Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
|
|
|
- Jerome Barton
- 5 years ago
- Views:
Transcription
1 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2
2 If you throw up a stone, it is Physics.
3 If you throw up a stone, it is Physics. If it lands on your head, it is Biophysics.
4 If you throw up a stone, it is Physics. If it lands on your head, it is Biophysics. If you write a computer program, it is Informatics.
5 If you throw up a stone, it is Physics. If it lands on your head, it is Biophysics. If you write a computer program, it is Informatics. If there is a bug in it, it is Bioinformatics

6
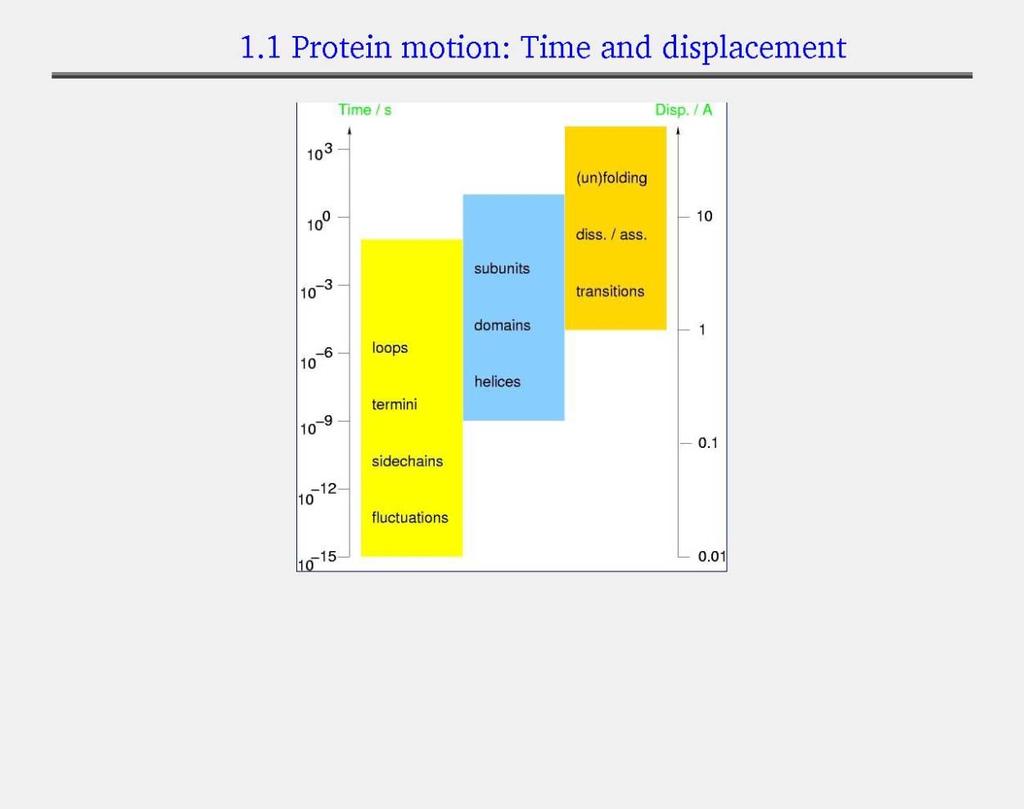
7

8 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided
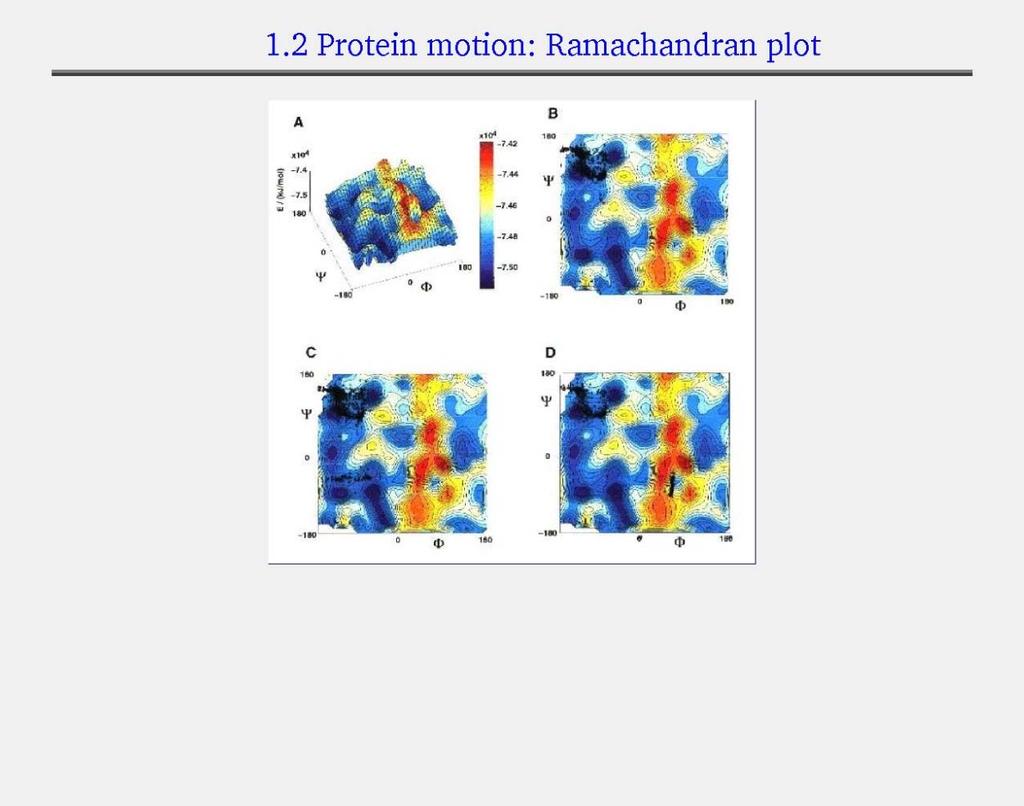
9
10

11

12
13 2.3a Hamiltonian equations of motion Hamiltonian equations (one degree of freedom): H Hamiltonian function, Hamiltonian, q, p Canonical variables: generalized coordinate (q) and momentum [impulses] (p).

14 q = coordinates p = momentum
 - r(t -")
15 v(t) = (r(t + t) - r(t - t))/2 t
16 Molecular systems strive to be in the lowest energy state

17 Molecular Dynamics Knowledge of the atomic forces and masses can be used to solve the position of each atom along a series of extremely small time steps (on the order of femtoseconds = seconds). The resulting series of snapshots of structural changes over time is called a trajectory. The use of this method to compute trajectories can be more easily seen when Newton's equation is expressed in the following form: The "leapfrog" method is a common numerical approach to calculating trajectories based on Newton's equation. The steps can be summarized as follows:

18
19 Force field The potential energy of a system can be expressed as a sum of valence (or bond), crossterm, and nonbond interactions: The energy of valence interactions comprises bond stretching (E bond ), valence angle bending (E angle ), dihedral angle torsion (E torsion ), and inversion (also called out-ofplane interactions) (E inversion or E oop ) terms, which are part of nearly all force fields for covalent systems. A Urey-Bradley term (E UB ) may be used to account for interactions between atom pairs involved in 1-3 configurations (i.e., atoms bound to a common atom): E valence = E bond + E angle + E torsion + E oop + E UB Modern (second-generation) forcefields include cross terms to account for such factors as bond or angle distortions caused by nearby atoms. Crossterms can include the following terms: stretch-stretch, stretch-bend-stretch, bend-bend, torsion-stretch, torsion-bend-bend, bend-torsion-bend, stretch-torsion-stretch. The energy of interactions between nonbonded atoms is accounted for by van der Waals (E vdw ), electrostatic (E Coulomb ), and (in some older forcefields) hydrogen bond (E hbond ) terms: E nonbond = E vdw + E Coulomb + E hbond
20 Force field

21 energy distance f = a/r 12 -b/r 6 Van der Waals forces The Lennard-Jones potential is mildly attractive as two uncharged molecules or atoms approach one another from a distance, but strongly repulsive when they approach too close. The resulting potential is shown. At equilibrium, the pair of atoms or molecules tend to go toward a separation corresponding to the minimum of the Lennard--Jones potential (a separation of 0.38 nanometers for the case shown in the Figure)
22 F = kqiqj/r2
23

24 Figure: Snapshots of ubiquitin pulling with constant velocity at three different time steps.
25 ERGODIC (l) of or relating to a process in which a sequence or sizable sample is equally representative of the whole (as in regard to a statistical parameter); (2) involving or relating to the probability that any state will recur, especially having zero probability that any state will never recur. (WEBSTER'S DICTIONARY) A collection of systems forms an ergodic ensemble if the modes of behavior found in any one system from time to time resemble its behavior at other temporal periods and if the behavior of any other system when chosen at random also is like the one system. We do not require identical performance, only quite similar time averages and number averages. (If you cannot tell one youth from another or one adult from another, they belong to an ergodic ensemble.) In an ergodic population, any single individual is representative of the entire population. The salient characteristics of this individual are essentially identical with any other member of the group. (Iberall)

26
27
28 #! /usr/bin/perl #=============================================================================== # # $Id: mcdemo.pl,v /03/12 16:13:28 jkleinj Exp $ # # mcdemo: Demo program for MC simulation of the number pi # # (C) 2003 Jens Kleinjung # # Dr Jens Kleinjung, Room P440 jkleinj@cs.vu.nl # Bioinformatics Unit, Faculty of Sciences Tel # Free University Amsterdam Fax # De Boelelaan 1081A, 1081 HV Amsterdam # #=============================================================================== # preset parameters $hits = 1; $miss = 1; for ($i=0; $i<100000; $i++) { # assign random x,y coordinates $x = rand; $y = rand; # calculate radius $r = sqrt(($x*$x)+($y*$y)); # sum up hits and misses if ($r <= 1) { $hits++; } else { $miss++; } # calculate pi $pi = (4*$hits)/($hits +$miss); # print pi if ($i%100 == 0) { print("$i $pi\n"); } } #===============================================================================

29
30 In many conformational search methods based on Monte Carlo (MC), after a MC move, the system is energy minimised, i.e. put in the lowest local energy conformation, for example by gradient descent (steepest descent).

31

32
33
34

35 antibody HyHEL-63 (cyan) complexed with Hen Egg White Lysozyme The X-ray structure of the antibody HyHEL-63 (cyan) uncomplexed and complexed with Hen Egg White Lysozyme (yellow) has shown that there are small but significant, local conformational changes in the antibody paratope on binding. The structure also reveals that most of the charged epitope residues face the antibody. Details are in Li YL, Li HM, Smith-Gill SJ and Mariuzza RA (2000) The conformations of the X-ray structure Three-dimensional structures of the free and antigen-bound Fab from monoclonal antilysozyme antibody HyHEL-63. Biochemistry 39: Salt links and electrostatic interactions provide much of the free energy of binding. Most of the charged residues face in interface in the X-ray structure. The importance of the salt link between Lys97 of HEL and Asp27 of the antibody heavy chain is revealed by molecular dynamics simulations. After 1NSec of MD simulation at 100 C the overall conformation of the complex has changed, but the salt link persists. Details are described in Sinha N and Smith-Gill SJ (2002) Electrostatics in protein binding and function. Current Protein & Peptide Science 3:
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
Today s lecture. Molecular Mechanics and docking. Lecture 22. Introduction to Bioinformatics Docking - ZDOCK. Protein-protein docking
 C N F O N G A V B O N F O M A C S V U Molecular Mechanics and docking Lecture 22 ntroduction to Bioinformatics 2007 oday s lecture 1. Protein interaction and docking a) Zdock method 2. Molecular motion
C N F O N G A V B O N F O M A C S V U Molecular Mechanics and docking Lecture 22 ntroduction to Bioinformatics 2007 oday s lecture 1. Protein interaction and docking a) Zdock method 2. Molecular motion
Experimentally solving protein structures, protein-protein interactions and simulating protein dynamics. Lecture 15
 Experimentally solving protein structures, protein-protein interactions and simulating protein dynamics C E N T R F O R I N T B I O I N F E Lecture 15 Introduction to Bioinformatics 2007 E G R A T I V
Experimentally solving protein structures, protein-protein interactions and simulating protein dynamics C E N T R F O R I N T B I O I N F E Lecture 15 Introduction to Bioinformatics 2007 E G R A T I V
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015,
 Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Biophysics II. Hydrophobic Bio-molecules. Key points to be covered. Molecular Interactions in Bio-molecular Structures - van der Waals Interaction
 Biophysics II Key points to be covered By A/Prof. Xiang Yang Liu Biophysics & Micro/nanostructures Lab Department of Physics, NUS 1. van der Waals Interaction 2. Hydrogen bond 3. Hydrophilic vs hydrophobic
Biophysics II Key points to be covered By A/Prof. Xiang Yang Liu Biophysics & Micro/nanostructures Lab Department of Physics, NUS 1. van der Waals Interaction 2. Hydrogen bond 3. Hydrophilic vs hydrophobic
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Free energy, electrostatics, and the hydrophobic effect
 Protein Physics 2016 Lecture 3, January 26 Free energy, electrostatics, and the hydrophobic effect Magnus Andersson magnus.andersson@scilifelab.se Theoretical & Computational Biophysics Recap Protein structure
Protein Physics 2016 Lecture 3, January 26 Free energy, electrostatics, and the hydrophobic effect Magnus Andersson magnus.andersson@scilifelab.se Theoretical & Computational Biophysics Recap Protein structure
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Biochemistry Prof. S. DasGupta Department of Chemistry Indian Institute of Technology Kharagpur. Lecture - 06 Protein Structure IV
 Biochemistry Prof. S. DasGupta Department of Chemistry Indian Institute of Technology Kharagpur Lecture - 06 Protein Structure IV We complete our discussion on Protein Structures today. And just to recap
Biochemistry Prof. S. DasGupta Department of Chemistry Indian Institute of Technology Kharagpur Lecture - 06 Protein Structure IV We complete our discussion on Protein Structures today. And just to recap
Gerd Krause, Structural bioinformatics and protein design Leibniz-Institute of molecular Pharmacology
 Basics of Molecular modelling _2 History, Interdisciplinary classification, Goals, Model definition, Molecular mechanics forcefields, Energy minimization algorithms Molecular dynamics simulation Comparative
Basics of Molecular modelling _2 History, Interdisciplinary classification, Goals, Model definition, Molecular mechanics forcefields, Energy minimization algorithms Molecular dynamics simulation Comparative
Molecular Dynamics Simulations. Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia
 Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Methods of Computer Simulation. Molecular Dynamics and Monte Carlo
 Molecular Dynamics Time is of the essence in biological processes therefore how do we understand time-dependent processes at the molecular level? How do we do this experimentally? How do we do this computationally?
Molecular Dynamics Time is of the essence in biological processes therefore how do we understand time-dependent processes at the molecular level? How do we do this experimentally? How do we do this computationally?
Supplement information
 Electronic Supplementary Material (ESI) for Physil Chemistry Chemil Physics. This journal is the Owner Societies 216 Supplement information Fullerenol C 6 (OH) 16 prevents amyloid fibrillization of Aβ
Electronic Supplementary Material (ESI) for Physil Chemistry Chemil Physics. This journal is the Owner Societies 216 Supplement information Fullerenol C 6 (OH) 16 prevents amyloid fibrillization of Aβ
3. An Introduction to Molecular Mechanics
 3. An Introduction to Molecular Mechanics Introduction When you use Chem3D to draw molecules, the program assigns bond lengths and bond angles based on experimental data. The program does not contain real
3. An Introduction to Molecular Mechanics Introduction When you use Chem3D to draw molecules, the program assigns bond lengths and bond angles based on experimental data. The program does not contain real
Proteins polymer molecules, folded in complex structures. Konstantin Popov Department of Biochemistry and Biophysics
 Proteins polymer molecules, folded in complex structures Konstantin Popov Department of Biochemistry and Biophysics Outline General aspects of polymer theory Size and persistent length of ideal linear
Proteins polymer molecules, folded in complex structures Konstantin Popov Department of Biochemistry and Biophysics Outline General aspects of polymer theory Size and persistent length of ideal linear
Molecular Modelling. part of Bioinformatik von RNA- und Proteinstrukturen. Sonja Prohaska. Leipzig, SS Computational EvoDevo University Leipzig
 part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
Molecular Dynamics Simulation of HIV-1 Reverse. Transcriptase
 Molecular Dynamics Simulation of HIV-1 Reverse Transcriptase Abderrahmane Benghanem 1 Maria Kurnikova 2 1 Rensselaer Polytechnic Institute, Troy, NY 2 Carnegie Mellon University, Pittsburgh, PA June 16,
Molecular Dynamics Simulation of HIV-1 Reverse Transcriptase Abderrahmane Benghanem 1 Maria Kurnikova 2 1 Rensselaer Polytechnic Institute, Troy, NY 2 Carnegie Mellon University, Pittsburgh, PA June 16,
Why Proteins Fold. How Proteins Fold? e - ΔG/kT. Protein Folding, Nonbonding Forces, and Free Energy
 Why Proteins Fold Proteins are the action superheroes of the body. As enzymes, they make reactions go a million times faster. As versatile transport vehicles, they carry oxygen and antibodies to fight
Why Proteins Fold Proteins are the action superheroes of the body. As enzymes, they make reactions go a million times faster. As versatile transport vehicles, they carry oxygen and antibodies to fight
Biomolecules are dynamic no single structure is a perfect model
 Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Computational Biology & Computational Medicine
 Computational Biology & Computational Medicine Homayoun Valafar Outline Why proteins? What are proteins? How do we compute them? How do we use computational approaches? Why Proteins? Molecular basis of
Computational Biology & Computational Medicine Homayoun Valafar Outline Why proteins? What are proteins? How do we compute them? How do we use computational approaches? Why Proteins? Molecular basis of
Other Cells. Hormones. Viruses. Toxins. Cell. Bacteria
 Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Bulk behaviour. Alanine. FIG. 1. Chemical structure of the RKLPDA peptide. Numbers on the left mark alpha carbons.
 Bulk behaviour To characterise the conformational behaviour of the peptide, first we looked at the statistics of alpha carbons and the torsion angles. Then they were correlated with positions of positively
Bulk behaviour To characterise the conformational behaviour of the peptide, first we looked at the statistics of alpha carbons and the torsion angles. Then they were correlated with positions of positively
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
3. An Introduction to Molecular Mechanics
 3. An Introduction to Molecular Mechanics Introduction When you use Chem3D to draw molecules, the program assigns bond lengths and bond angles based on experimental data. The program does not contain real
3. An Introduction to Molecular Mechanics Introduction When you use Chem3D to draw molecules, the program assigns bond lengths and bond angles based on experimental data. The program does not contain real
Energy functions and their relationship to molecular conformation. CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror
 Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Yesterday s Nobel Prize: single-particle cryoelectron microscopy 2 Outline Energy
Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Yesterday s Nobel Prize: single-particle cryoelectron microscopy 2 Outline Energy
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
Semi Empirical Force Fields and Their Limitations. Potential Energy Surface (PES)
") Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
Using Higher Calculus to Study Biologically Important Molecules Julie C. Mitchell
 Using Higher Calculus to Study Biologically Important Molecules Julie C. Mitchell Mathematics and Biochemistry University of Wisconsin - Madison 0 There Are Many Kinds Of Proteins The word protein comes
Using Higher Calculus to Study Biologically Important Molecules Julie C. Mitchell Mathematics and Biochemistry University of Wisconsin - Madison 0 There Are Many Kinds Of Proteins The word protein comes
Molecular Mechanics. Yohann Moreau. November 26, 2015
 Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Differences in Electrostatic Properties at Antibody Antigen Binding Sites: Implications for Specificity and Cross-Reactivity
 2946 Biophysical Journal Volume 83 December 2002 2946 2968 Differences in Electrostatic Properties at Antibody Antigen Binding Sites: Implications for Specificity and Cross-Reactivity Neeti Sinha, Srinivasan
2946 Biophysical Journal Volume 83 December 2002 2946 2968 Differences in Electrostatic Properties at Antibody Antigen Binding Sites: Implications for Specificity and Cross-Reactivity Neeti Sinha, Srinivasan
Reactive potentials and applications
 1.021, 3.021, 10.333, 22.00 Introduction to Modeling and Simulation Spring 2011 Part I Continuum and particle methods Reactive potentials and applications Lecture 8 Markus J. Buehler Laboratory for Atomistic
1.021, 3.021, 10.333, 22.00 Introduction to Modeling and Simulation Spring 2011 Part I Continuum and particle methods Reactive potentials and applications Lecture 8 Markus J. Buehler Laboratory for Atomistic
arxiv: v1 [cond-mat.stat-mech] 6 Jan 2014
![arxiv: v1 [cond-mat.stat-mech] 6 Jan 2014](/thumbs/72/67212951.jpg "arxiv: v1 [cond-mat.stat-mech] 6 Jan 2014") arxiv:1401.1181v1 [cond-mat.stat-mech] 6 Jan 2014 Determination of Forces from a Potential in Molecular Dynamics (note) Bernard Monasse Mines-ParisTech, Cemef bernard.monasse@mines-paristech.fr January
arxiv:1401.1181v1 [cond-mat.stat-mech] 6 Jan 2014 Determination of Forces from a Potential in Molecular Dynamics (note) Bernard Monasse Mines-ParisTech, Cemef bernard.monasse@mines-paristech.fr January
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
k θ (θ θ 0 ) 2 angles r i j r i j
 2 angles r i j r i j") 1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
BIOC : Homework 1 Due 10/10
 Contact information: Name: Student # BIOC530 2012: Homework 1 Due 10/10 Department Email address The following problems are based on David Baker s lectures of forces and protein folding. When numerical
Contact information: Name: Student # BIOC530 2012: Homework 1 Due 10/10 Department Email address The following problems are based on David Baker s lectures of forces and protein folding. When numerical
This semester. Books
 Models mostly proteins from detailed to more abstract models Some simulation methods This semester Books None necessary for my group and Prof Rarey Molecular Modelling: Principles and Applications Leach,
Models mostly proteins from detailed to more abstract models Some simulation methods This semester Books None necessary for my group and Prof Rarey Molecular Modelling: Principles and Applications Leach,
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 1 David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Time/s Multi-Scale Modeling Based on SDSC Blue Horizon (SP3) 1.728 Tflops
1 CE 530 Molecular Simulation Lecture 1 David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Time/s Multi-Scale Modeling Based on SDSC Blue Horizon (SP3) 1.728 Tflops
Monte Carlo (MC) Simulation Methods. Elisa Fadda
 Simulation Methods. Elisa Fadda") Monte Carlo (MC) Simulation Methods Elisa Fadda 1011-CH328, Molecular Modelling & Drug Design 2011 Experimental Observables A system observable is a property of the system state. The system state i is
Monte Carlo (MC) Simulation Methods Elisa Fadda 1011-CH328, Molecular Modelling & Drug Design 2011 Experimental Observables A system observable is a property of the system state. The system state i is
General Physical Chemistry II
 General Physical Chemistry II Lecture 13 Aleksey Kocherzhenko October 16, 2014" Last time " The Hückel method" Ø Used to study π systems of conjugated molecules" Ø π orbitals are treated separately from
General Physical Chemistry II Lecture 13 Aleksey Kocherzhenko October 16, 2014" Last time " The Hückel method" Ø Used to study π systems of conjugated molecules" Ø π orbitals are treated separately from
Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions Van der Waals Interactions
Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions Van der Waals Interactions
1 of 43 Boardworks Ltd Chemistry 11. Chemical Bonding
 1 of 43 Boardworks Ltd 2009 Chemistry 11 Chemical Bonding 2 of 43 Boardworks Ltd 2009 Electrostatic Forces An electrostatic force is a forces existing as a result of the attraction or repulsion between
1 of 43 Boardworks Ltd 2009 Chemistry 11 Chemical Bonding 2 of 43 Boardworks Ltd 2009 Electrostatic Forces An electrostatic force is a forces existing as a result of the attraction or repulsion between
Lecture C2 Microscopic to Macroscopic, Part 2: Intermolecular Interactions. Let's get together.
 Lecture C2 Microscopic to Macroscopic, Part 2: Intermolecular Interactions Let's get together. Most gases are NOT ideal except at very low pressures: Z=1 for ideal gases Intermolecular interactions come
Lecture C2 Microscopic to Macroscopic, Part 2: Intermolecular Interactions Let's get together. Most gases are NOT ideal except at very low pressures: Z=1 for ideal gases Intermolecular interactions come
Protein Structure Basics
 Protein Structure Basics Presented by Alison Fraser, Christine Lee, Pradhuman Jhala, Corban Rivera Importance of Proteins Muscle structure depends on protein-protein interactions Transport across membranes
Protein Structure Basics Presented by Alison Fraser, Christine Lee, Pradhuman Jhala, Corban Rivera Importance of Proteins Muscle structure depends on protein-protein interactions Transport across membranes
What are covalent bonds?
 Covalent Bonds What are covalent bonds? Covalent Bonds A covalent bond is formed when neutral atoms share one or more pairs of electrons. Covalent Bonds Covalent bonds form between two or more non-metal
Covalent Bonds What are covalent bonds? Covalent Bonds A covalent bond is formed when neutral atoms share one or more pairs of electrons. Covalent Bonds Covalent bonds form between two or more non-metal
Energy functions and their relationship to molecular conformation. CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror
 Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Outline Energy functions for proteins (or biomolecular systems more generally)
Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Outline Energy functions for proteins (or biomolecular systems more generally)
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Elbow Flexibility and Ligand-Induced Domain Rearrangements in Antibody Fab NC6.8: Large Effects of a Small Hapten
 614 Biophysical Journal Volume 79 August 2000 614 628 Elbow Flexibility and Ligand-Induced Domain Rearrangements in Antibody Fab NC6.8: Large Effects of a Small Hapten Christoph A. Sotriffer, Bernd M.
614 Biophysical Journal Volume 79 August 2000 614 628 Elbow Flexibility and Ligand-Induced Domain Rearrangements in Antibody Fab NC6.8: Large Effects of a Small Hapten Christoph A. Sotriffer, Bernd M.
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Lecture 5: Electrostatic Interactions & Screening
 Lecture 5: Electrostatic Interactions & Screening Lecturer: Prof. Brigita Urbanc (brigita@drexel.edu) PHYS 461 & 561, Fall 2009-2010 1 A charged particle (q=+1) in water, at the interface between water
Lecture 5: Electrostatic Interactions & Screening Lecturer: Prof. Brigita Urbanc (brigita@drexel.edu) PHYS 461 & 561, Fall 2009-2010 1 A charged particle (q=+1) in water, at the interface between water
Molecular Mechanics. C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology. January 2001
 Molecular Mechanics C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology January 2001 Introduction Molecular Mechanics uses classical type models to predict the energy
Molecular Mechanics C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology January 2001 Introduction Molecular Mechanics uses classical type models to predict the energy
Modelação e Simulação de Sistemas para Micro/Nano Tecnologias
 Modelação e Simulação de Sistemas para Micro/Nano Tecnologias http://gec.di.uminho.pt/mmnt/modsim/ Alberto José Proença, António Joaquim Esteves 2011/12 Mestrado em Micro/Nano Tecnologias ESCOLA DE ENGENHARIA
Modelação e Simulação de Sistemas para Micro/Nano Tecnologias http://gec.di.uminho.pt/mmnt/modsim/ Alberto José Proença, António Joaquim Esteves 2011/12 Mestrado em Micro/Nano Tecnologias ESCOLA DE ENGENHARIA
Protein Folding experiments and theory
 Protein Folding experiments and theory 1, 2,and 3 Protein Structure Fig. 3-16 from Lehninger Biochemistry, 4 th ed. The 3D structure is not encoded at the single aa level Hydrogen Bonding Shared H atom
Protein Folding experiments and theory 1, 2,and 3 Protein Structure Fig. 3-16 from Lehninger Biochemistry, 4 th ed. The 3D structure is not encoded at the single aa level Hydrogen Bonding Shared H atom
Medical Research, Medicinal Chemistry, University of Leuven, Leuven, Belgium.
 Supporting Information Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins Muhammad Usman Mirza
Supporting Information Towards peptide vaccines against Zika virus: Immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins Muhammad Usman Mirza
Chapter 3. Crystal Binding
 Chapter 3. Crystal Binding Energy of a crystal and crystal binding Cohesive energy of Molecular crystals Ionic crystals Metallic crystals Elasticity What causes matter to exist in three different forms?
Chapter 3. Crystal Binding Energy of a crystal and crystal binding Cohesive energy of Molecular crystals Ionic crystals Metallic crystals Elasticity What causes matter to exist in three different forms?
Parameterization of a reactive force field using a Monte Carlo algorithm
 Parameterization of a reactive force field using a Monte Carlo algorithm Eldhose Iype (e.iype@tue.nl) November 19, 2015 Where innovation starts Thermochemical energy storage 2/1 MgSO 4.xH 2 O+Q MgSO 4
Parameterization of a reactive force field using a Monte Carlo algorithm Eldhose Iype (e.iype@tue.nl) November 19, 2015 Where innovation starts Thermochemical energy storage 2/1 MgSO 4.xH 2 O+Q MgSO 4
Multiscale Materials Modeling
 Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Simulations with MM Force Fields. Monte Carlo (MC) and Molecular Dynamics (MD) Video II.vi
 and Molecular Dynamics (MD) Video II.vi") Simulations with MM Force Fields Monte Carlo (MC) and Molecular Dynamics (MD) Video II.vi Some slides taken with permission from Howard R. Mayne Department of Chemistry University of New Hampshire Walking
Simulations with MM Force Fields Monte Carlo (MC) and Molecular Dynamics (MD) Video II.vi Some slides taken with permission from Howard R. Mayne Department of Chemistry University of New Hampshire Walking
MOLECULAR MODELING IN BIOLOGY (BIO 3356) SYLLABUS
 SYLLABUS") New York City College of Technology School of Arts and Sciences Department of Biological Sciences MOLECULAR MODELING IN BIOLOGY (BIO 3356) SYLLABUS Course Information Course Title: Molecular Modeling in
New York City College of Technology School of Arts and Sciences Department of Biological Sciences MOLECULAR MODELING IN BIOLOGY (BIO 3356) SYLLABUS Course Information Course Title: Molecular Modeling in
The protein folding problem consists of two parts:
 Energetics and kinetics of protein folding The protein folding problem consists of two parts: 1)Creating a stable, well-defined structure that is significantly more stable than all other possible structures.
Energetics and kinetics of protein folding The protein folding problem consists of two parts: 1)Creating a stable, well-defined structure that is significantly more stable than all other possible structures.
Conformational Geometry of Peptides and Proteins:
 Conformational Geometry of Peptides and Proteins: Before discussing secondary structure, it is important to appreciate the conformational plasticity of proteins. Each residue in a polypeptide has three
Conformational Geometry of Peptides and Proteins: Before discussing secondary structure, it is important to appreciate the conformational plasticity of proteins. Each residue in a polypeptide has three
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation
 Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Imperfect Gases. NC State University
 Chemistry 431 Lecture 3 Imperfect Gases NC State University The Compression Factor One way to represent the relationship between ideal and real gases is to plot the deviation from ideality as the gas is
Chemistry 431 Lecture 3 Imperfect Gases NC State University The Compression Factor One way to represent the relationship between ideal and real gases is to plot the deviation from ideality as the gas is
The Chemical Basis of Life
 The Chemical Basis of Life Chapter 2 Objectives Identify the four elements that make up 96% of living matter. Distinguish between the following pairs of terms: neutron and proton, atomic number and mass
The Chemical Basis of Life Chapter 2 Objectives Identify the four elements that make up 96% of living matter. Distinguish between the following pairs of terms: neutron and proton, atomic number and mass
There are two main requirements for atoms to form a covalent bond and make a molecule:
 HOW ATOMS BOND TO EACH OTHER Covalent bonding Remember that a hydrogen atom has 1 proton and 1 electron and that the electron and the proton are attracted to each other. But if the atoms get close enough
HOW ATOMS BOND TO EACH OTHER Covalent bonding Remember that a hydrogen atom has 1 proton and 1 electron and that the electron and the proton are attracted to each other. But if the atoms get close enough
Analysis of MD trajectories in GROMACS David van der Spoel
 Analysis of MD trajectories in GROMACS David van der Spoel What does MD produce? Energy terms E(t) Coordinates x(t) Velocities v(t) Forces f(t) Managing your files trjcat - merging trajectories concatenating
Analysis of MD trajectories in GROMACS David van der Spoel What does MD produce? Energy terms E(t) Coordinates x(t) Velocities v(t) Forces f(t) Managing your files trjcat - merging trajectories concatenating
Biomolecules: lecture 9
 Biomolecules: lecture 9 - understanding further why amino acids are the building block for proteins - understanding the chemical properties amino acids bring to proteins - realizing that many proteins
Biomolecules: lecture 9 - understanding further why amino acids are the building block for proteins - understanding the chemical properties amino acids bring to proteins - realizing that many proteins
CHEMISTRY 4021/8021 MIDTERM EXAM 1 SPRING 2014
 CHEMISTRY 4021/8021 Q1) Propose a simple, united-atom molecular mechanics force-field needed to generate a potential energy surface for an isolated molecule of acetone (Me 2 CO). I.e., provide an energy
CHEMISTRY 4021/8021 Q1) Propose a simple, united-atom molecular mechanics force-field needed to generate a potential energy surface for an isolated molecule of acetone (Me 2 CO). I.e., provide an energy
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
NWChem: Molecular Dynamics and QM/MM
 NWChem: Molecular Dynamics and QM/MM Molecular Dynamics Functionality Target systems: General features: biomolecules (proteins, DNA/RNA, biomembranes) energy evaluation (SP) energy minimization (EM) molecular
NWChem: Molecular Dynamics and QM/MM Molecular Dynamics Functionality Target systems: General features: biomolecules (proteins, DNA/RNA, biomembranes) energy evaluation (SP) energy minimization (EM) molecular
510 Subject Index. Hamiltonian 33, 86, 88, 89 Hamilton operator 34, 164, 166
 Subject Index Ab-initio calculation 24, 122, 161. 165 Acentric factor 279, 338 Activity absolute 258, 295 coefficient 7 definition 7 Atom 23 Atomic units 93 Avogadro number 5, 92 Axilrod-Teller-forces
Subject Index Ab-initio calculation 24, 122, 161. 165 Acentric factor 279, 338 Activity absolute 258, 295 coefficient 7 definition 7 Atom 23 Atomic units 93 Avogadro number 5, 92 Axilrod-Teller-forces
Protein Structure Analysis
 BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
Computer simulation methods (1) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (1) Dr. Vania Calandrini Why computational methods To understand and predict the properties of complex systems (many degrees of freedom): liquids, solids, adsorption of molecules
Computer simulation methods (1) Dr. Vania Calandrini Why computational methods To understand and predict the properties of complex systems (many degrees of freedom): liquids, solids, adsorption of molecules
Supplementary Information
 Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
BCMP 201 Protein biochemistry
 BCMP 201 Protein biochemistry BCMP 201 Protein biochemistry with emphasis on the interrelated roles of protein structure, catalytic activity, and macromolecular interactions in biological processes. The
BCMP 201 Protein biochemistry BCMP 201 Protein biochemistry with emphasis on the interrelated roles of protein structure, catalytic activity, and macromolecular interactions in biological processes. The
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Computer simulation methods (2) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Analysis of the simulation
 Analysis of the simulation Marcus Elstner and Tomáš Kubař January 7, 2014 Thermodynamic properties time averages of thermodynamic quantites correspond to ensemble averages (ergodic theorem) some quantities
Analysis of the simulation Marcus Elstner and Tomáš Kubař January 7, 2014 Thermodynamic properties time averages of thermodynamic quantites correspond to ensemble averages (ergodic theorem) some quantities
Introduction to Classical Molecular Dynamics. Giovanni Chillemi HPC department, CINECA
 Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii
 And the Basic Force Field Chem 4021/8021 Video II.iii") The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii Fundamental Points About Which to Be Thinking It s clear the PES is useful, so how can I construct it for an arbitrary
The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii Fundamental Points About Which to Be Thinking It s clear the PES is useful, so how can I construct it for an arbitrary
Exploring the Free Energy Surface of Short Peptides by Using Metadynamics
 John von Neumann Institute for Computing Exploring the Free Energy Surface of Short Peptides by Using Metadynamics C. Camilloni, A. De Simone published in From Computational Biophysics to Systems Biology
John von Neumann Institute for Computing Exploring the Free Energy Surface of Short Peptides by Using Metadynamics C. Camilloni, A. De Simone published in From Computational Biophysics to Systems Biology
Statistical Mechanics for Proteins
 The Partition Function From Q all relevant thermodynamic properties can be obtained by differentiation of the free energy F: = kt q p E q pd d h T V Q ), ( exp 1! 1 ),, ( 3 3 3 ),, ( ln ),, ( T V Q kt
The Partition Function From Q all relevant thermodynamic properties can be obtained by differentiation of the free energy F: = kt q p E q pd d h T V Q ), ( exp 1! 1 ),, ( 3 3 3 ),, ( ln ),, ( T V Q kt
ENERGY MINIMIZATION AND CONFORMATION SEARCH ANALYSIS OF TYPE-2 ANTI-DIABETES DRUGS
 Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Basics of Statistical Mechanics
 Basics of Statistical Mechanics Review of ensembles Microcanonical, canonical, Maxwell-Boltzmann Constant pressure, temperature, volume, Thermodynamic limit Ergodicity (see online notes also) Reading assignment:
Basics of Statistical Mechanics Review of ensembles Microcanonical, canonical, Maxwell-Boltzmann Constant pressure, temperature, volume, Thermodynamic limit Ergodicity (see online notes also) Reading assignment:
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
= (-22) = +2kJ /mol
 = +2kJ /mol") Lecture 8: Thermodynamics & Protein Stability Assigned reading in Campbell: Chapter 4.4-4.6 Key Terms: DG = -RT lnk eq = DH - TDS Transition Curve, Melting Curve, Tm DH calculation DS calculation van der
Lecture 8: Thermodynamics & Protein Stability Assigned reading in Campbell: Chapter 4.4-4.6 Key Terms: DG = -RT lnk eq = DH - TDS Transition Curve, Melting Curve, Tm DH calculation DS calculation van der
Multiscale Materials Modeling
 Multiscale Materials Modeling Lecture 02 Capabilities of Classical Molecular Simulation These notes created by David Keffer, University of Tennessee, Knoxville, 2009. Outline Capabilities of Classical
Multiscale Materials Modeling Lecture 02 Capabilities of Classical Molecular Simulation These notes created by David Keffer, University of Tennessee, Knoxville, 2009. Outline Capabilities of Classical
Biomolecular modeling I
 2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
Molecular Simulation II. Classical Mechanical Treatment
 Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences