Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
|
|
|
- Darrell Ramsey
- 6 years ago
- Views:
Transcription
1 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction
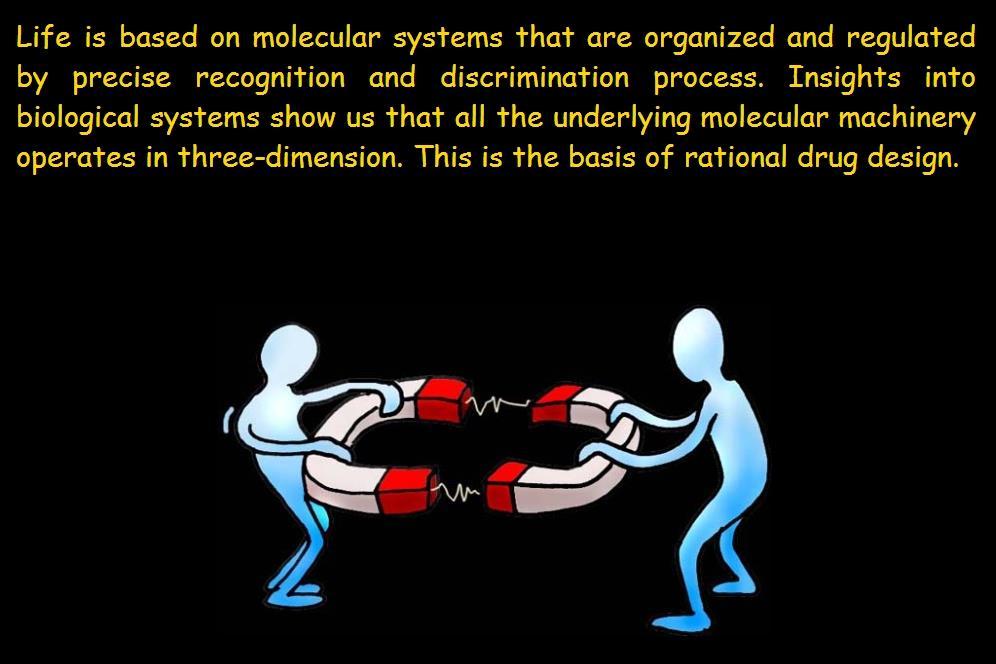
2 Introduction

3 Introduction
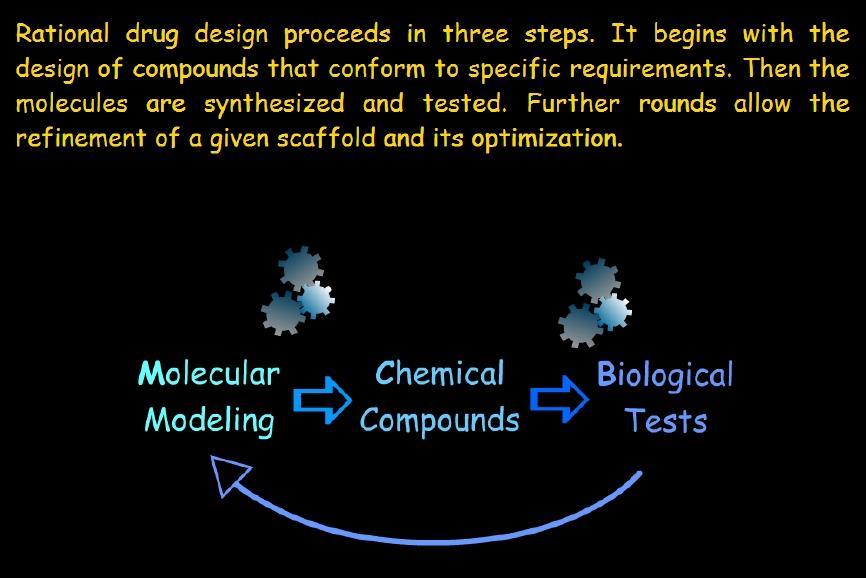
4 Introduction
5 Computer Chemistry in Drug Design Drug Discovery: Target identification Lead discovery Lead optimization Computer chemistry? Drug Development Pre-clinical development Clinical development Post-marketing surveillance
6 Computer Chemistry in Drug Design How a lead for a new drug can be discovered? Serendipitous pathway Screening pathway Chemical modification pathway Rational pathway
7 Rational Drug Design Unlike the historical method of drug discovery, by trial-and-error testing of chemical substances on animals, and matching the apparent effects to treatments, rational drug design begins with a knowledge of specific chemical responses in the body or target organism, and tailoring combinations of these to fit a treatment profile.
8 Computer Aided Drug Design Drug design is an empirical procedure Rational drug design is the rational use of as more experimental data as possible Computer chemistry is a tool for rationally extracting all information from data Computer chemistry aids (does not substitute) the experimental procedures At present drug design without computer chemistry is largely inefficient
9 Computer Chemistry in Drug Design Computer chemistry tools for drug design Molecular modeling Structure-based Drug Design Ligand-based Drug Design Chemiometrics approaches
10 Computer Chemistry in Drug Design Molecular modeling Model building Interactive molecular graphics Molecule building through sketcher or fragments Conformational minimization Molecular mechanics Quantum mechanics Conformational analysis Sistematic search Montecarlo methods Molecular dynamics
11 Computer Chemistry in Drug Design Structure (Receptor)-based drug design Molecular docking Ligand (Pharmacophore)-based drug design Pharmacophore modeling Chemiometrics approaches QSAR 3D-QSAR
12 Molecular modeling Molecular model Computer graphics atomic coordinates cartesian coordinates (good for computers) x, y, z coordinates of atoms PDB, MOL2 files internal coordinates (good for men) bond lengths and angles, torsions Z-matrix

13 Molecular graphics Molecular modeling

14 Molecular modeling Model building (by fragment)

15 Molecular modeling Model building (sketcher)
16 Molecular modeling Conformations stable conformation local minimum preferred conformation absolute minimum bioactive conformation?????? Building by fragment poor conformation Sketcher wrong conformation It needs some optimization

17 Molecular Energies

18 Molecular Energies

19 Molecular Energies
20 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Molecular Mechanics

21 WHAT IS MOLECULAR MECHANICS (MM) The mechanical molecular model (or force field model) was developed to describe molecular structures and properties in as practical a manner as possible, ignoring the electronic motions and calculating the energy of a system as a function of the nuclear position only.
22 WHAT IS A FORCE -FIELD A force field is the whole of the functional form and parameter sets used to describe the potential energy of a system of particles. Energies are associated with the deviation of bonds and angles away from their reference values There is a function that describes how the energy changes as bonds are rotated Contains terms that describes the interaction between non bonded parts of the system

23 N 1 i N 1 i j ij r 0 4π j q i q 6 ij r ij σ 12 ij r ij σ ij 4ε cos bonds 2 γ nω torsions V ι, θ ι θ angles k ι, l ι l k Ν r Ε n i i function that describes how the energy changes as bonds changes..
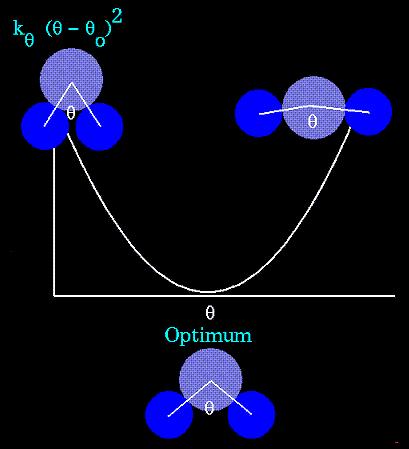
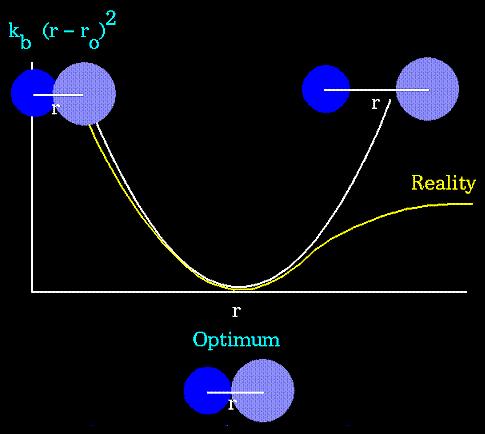
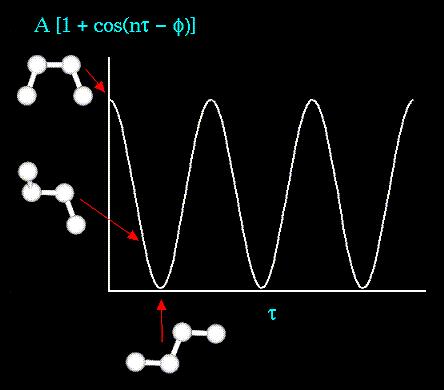
24 STRETCHING, BENDING, TORSION TERMS

25 N 1 i N 1 i j ij r 0 4π j q i q 6 ij r ij σ 12 ij r ij σ ij 4ε NON BONDED TERMS

26 Empirical force-field methodology is based on classical mechanics and on the assumption that the total steric energy of a structure can be expressed as sum of contributions from many interaction types.
27 EXACT LOCATION OF MINIMA When we reach a minimum? It is rarely possible to identify the real minimum, relative or absolute; we can only hope to find an approssimation to the true minimum. A simple strategy to decide when the calculation is sufficiently close to the minimum is to monitor the energy from one iteration to the next, and to stop when the difference in energy between successive steps falls below a specified threshold. So, we can say that the minimization reach the convergence.

28 MINIMIZATION METHODS

29 Molecular dynamics

30 Molecular dynamics MD: a simulation of the particle motion


31 Molecular dynamics The motion (determined by the temperature) allows conformational changes
32 Molecular dynamics Calculates the time dependent behaviour of a molecular system Provides detailed information on the fluctuations and conformational changes of macromolecules Routinely used to investigate the structure, dynamics and thermodynamics of biological molecules Used in the determination of structures from x- ray and NMR experiments
33 Molecular dynamics In a molecular dynamics (MD) simulation it is possible to explore the macroscopic properties of a system The connection between microscopic simulation and macroscopic properties is made through statistical mechanics Allows to study both thermodynamic properties and time dependent (kinetic) phenomenon
34 Molecular dynamics A MD simulation is practically carried out through the application of the Newton law: f = m x a The motion of each particle of the system is calculated from a a is calculated from f f is calculated from the potential V
35 Molecular dynamics A very simplified MD run
36 Molecular dynamics The potential V can be calculated at different accuracy level (from MM to QM) In biology the potential V is generally obtained by a MM force field This is a classical treatment allowing the calculation of conformational changes but usually it is not able to reproduce chemical reactions
37 Molecular dynamics t cannot be longer than the fastest atomic motion, therefore: t = consequently a simulation of a microsecond needs one billion steps
38 Molecular dynamics Temperature is directly correlated with kynetic energy: Generally a free evolution of the system is not allowed. Constraints on temperature and/or pressure are imposed in order to reproduce a particular ensemble.
39 Molecular dynamics Environment simulation The solvent can be simulated in an implicit and in an explicit manner. Implicit solvent (in most cases the continuum approximation is used): fast calculation but poor results Explicit solvent (periodic boundary conditions are generally used): accurate results but time consuming
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,..
 3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Principles of Drug Design
 Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Principles of Drug Design
 (16:663:502) Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu For more current information, please check WebCT at https://webct.rutgers.edu
(16:663:502) Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu For more current information, please check WebCT at https://webct.rutgers.edu
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS
 EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS PETER GUND Pharmacopeia Inc., CN 5350 Princeton, NJ 08543, USA pgund@pharmacop.com Empirical and theoretical approaches to drug discovery have often
EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS PETER GUND Pharmacopeia Inc., CN 5350 Princeton, NJ 08543, USA pgund@pharmacop.com Empirical and theoretical approaches to drug discovery have often
Computer Graphics Applications on Molecular Biology and Drug Design
 Computer Graphics Applications on Molecular Biology and Drug Design Katerina PERDIKURI 1, Athanasios TSAKALIDIS 1 1 Department of Computer Engineering and Informatics University of Patras,26500 Patras,
Computer Graphics Applications on Molecular Biology and Drug Design Katerina PERDIKURI 1, Athanasios TSAKALIDIS 1 1 Department of Computer Engineering and Informatics University of Patras,26500 Patras,
1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?!
 for structure refinement from X-ray diffraction data?!") 1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?! Visual analysis: Identification of ligand density from
1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?! Visual analysis: Identification of ligand density from
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
ENERGY MINIMIZATION AND CONFORMATION SEARCH ANALYSIS OF TYPE-2 ANTI-DIABETES DRUGS
 Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Energy functions and their relationship to molecular conformation. CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror
 Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Outline Energy functions for proteins (or biomolecular systems more generally)
Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Outline Energy functions for proteins (or biomolecular systems more generally)
I690/B680 Structural Bioinformatics Spring Protein Structure Determination by NMR Spectroscopy
 I690/B680 Structural Bioinformatics Spring 2006 Protein Structure Determination by NMR Spectroscopy Suggested Reading (1) Van Holde, Johnson, Ho. Principles of Physical Biochemistry, 2 nd Ed., Prentice
I690/B680 Structural Bioinformatics Spring 2006 Protein Structure Determination by NMR Spectroscopy Suggested Reading (1) Van Holde, Johnson, Ho. Principles of Physical Biochemistry, 2 nd Ed., Prentice
Homework Problem Set 1 Solutions
 Chemistry 380.37 Dr. Jean M. Standard omework Problem Set 1 Solutions 1. A student investigates a bond between atoms A and B in a molecule using a software package for molecular mechanics. The student
Chemistry 380.37 Dr. Jean M. Standard omework Problem Set 1 Solutions 1. A student investigates a bond between atoms A and B in a molecule using a software package for molecular mechanics. The student
Methods of Computer Simulation. Molecular Dynamics and Monte Carlo
 Molecular Dynamics Time is of the essence in biological processes therefore how do we understand time-dependent processes at the molecular level? How do we do this experimentally? How do we do this computationally?
Molecular Dynamics Time is of the essence in biological processes therefore how do we understand time-dependent processes at the molecular level? How do we do this experimentally? How do we do this computationally?
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
Modeling Biological Systems Opportunities for Computer Scientists
 Modeling Biological Systems Opportunities for Computer Scientists Filip Jagodzinski RBO Tutorial Series 25 June 2007 Computer Science Robotics & Biology Laboratory Protein: πρώτα, "prota, of Primary Importance
Modeling Biological Systems Opportunities for Computer Scientists Filip Jagodzinski RBO Tutorial Series 25 June 2007 Computer Science Robotics & Biology Laboratory Protein: πρώτα, "prota, of Primary Importance
Scuola di Chimica Computazionale
 Societa Chimica Italiana Gruppo Interdivisionale di Chimica Computazionale Scuola di Chimica Computazionale Introduzione, per Esercizi, all Uso del Calcolatore in Chimica Organica e Biologica Modellistica
Societa Chimica Italiana Gruppo Interdivisionale di Chimica Computazionale Scuola di Chimica Computazionale Introduzione, per Esercizi, all Uso del Calcolatore in Chimica Organica e Biologica Modellistica
Modeling Biomolecular Systems II. BME 540 David Sept
 Modeling Biomolecular Systems II BME 540 David Sept Introduction Why do we perform simulations? What makes simulations possible? How do we perform simulations? What types of things/systems do we simulate?
Modeling Biomolecular Systems II BME 540 David Sept Introduction Why do we perform simulations? What makes simulations possible? How do we perform simulations? What types of things/systems do we simulate?
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Protein Simulations in Confined Environments
 Critical Review Lecture Protein Simulations in Confined Environments Murat Cetinkaya 1, Jorge Sofo 2, Melik C. Demirel 1 1. 2. College of Engineering, Pennsylvania State University, University Park, 16802,
Critical Review Lecture Protein Simulations in Confined Environments Murat Cetinkaya 1, Jorge Sofo 2, Melik C. Demirel 1 1. 2. College of Engineering, Pennsylvania State University, University Park, 16802,
Molecular Modelling for Medicinal Chemistry (F13MMM) Room A36
 Room A36") Molecular Modelling for Medicinal Chemistry (F13MMM) jonathan.hirst@nottingham.ac.uk Room A36 http://comp.chem.nottingham.ac.uk Assisted reading Molecular Modelling: Principles and Applications. Andrew
Molecular Modelling for Medicinal Chemistry (F13MMM) jonathan.hirst@nottingham.ac.uk Room A36 http://comp.chem.nottingham.ac.uk Assisted reading Molecular Modelling: Principles and Applications. Andrew
Improving Protein Function Prediction with Molecular Dynamics Simulations. Dariya Glazer Russ Altman
 Improving Protein Function Prediction with Molecular Dynamics Simulations Dariya Glazer Russ Altman Motivation Sometimes the 3D structure doesn t score well for a known function. The experimental structure
Improving Protein Function Prediction with Molecular Dynamics Simulations Dariya Glazer Russ Altman Motivation Sometimes the 3D structure doesn t score well for a known function. The experimental structure
The Conformation Search Problem
 Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Protein Dynamics, Allostery and Function
 Protein Dynamics, Allostery and Function Lecture 3. Protein Dynamics Xiaolin Cheng UT/ORNL Center for Molecular Biophysics SJTU Summer School 2017 1 Obtaining Dynamic Information Experimental Approaches
Protein Dynamics, Allostery and Function Lecture 3. Protein Dynamics Xiaolin Cheng UT/ORNL Center for Molecular Biophysics SJTU Summer School 2017 1 Obtaining Dynamic Information Experimental Approaches
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Molecular Mechanics. Yohann Moreau. November 26, 2015
 Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit
 October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
Force fields in computer simulation of soft nanomaterials
 Lecture 2 Part B Force fields in computer simulation of soft nanomaterials Recommended reading: Leach Chapter 4 1 Force Field Methods Force field methods are simulation methods that use classical force
Lecture 2 Part B Force fields in computer simulation of soft nanomaterials Recommended reading: Leach Chapter 4 1 Force Field Methods Force field methods are simulation methods that use classical force
Structure-Based Drug Discovery An Overview
 Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane
 A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane Supplementary Information: Simulation Procedure and Physical Property Analysis Simulation Procedure The molecular
A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane Supplementary Information: Simulation Procedure and Physical Property Analysis Simulation Procedure The molecular
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Energy functions and their relationship to molecular conformation. CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror
 Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Yesterday s Nobel Prize: single-particle cryoelectron microscopy 2 Outline Energy
Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Yesterday s Nobel Prize: single-particle cryoelectron microscopy 2 Outline Energy
Introduction to Computational Structural Biology
 Introduction to Computational Structural Biology Part I 1. Introduction The disciplinary character of Computational Structural Biology The mathematical background required and the topics covered Bibliography
Introduction to Computational Structural Biology Part I 1. Introduction The disciplinary character of Computational Structural Biology The mathematical background required and the topics covered Bibliography
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Chemistry 4560/5560 Molecular Modeling Fall 2014
 Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors
 MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
Statistical Mechanics for Proteins
 The Partition Function From Q all relevant thermodynamic properties can be obtained by differentiation of the free energy F: = kt q p E q pd d h T V Q ), ( exp 1! 1 ),, ( 3 3 3 ),, ( ln ),, ( T V Q kt
The Partition Function From Q all relevant thermodynamic properties can be obtained by differentiation of the free energy F: = kt q p E q pd d h T V Q ), ( exp 1! 1 ),, ( 3 3 3 ),, ( ln ),, ( T V Q kt
Molecular Modelling. Computational Chemistry Demystified. RSC Publishing. Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK
 Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Exercise 2: Solvating the Structure Before you continue, follow these steps: Setting up Periodic Boundary Conditions
 Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
This semester. Books
 Models mostly proteins from detailed to more abstract models Some simulation methods This semester Books None necessary for my group and Prof Rarey Molecular Modelling: Principles and Applications Leach,
Models mostly proteins from detailed to more abstract models Some simulation methods This semester Books None necessary for my group and Prof Rarey Molecular Modelling: Principles and Applications Leach,
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
Toward an Understanding of GPCR-ligand Interactions. Alexander Heifetz
 Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
An Informal AMBER Small Molecule Force Field :
 An Informal AMBER Small Molecule Force Field : parm@frosst Credit Christopher Bayly (1992-2010) initiated, contributed and lead the efforts Daniel McKay (1997-2010) and Jean-François Truchon (2002-2010)
An Informal AMBER Small Molecule Force Field : parm@frosst Credit Christopher Bayly (1992-2010) initiated, contributed and lead the efforts Daniel McKay (1997-2010) and Jean-François Truchon (2002-2010)
Javier Junquera. Statistical mechanics
 Javier Junquera Statistical mechanics From the microscopic to the macroscopic level: the realm of statistical mechanics Computer simulations Thermodynamic state Generates information at the microscopic
Javier Junquera Statistical mechanics From the microscopic to the macroscopic level: the realm of statistical mechanics Computer simulations Thermodynamic state Generates information at the microscopic
Computational structural biology and bioinformatics
 Computational structural biology and bioinformatics What is it all about? Why take it? What are we going to be doing? Organizational notes. Grades etc. Books. CS6104. Spring CS6104. 04. Spring Alexey 04.
Computational structural biology and bioinformatics What is it all about? Why take it? What are we going to be doing? Organizational notes. Grades etc. Books. CS6104. Spring CS6104. 04. Spring Alexey 04.
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
Advanced Molecular Dynamics
 Advanced Molecular Dynamics Introduction May 2, 2017 Who am I? I am an associate professor at Theoretical Physics Topics I work on: Algorithms for (parallel) molecular simulations including GPU acceleration
Advanced Molecular Dynamics Introduction May 2, 2017 Who am I? I am an associate professor at Theoretical Physics Topics I work on: Algorithms for (parallel) molecular simulations including GPU acceleration
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining
![Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining](/thumbs/91/107031261.jpg "Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining") Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Molecular Simulation II. Classical Mechanical Treatment
 Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
CHAPTER 6 QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIP (QSAR) ANALYSIS
 ANALYSIS") 159 CHAPTER 6 QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIP (QSAR) ANALYSIS 6.1 INTRODUCTION The purpose of this study is to gain on insight into structural features related the anticancer, antioxidant
159 CHAPTER 6 QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIP (QSAR) ANALYSIS 6.1 INTRODUCTION The purpose of this study is to gain on insight into structural features related the anticancer, antioxidant
Keywords: anti-coagulants, factor Xa, QSAR, Thrombosis. Introduction
 PostDoc Journal Vol. 2, No. 3, March 2014 Journal of Postdoctoral Research www.postdocjournal.com QSAR Study of Thiophene-Anthranilamides Based Factor Xa Direct Inhibitors Preetpal S. Sidhu Department
PostDoc Journal Vol. 2, No. 3, March 2014 Journal of Postdoctoral Research www.postdocjournal.com QSAR Study of Thiophene-Anthranilamides Based Factor Xa Direct Inhibitors Preetpal S. Sidhu Department
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Biomolecules are dynamic no single structure is a perfect model
 Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Monte Carlo (MC) Simulation Methods. Elisa Fadda
 Simulation Methods. Elisa Fadda") Monte Carlo (MC) Simulation Methods Elisa Fadda 1011-CH328, Molecular Modelling & Drug Design 2011 Experimental Observables A system observable is a property of the system state. The system state i is
Monte Carlo (MC) Simulation Methods Elisa Fadda 1011-CH328, Molecular Modelling & Drug Design 2011 Experimental Observables A system observable is a property of the system state. The system state i is
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Nonlinear QSAR and 3D QSAR
 onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
Computational Studies of the Photoreceptor Rhodopsin. Scott E. Feller Wabash College
 Computational Studies of the Photoreceptor Rhodopsin Scott E. Feller Wabash College Rhodopsin Photocycle Dark-adapted Rhodopsin hn Isomerize retinal Photorhodopsin ~200 fs Bathorhodopsin Meta-II ms timescale
Computational Studies of the Photoreceptor Rhodopsin Scott E. Feller Wabash College Rhodopsin Photocycle Dark-adapted Rhodopsin hn Isomerize retinal Photorhodopsin ~200 fs Bathorhodopsin Meta-II ms timescale
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
Virtual screening for drug discovery. Markus Lill Purdue University
 Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Molecular Dynamics Simulations. Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia
 Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Statistical concepts in QSAR.
 Statistical concepts in QSAR. Computational chemistry represents molecular structures as a numerical models and simulates their behavior with the equations of quantum and classical physics. Available programs
Statistical concepts in QSAR. Computational chemistry represents molecular structures as a numerical models and simulates their behavior with the equations of quantum and classical physics. Available programs
Biophysical and Bioengineering Methods for the Study of the Complement System at Atomic Resolution
 Biophysical and Bioengineering Methods for the Study of the Complement System at Atomic Resolution dmorikis@engr.ucr.edu DIMITRIOS MORIKIS Department of Bioengineering University of California, Riverside
Biophysical and Bioengineering Methods for the Study of the Complement System at Atomic Resolution dmorikis@engr.ucr.edu DIMITRIOS MORIKIS Department of Bioengineering University of California, Riverside
The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii
 And the Basic Force Field Chem 4021/8021 Video II.iii") The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii Fundamental Points About Which to Be Thinking It s clear the PES is useful, so how can I construct it for an arbitrary
The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii Fundamental Points About Which to Be Thinking It s clear the PES is useful, so how can I construct it for an arbitrary
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Ping-Chiang Lyu. Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University.
 Pharmacophore-based Drug design Ping-Chiang Lyu Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University 96/08/07 Outline Part I: Analysis The analytical
Pharmacophore-based Drug design Ping-Chiang Lyu Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University 96/08/07 Outline Part I: Analysis The analytical
Modelação e Simulação de Sistemas para Micro/Nano Tecnologias
 Modelação e Simulação de Sistemas para Micro/Nano Tecnologias http://gec.di.uminho.pt/mmnt/modsim/ Alberto José Proença, António Joaquim Esteves 2011/12 Mestrado em Micro/Nano Tecnologias ESCOLA DE ENGENHARIA
Modelação e Simulação de Sistemas para Micro/Nano Tecnologias http://gec.di.uminho.pt/mmnt/modsim/ Alberto José Proença, António Joaquim Esteves 2011/12 Mestrado em Micro/Nano Tecnologias ESCOLA DE ENGENHARIA
COMBINATORIAL CHEMISTRY: CURRENT APPROACH
 COMBINATORIAL CHEMISTRY: CURRENT APPROACH Dwivedi A. 1, Sitoke A. 2, Joshi V. 3, Akhtar A.K. 4* and Chaturvedi M. 1, NRI Institute of Pharmaceutical Sciences, Bhopal, M.P.-India 2, SRM College of Pharmacy,
COMBINATORIAL CHEMISTRY: CURRENT APPROACH Dwivedi A. 1, Sitoke A. 2, Joshi V. 3, Akhtar A.K. 4* and Chaturvedi M. 1, NRI Institute of Pharmaceutical Sciences, Bhopal, M.P.-India 2, SRM College of Pharmacy,
Applications of Molecular Dynamics
 June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
Solvatochromic shifts of a polarity probe implicit and explicit solvent modeling
 Solvatochromic shifts of a polarity probe implicit and explicit solvent modeling Andrzej Eilmes Faculty of Chemistry, Jagiellonian University, Kraków solvent effects in absorption spectra Absorption spectra
Solvatochromic shifts of a polarity probe implicit and explicit solvent modeling Andrzej Eilmes Faculty of Chemistry, Jagiellonian University, Kraków solvent effects in absorption spectra Absorption spectra
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
T6.2 Molecular Mechanics
 T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
FRAUNHOFER IME SCREENINGPORT
 FRAUNHOFER IME SCREENINGPORT Design of screening projects General remarks Introduction Screening is done to identify new chemical substances against molecular mechanisms of a disease It is a question of
FRAUNHOFER IME SCREENINGPORT Design of screening projects General remarks Introduction Screening is done to identify new chemical substances against molecular mechanisms of a disease It is a question of
Quantum Chemical Simulations and Descriptors. Dr. Antonio Chana, Dr. Mosè Casalegno
 Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation
 Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation Charles Blundell charles.blundell@c4xdiscovery.com www.c4xdiscovery.com Rigid: single
Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation Charles Blundell charles.blundell@c4xdiscovery.com www.c4xdiscovery.com Rigid: single
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions