages Molecular
Random Walks A&T and F&S 3.1.2
 Random Walks A&T 110-123 and F&S 3.1.2 As we explained last time, it is very difficult to sample directly a general probability distribution. - If we sample from another distribution, the overlap will
Random Walks A&T 110-123 and F&S 3.1.2 As we explained last time, it is very difficult to sample directly a general probability distribution. - If we sample from another distribution, the overlap will
Scientific Computing II
 Scientific Computing II Molecular Dynamics Simulation Michael Bader SCCS Summer Term 2015 Molecular Dynamics Simulation, Summer Term 2015 1 Continuum Mechanics for Fluid Mechanics? Molecular Dynamics the
Scientific Computing II Molecular Dynamics Simulation Michael Bader SCCS Summer Term 2015 Molecular Dynamics Simulation, Summer Term 2015 1 Continuum Mechanics for Fluid Mechanics? Molecular Dynamics the
Optimization Methods via Simulation
 Optimization Methods via Simulation Optimization problems are very important in science, engineering, industry,. Examples: Traveling salesman problem Circuit-board design Car-Parrinello ab initio MD Protein
Optimization Methods via Simulation Optimization problems are very important in science, engineering, industry,. Examples: Traveling salesman problem Circuit-board design Car-Parrinello ab initio MD Protein
Limitations of temperature replica exchange (T-REMD) for protein folding simulations
 for protein folding simulations") Limitations of temperature replica exchange (T-REMD) for protein folding simulations Jed W. Pitera, William C. Swope IBM Research pitera@us.ibm.com Anomalies in protein folding kinetic thermodynamic 322K
Limitations of temperature replica exchange (T-REMD) for protein folding simulations Jed W. Pitera, William C. Swope IBM Research pitera@us.ibm.com Anomalies in protein folding kinetic thermodynamic 322K
Computational Modeling of Protein Dynamics. Yinghao Wu Department of Systems and Computational Biology Albert Einstein College of Medicine Fall 2014
 Computational Modeling of Protein Dynamics Yinghao Wu Department of Systems and Computational Biology Albert Einstein College of Medicine Fall 2014 Outline Background of protein dynamics Basic computational
Computational Modeling of Protein Dynamics Yinghao Wu Department of Systems and Computational Biology Albert Einstein College of Medicine Fall 2014 Outline Background of protein dynamics Basic computational
Calculation of point-to-point short time and rare. trajectories with boundary value formulation
 Calculation of point-to-point short time and rare trajectories with boundary value formulation Dov Bai and Ron Elber Department of Computer Science, Upson Hall 413 Cornell University, Ithaca, NY 14853
Calculation of point-to-point short time and rare trajectories with boundary value formulation Dov Bai and Ron Elber Department of Computer Science, Upson Hall 413 Cornell University, Ithaca, NY 14853
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Hyeyoung Shin a, Tod A. Pascal ab, William A. Goddard III abc*, and Hyungjun Kim a* Korea
 The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
Phase Equilibria and Molecular Solutions Jan G. Korvink and Evgenii Rudnyi IMTEK Albert Ludwig University Freiburg, Germany
 Phase Equilibria and Molecular Solutions Jan G. Korvink and Evgenii Rudnyi IMTEK Albert Ludwig University Freiburg, Germany Preliminaries Learning Goals Phase Equilibria Phase diagrams and classical thermodynamics
Phase Equilibria and Molecular Solutions Jan G. Korvink and Evgenii Rudnyi IMTEK Albert Ludwig University Freiburg, Germany Preliminaries Learning Goals Phase Equilibria Phase diagrams and classical thermodynamics
January 29, Non-linear conjugate gradient method(s): Fletcher Reeves Polak Ribière January 29, 2014 Hestenes Stiefel 1 / 13
: Fletcher Reeves Polak Ribière January 29, 2014 Hestenes Stiefel 1 / 13") Non-linear conjugate gradient method(s): Fletcher Reeves Polak Ribière Hestenes Stiefel January 29, 2014 Non-linear conjugate gradient method(s): Fletcher Reeves Polak Ribière January 29, 2014 Hestenes
Non-linear conjugate gradient method(s): Fletcher Reeves Polak Ribière Hestenes Stiefel January 29, 2014 Non-linear conjugate gradient method(s): Fletcher Reeves Polak Ribière January 29, 2014 Hestenes
Protein Structure Analysis
 BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
Statistical Mechanics. Atomistic view of Materials
 Statistical Mechanics Atomistic view of Materials What is statistical mechanics? Microscopic (atoms, electrons, etc.) Statistical mechanics Macroscopic (Thermodynamics) Sample with constrains Fixed thermodynamics
Statistical Mechanics Atomistic view of Materials What is statistical mechanics? Microscopic (atoms, electrons, etc.) Statistical mechanics Macroscopic (Thermodynamics) Sample with constrains Fixed thermodynamics
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
Brief Review of Statistical Mechanics
 Brief Review of Statistical Mechanics Introduction Statistical mechanics: a branch of physics which studies macroscopic systems from a microscopic or molecular point of view (McQuarrie,1976) Also see (Hill,1986;
Brief Review of Statistical Mechanics Introduction Statistical mechanics: a branch of physics which studies macroscopic systems from a microscopic or molecular point of view (McQuarrie,1976) Also see (Hill,1986;
Crystal Structure Prediction using CRYSTALG program
 Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Statistical Thermodynamics and Monte-Carlo Evgenii B. Rudnyi and Jan G. Korvink IMTEK Albert Ludwig University Freiburg, Germany
 Statistical Thermodynamics and Monte-Carlo Evgenii B. Rudnyi and Jan G. Korvink IMTEK Albert Ludwig University Freiburg, Germany Preliminaries Learning Goals From Micro to Macro Statistical Mechanics (Statistical
Statistical Thermodynamics and Monte-Carlo Evgenii B. Rudnyi and Jan G. Korvink IMTEK Albert Ludwig University Freiburg, Germany Preliminaries Learning Goals From Micro to Macro Statistical Mechanics (Statistical
Biomolecular modeling I
 2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
CE 530 Molecular Simulation
 CE 530 Molecular Simulation Lecture Molecular Dynamics Simulation David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu MD of hard disks intuitive Review and Preview collision
CE 530 Molecular Simulation Lecture Molecular Dynamics Simulation David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu MD of hard disks intuitive Review and Preview collision
Intermolecular Forces and Monte-Carlo Integration 열역학특수연구
 Intermolecular Forces and Monte-Carlo Integration 열역학특수연구 2003.3.28 Source of the lecture note. J.M.Prausnitz and others, Molecular Thermodynamics of Fluid Phase Equiliria Atkins, Physical Chemistry Lecture
Intermolecular Forces and Monte-Carlo Integration 열역학특수연구 2003.3.28 Source of the lecture note. J.M.Prausnitz and others, Molecular Thermodynamics of Fluid Phase Equiliria Atkins, Physical Chemistry Lecture
MOLECULAR DYNAMIC SIMULATION OF WATER VAPOR INTERACTION WITH VARIOUS TYPES OF PORES USING HYBRID COMPUTING STRUCTURES
 MOLECULAR DYNAMIC SIMULATION OF WATER VAPOR INTERACTION WITH VARIOUS TYPES OF PORES USING HYBRID COMPUTING STRUCTURES V.V. Korenkov 1,3, a, E.G. Nikonov 1, b, M. Popovičová 2, с 1 Joint Institute for Nuclear
MOLECULAR DYNAMIC SIMULATION OF WATER VAPOR INTERACTION WITH VARIOUS TYPES OF PORES USING HYBRID COMPUTING STRUCTURES V.V. Korenkov 1,3, a, E.G. Nikonov 1, b, M. Popovičová 2, с 1 Joint Institute for Nuclear
Enhanced sampling via molecular dynamics II: Unbiased approaches
 Enhanced sampling via molecular dynamics II: Unbiased approaches Mark E. Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Sciences New York University, 100 Washington Square East, NY
Enhanced sampling via molecular dynamics II: Unbiased approaches Mark E. Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Sciences New York University, 100 Washington Square East, NY
Thermodynamic behaviour of mixtures containing CO 2. A molecular simulation study
 Thermodynamic behaviour of mixtures containing. A molecular simulation study V. Lachet, C. Nieto-Draghi, B. Creton (IFPEN) Å. Ervik, G. Skaugen, Ø. Wilhelmsen, M. Hammer (SINTEF) Introduction quality issues
Thermodynamic behaviour of mixtures containing. A molecular simulation study V. Lachet, C. Nieto-Draghi, B. Creton (IFPEN) Å. Ervik, G. Skaugen, Ø. Wilhelmsen, M. Hammer (SINTEF) Introduction quality issues
Free energy calculations using molecular dynamics simulations. Anna Johansson
 Free energy calculations using molecular dynamics simulations Anna Johansson 2007-03-13 Outline Introduction to concepts Why is free energy important? Calculating free energy using MD Thermodynamical Integration
Free energy calculations using molecular dynamics simulations Anna Johansson 2007-03-13 Outline Introduction to concepts Why is free energy important? Calculating free energy using MD Thermodynamical Integration
Molecular mechanics. classical description of molecules. Marcus Elstner and Tomáš Kubař. April 29, 2016
 classical description of molecules April 29, 2016 Chemical bond Conceptual and chemical basis quantum effect solution of the SR numerically expensive (only small molecules can be treated) approximations
classical description of molecules April 29, 2016 Chemical bond Conceptual and chemical basis quantum effect solution of the SR numerically expensive (only small molecules can be treated) approximations
Thus, the volume element remains the same as required. With this transformation, the amiltonian becomes = p i m i + U(r 1 ; :::; r N ) = and the canon
 = and the canon") G5.651: Statistical Mechanics Notes for Lecture 5 From the classical virial theorem I. TEMPERATURE AND PRESSURE ESTIMATORS hx i x j i = kt ij we arrived at the equipartition theorem: * + p i = m i NkT
G5.651: Statistical Mechanics Notes for Lecture 5 From the classical virial theorem I. TEMPERATURE AND PRESSURE ESTIMATORS hx i x j i = kt ij we arrived at the equipartition theorem: * + p i = m i NkT
Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012
 Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012 K. Kremer Max Planck Institute for Polymer Research, Mainz Overview Simulations, general considerations
Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012 K. Kremer Max Planck Institute for Polymer Research, Mainz Overview Simulations, general considerations