Molecular Dynamics Simulations. Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia
|
|
|
- Merilyn Golden
- 5 years ago
- Views:
Transcription
1 Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1
2 An Introduction to Molecular Dynamics Simulations Macroscopic properties are often determined by molecule-level behavior. Quantitative and/or qualitative information about macroscopic behavior of macromolecules can be obtained from simulation of a system at atomistic level. Molecular dynamics simulations calculate the motion of the atoms in a molecular assembly using Newtonian dynamics to determine the net force and acceleration experienced by each atom. Each atom i at position r i, is treated as a point with a mass m i and a fixed charge q i. 2

3 The model system 3

4 Periodic Boundary conditions 4
5 Molecular Dynamics Simulations! Periodic boundary conditions! Our systems are very small 10 5 or 10 6 << Avogadro number! Boundaries will have a large effect on the calculated properties! Use periodic boundary conditions. The system is surrounded by translated copies of itself van Gunsteren, W.F., et al., Biomolecular modeling: Goals, problems, perspectives. Angew. Chem. Int. Edit., (25): p
6 Cutt-off and minimun image convection 6
7 Steps in Molecular Dynamics Simulations 1) Build realistic atomistic model of the system 2) Simulate the behavior of your system over time using specific conditions (temperature, pressure, volume, etc) 3) Analyze the results obtained from MD and relate to macroscopic level properties 7
8 What is a Forcefield? In molecular dynamics a molecule is described as a series of charged points (atoms) linked by springs (bonds). To describe the time evolution of bond lengths, bond angles and torsions, also the nonbond van der Waals and elecrostatic interactions between atoms, one uses a forcefield. The forcefield is a collection of equations and associated constants designed to reproduce molecular geometry and selected properties of tested structures. 8

9 9
10 Energy Terms Described in the... forcefield Bond Angle Dihedral Improper 10
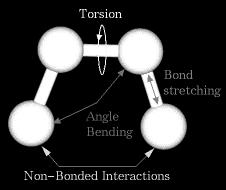
11 11
 = ½ k s,ij ( r ij - r")
12 Bonds: Harmonic bond Stretching V(rij) represents the energy required to stretch or compress a covalent bond: A bond can be thought of as a spring having its own equilibrium length, r o, and the energy required to stretch or compress it can be approximated by the Hookean potential for an ideal spring: V(rij) = ½ k s,ij ( r ij - r o ) 2 12

")
13 Angles: Harmonic angle bending E bend is the energy required to bend a bond from its equilibrium angle, θ o : Again this system can be modeled by a spring, and the energy is given by the Hookean potential with respect to angle: E bend = ½ kb,ijk (θ ijk - θ o ) 2 13
14 Improper dihedrals: planarity and harmonic E improper is the energy required to deform a planar group of atoms from its equilibrium angle, ω o, usually equal to zero: k i l j ω Thomas W. Shattuck Again this system can be modeled by a spring, and the energy is given by the Hookean potential with respect to planar angle: E improper = ½ k o,ijkl (ω ijkl - ω o ) 2 14
15 Dihedral Angle potentials The dihedral angle potentials describe the interaction arising from torsional forces in molecules. (They are sometimes referred to as torsion potentials.) Formally the dihedral angle (also known as a torsion angle) between four atoms A-B-C-D is defined as the angle between the the planes ABC (marked by red lines) and BCD (marked in purple): 15
16 single cosine E tor is the energy of torsion needed to rotate about bonds: Torsional interactions are modeled by the potential: E tor = ½ k tor,1 (1 - cos φ ) + ½ k tor,2 (1 - cos 2 φ ) + ½ k tor,3 ( 1 - cos 3 φ ) asymmetry (butane) 2-fold groups e.g. COO- standard tetrahedral torsions 16
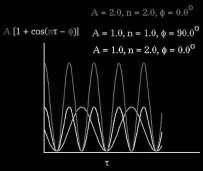
17 17
18 The intermolecular potential function (NON- bonded) short ranged interactions: Short ranged (van der Waals) potentials The three body potentials The Tersoff Covalent potentials Four Body potentials Metal potentials External fields 18

19 Short Ranged (van der Waals) Potentials 12-6 potential E vdw is the steric exclusion and long-range attraction energy (QM origins): 19
20 DL_POLY uses two cut-offs (8 Å and 1.4 Å) 20
21 ILJ vs. LJ m V ILJ n r m r 0 r n(r) n r n(r) m r 0 r m V LJ r 0 r 12 2 r 0 r r r 0 n r 2 C 6 n(r) n(r) m r o 6 less attractive long range C 6 2r o r r 0 n r 2 less repulsive short range 21
22 + part of the formulation: represents the size repulsion contribution - part of the formulation: represents the effective dispersion attractive to the same pair β is an adjustable parameter (like ambient characteristic) use the same value of ε, r 0 for different molecules => structural meaning 22
23 β is sensible to the charge distribution and to the choice of the interaction of the reference centers. The initial value of β can be estimated from the cubic root of the polarizability of the involved partners.[1] due to the (i.e H bonding) for the charge transfer effects the values of β need to be varied within a well defined interval for optimization. [1] Capittelli et al., Chem. Phys., 2007, 338,
24 Long ranged Electrostatic (Coulombic)interactions Direct Coulomb Sum Truncated and Shifted Coulomb sum Ewald Sum Smoothed Particle Mesh Ewald (SPME) Hautman Klein Ewald (HKE)for systems with 2D periodicity Reaction Field Dynamics shell model Relaxed shell model 24
25 25

26 Ewald sum A Probe Unit Positive Charge in a Periodic Lattice of Ions. 26

27 van Gunsteren, W.F., et al., Biomolecular modeling: Goals, problems, perspectives. Angew. Chem. Int. Edit., (25): p
28 Microcanonical emsembles
29
30 Molecular Dynamics Simulations! Ensembles! Isoenergetic " System in a closed, rigid and insulated container (NVE) " The system has constant energy (E) " All states are equally likely where #(E) are the number of states with energy E Baptista, AM. Molecular Simulation Group,Instituto de Tecnologia Química e Biológica. Personal communication 30
31 Molecular Dynamics Simulations! Ensembles! Isothermal " Closed system at constant temperature and volume (NVT) " Energy can be exchanged with the bath at temperature T " A state with low energy is more likely than a state with high energy Baptista, AM. Molecular Simulation Group,Instituto de Tecnologia Química e Biológica. Personal communication 31
32 Molecular Dynamics Simulations! Ensembles! Isothermal-isobaric " Closed system connected to a piston and at constant temperature (NPT) " Energy can be exchanged with the bath at temperature T " Volume (V) can be exchanged with the bath at pressure P " A state with low E+pV is more likely than a state with high E+pV Baptista, AM. Molecular Simulation Group,Instituto de Tecnologia Química e Biológica. Personal communication 32
33 Molecular Dynamics Simulations! Ensembles! Grand canonical ensemble " System in a rigid container able to exchange particles with a reservoir and, in contact with a heat bath (NT) " Energy can be exchanged with the bath at temperature T " The number of molecules (N) can vary by exchange with the reservoir at chemical potential Baptista, AM. Molecular Simulation Group,Instituto de Tecnologia Química e Biológica. Personal communication 33
34 Molecular Dynamics Simulations! Thermostats! Berendsen " The system is coupled to an external heat bath with fixed T bath " The heat bath supplies/removes energy to/from the system " Velocities are scaled proportionally to T bath - T(t) $ determines the how tightly the bath and the system are coupled together (ps). Usually $ = 0.4 ps. " Scaling factor for the velocities Leach, A.R., Molecular modelling: principles and applications. 2nd ed. 2001: Prentice Hall. 34
35 Molecular Dynamics Simulations! Thermostats! Nosé-Hoover " Equations of motion are changed by introducing a friction force " The friction force is proportional to the particle velocity and friction parameter % where the friction parameter % is given by Leach, A.R., Molecular modelling: principles and applications. 2nd ed. 2001: Prentice Hall. 35
36 Molecular Dynamics Simulations! Difference between Berendsen and Nosé thermostats! Berendsen gives exponential decay of the system towards de desired temperature. Does not generate a proper ensemble! Nosé can induce oscillations on the system. Gives a correct ensemble! Example: Temperature increase of a water box (300 K to 330 K) Hess, B. Gromacs Workshop at CSC Personal communication 36
37 Molecular Dynamics Simulations! Thermostats! Heat groups " Energy exchange between different components is not perfect " Different components can have different temperatures but the global temperature system is correct " Heat group concept Water 400 K Protein 200 K Water 300 K Protein 300 K 1 heat group 2 heat groups 37
38 Molecular Dynamics Simulations! Barostats! Berendsen " Analogous to the Berendsen thermostat " The system maintains constant pressure by changing its volume " The rate of change of the pressure is given by: $ P determines the how tightly the bath and the system are coupled together (ps). Usually $ P = 1.0 ps. " Scaling factor for the box. Atomic coordinates are scaled by & 1/3 ' is the experimental isothermal compressibility. Leach, A.R., Molecular modelling: principles and applications. 2nd ed. 2001: Prentice Hall. 38
39 Molecular Dynamics Simulations! Integration of the equations of motion! Integration is broken in many small fixed time )t steps! The algorithms must: " Conserve energy and momentum " Be time-reversible " Permit a long time step )t! Algorithms used: " Verlet algorithm " Leap-frog algorithm Leach, A.R., Molecular modelling: principles and applications. 2nd ed. 2001: Prentice Hall. 39
40 Molecular Dynamics Simulations! Verlet algorithm! Uses the positions and accelerations at time t, and the positions from the previous step, r(t-)t), to calculate the new positions at t +)t, r(t+)t).! Velocities are obtained by: Leach, A.R., Molecular modelling: principles and applications. 2nd ed. 2001: Prentice Hall. 40
41 Molecular Dynamics Simulations! Verlet algorithm! Positions and velocities are not synchronized! Numerically more stable Leach, A.R., Molecular modelling: principles and applications. 2nd ed. 2001: Prentice Hall. 41

42 The difference between the Verlet algorithm and the leapfrog algorithm is that the latter evaluates the velocities at half-integer time steps, which are artificial points in time, exactly half way between two time steps. The algorithm uses these half-integer time steps velocities to compute the new positions. To derive the leapfrog algorithm from the Verlet algorithm let us first define the velocity at half-integer time steps 42
43 The steps to take in solving the equations of motions using the leapfrog algorithm are summarized in the list below. It is assumed that the initial positions, r(t), and velocities, v(t Δt/2), are known. 1 Calculate F(t). 2 Calculate new velocities v(t+δt/2) using v(t Δt/2) and F(t). 3 Calculate new positions r(t Δt/2) using r(t) and v(t Δt/2). 4 Repeat these steps until a certain stop criterium. 43
44 Molecular Dynamics Simulations! Constrains! High frequency motions misbehave in classical simulation because they are of quantum nature! Most of the times, high frequency motions are not essential to the properties that we want to study! Bond constraints and, in some cases, angle constrains should be used! Removing the bond vibrations with constraints allows one to increase the time step from 0.5 or 1 fs to 2 or 4 fs Leach, A.R., Molecular modelling: principles and applications. 2nd ed. 2001: Prentice Hall. 44
45 Molecular Dynamics Simulations! Constraint algorithms! SHAKE " The SHAKE algorithm changes a set of unconstrained coordinates r to a set of coordinates r that fulfil a list of distance constraints, using a set r as reference. " Iterative method r r r van der Spoel, D., et al., Gromacs user manual. Version
46 Molecular Dynamics Simulations! Constraint algorithms! LINCS " Non-iterative method (faster than SHAKE) " it can only be used with bond constraints and isolated angle constraints! SETTLE " Analytical solution of SHAKE specifically for water molecules 46 van der Spoel, D., et al., Gromacs user manual. Version
47 Molecular Dynamics Simulations! Restraints! Sometimes are confused with constrains! Particles remain close to a reference position! Positions, distances, angles, dihedral! Avoid disastrous deviations during equilibrations! Include knowledge from experimental data (ex: NMR) 47 van der Spoel, D., et al., Gromacs user manual. Version
48 Molecular Dynamics Simulations! Basic MD algorithm Input: Position of all atoms of the system START 1. Generated velocities by a Maxwell random distribution 2. Compute forces on each particle 3. Compute the scaling factors & and µ* 4. Update and scale velocities (Temperature) 5. Update new unconstrained coordinates (Move the particles) 6. Apply constraint algorithm to coordinates (SHAKE) 7. Correct velocities for constraints (due to the constraints used) 8. Scale coordinates and box (Pressure) 9. Write output Repeat steps 2-9 N times END 48 van der Spoel, D., et al., Gromacs user manual. Version

49 DL_POLY 49
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Introduction to Classical Molecular Dynamics. Giovanni Chillemi HPC department, CINECA
 Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
 A Nobel Prize for Molecular Dynamics and QM/MM What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential
A Nobel Prize for Molecular Dynamics and QM/MM What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Biomolecules are dynamic no single structure is a perfect model
 Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Biomolecular modeling I
 2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
What is Classical Molecular Dynamics?
 What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Force fields, thermo- and barostats. Berk Hess
 Force fields, thermo- and barostats Berk Hess What is a force field? A force field usually consists of three parts: a set of functional forms parameters for the functional forms that, usually, depend on
Force fields, thermo- and barostats Berk Hess What is a force field? A force field usually consists of three parts: a set of functional forms parameters for the functional forms that, usually, depend on
MD Thermodynamics. Lecture 12 3/26/18. Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky
 MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
Methods of Computer Simulation. Molecular Dynamics and Monte Carlo
 Molecular Dynamics Time is of the essence in biological processes therefore how do we understand time-dependent processes at the molecular level? How do we do this experimentally? How do we do this computationally?
Molecular Dynamics Time is of the essence in biological processes therefore how do we understand time-dependent processes at the molecular level? How do we do this experimentally? How do we do this computationally?
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii
 And the Basic Force Field Chem 4021/8021 Video II.iii") The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii Fundamental Points About Which to Be Thinking It s clear the PES is useful, so how can I construct it for an arbitrary
The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii Fundamental Points About Which to Be Thinking It s clear the PES is useful, so how can I construct it for an arbitrary
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
Principles and Applications of Molecular Dynamics Simulations with NAMD
 Principles and Applications of Molecular Dynamics Simulations with NAMD Nov. 14, 2016 Computational Microscope NCSA supercomputer JC Gumbart Assistant Professor of Physics Georgia Institute of Technology
Principles and Applications of Molecular Dynamics Simulations with NAMD Nov. 14, 2016 Computational Microscope NCSA supercomputer JC Gumbart Assistant Professor of Physics Georgia Institute of Technology
Molecular Dynamics Simulation of a Nanoconfined Water Film
 Molecular Dynamics Simulation of a Nanoconfined Water Film Kyle Lindquist, Shu-Han Chao May 7, 2013 1 Introduction The behavior of water confined in nano-scale environment is of interest in many applications.
Molecular Dynamics Simulation of a Nanoconfined Water Film Kyle Lindquist, Shu-Han Chao May 7, 2013 1 Introduction The behavior of water confined in nano-scale environment is of interest in many applications.
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Hands-on : Model Potential Molecular Dynamics
 Hands-on : Model Potential Molecular Dynamics OUTLINE 0. DL_POLY code introduction 0.a Input files 1. THF solvent molecule 1.a Geometry optimization 1.b NVE/NVT dynamics 2. Liquid THF 2.a Equilibration
Hands-on : Model Potential Molecular Dynamics OUTLINE 0. DL_POLY code introduction 0.a Input files 1. THF solvent molecule 1.a Geometry optimization 1.b NVE/NVT dynamics 2. Liquid THF 2.a Equilibration
Supporting Information
 Supporting Information Structure and Dynamics of Uranyl(VI) and Plutonyl(VI) Cations in Ionic Liquid/Water Mixtures via Molecular Dynamics Simulations Katie A. Maerzke, George S. Goff, Wolfgang H. Runde,
Supporting Information Structure and Dynamics of Uranyl(VI) and Plutonyl(VI) Cations in Ionic Liquid/Water Mixtures via Molecular Dynamics Simulations Katie A. Maerzke, George S. Goff, Wolfgang H. Runde,
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
Supporting Information for: Physics Behind the Water Transport through. Nanoporous Graphene and Boron Nitride
 Supporting Information for: Physics Behind the Water Transport through Nanoporous Graphene and Boron Nitride Ludovic Garnier, Anthony Szymczyk, Patrice Malfreyt, and Aziz Ghoufi, Institut de Physique de
Supporting Information for: Physics Behind the Water Transport through Nanoporous Graphene and Boron Nitride Ludovic Garnier, Anthony Szymczyk, Patrice Malfreyt, and Aziz Ghoufi, Institut de Physique de
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
Simulation of molecular systems by molecular dynamics
 Simulation of molecular systems by molecular dynamics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Dynamics, Label RFCT 2015 November 26, 2015 1 / 35 Introduction
Simulation of molecular systems by molecular dynamics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Dynamics, Label RFCT 2015 November 26, 2015 1 / 35 Introduction
Scientific Computing II
 Scientific Computing II Molecular Dynamics Simulation Michael Bader SCCS Summer Term 2015 Molecular Dynamics Simulation, Summer Term 2015 1 Continuum Mechanics for Fluid Mechanics? Molecular Dynamics the
Scientific Computing II Molecular Dynamics Simulation Michael Bader SCCS Summer Term 2015 Molecular Dynamics Simulation, Summer Term 2015 1 Continuum Mechanics for Fluid Mechanics? Molecular Dynamics the
Molecular Dynamics Simulation of Methanol-Water Mixture
 Molecular Dynamics Simulation of Methanol-Water Mixture Palazzo Mancini, Mara Cantoni University of Urbino Carlo Bo Abstract In this study some properties of the methanol-water mixture such as diffusivity,
Molecular Dynamics Simulation of Methanol-Water Mixture Palazzo Mancini, Mara Cantoni University of Urbino Carlo Bo Abstract In this study some properties of the methanol-water mixture such as diffusivity,
A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane
 A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane Supplementary Information: Simulation Procedure and Physical Property Analysis Simulation Procedure The molecular
A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane Supplementary Information: Simulation Procedure and Physical Property Analysis Simulation Procedure The molecular
Computational Chemistry - MD Simulations
 Computational Chemistry - MD Simulations P. Ojeda-May pedro.ojeda-may@umu.se Department of Chemistry/HPC2N, Umeå University, 901 87, Sweden. May 2, 2017 Table of contents 1 Basics on MD simulations Accelerated
Computational Chemistry - MD Simulations P. Ojeda-May pedro.ojeda-may@umu.se Department of Chemistry/HPC2N, Umeå University, 901 87, Sweden. May 2, 2017 Table of contents 1 Basics on MD simulations Accelerated
Parallel Utility for Modeling of Molecular Aggregation
 Parallel Utility for Modeling of Molecular Aggregation Peter Spijker, Bart Markvoort and Peter Hilbers BioModeling and bioinformatics Eindhoven University of Technology Eindhoven,The Netherlands p.spijker@pumma.nl
Parallel Utility for Modeling of Molecular Aggregation Peter Spijker, Bart Markvoort and Peter Hilbers BioModeling and bioinformatics Eindhoven University of Technology Eindhoven,The Netherlands p.spijker@pumma.nl
Supporting Information
 Projection of atomistic simulation data for the dynamics of entangled polymers onto the tube theory: Calculation of the segment survival probability function and comparison with modern tube models Pavlos
Projection of atomistic simulation data for the dynamics of entangled polymers onto the tube theory: Calculation of the segment survival probability function and comparison with modern tube models Pavlos
Molecular dynamics simulations of a single stranded (ss) DNA
 DNA") Molecular dynamics simulations of a single stranded (ss) DNA Subhasish Chatterjee 1, Bonnie Gersten 1, Siddarth Thakur 2, Alexander Burin 2 1 Department of Chemistry, Queens College and the Graduate Center
Molecular dynamics simulations of a single stranded (ss) DNA Subhasish Chatterjee 1, Bonnie Gersten 1, Siddarth Thakur 2, Alexander Burin 2 1 Department of Chemistry, Queens College and the Graduate Center
Supplemental Material for Global Langevin model of multidimensional biomolecular dynamics
 Supplemental Material for Global Langevin model of multidimensional biomolecular dynamics Norbert Schaudinnus, Benjamin Lickert, Mithun Biswas and Gerhard Stock Biomolecular Dynamics, Institute of Physics,
Supplemental Material for Global Langevin model of multidimensional biomolecular dynamics Norbert Schaudinnus, Benjamin Lickert, Mithun Biswas and Gerhard Stock Biomolecular Dynamics, Institute of Physics,
The Effect of Model Internal Flexibility Upon NEMD Simulations of Viscosity
 Draft: September 29, 1999 The Effect of Model Internal Flexibility Upon NEMD Simulations of Viscosity N. G. Fuller 1 and R. L. Rowley 1,2 Abstract The influence of model flexibility upon simulated viscosity
Draft: September 29, 1999 The Effect of Model Internal Flexibility Upon NEMD Simulations of Viscosity N. G. Fuller 1 and R. L. Rowley 1,2 Abstract The influence of model flexibility upon simulated viscosity
Javier Junquera. Statistical mechanics
 Javier Junquera Statistical mechanics From the microscopic to the macroscopic level: the realm of statistical mechanics Computer simulations Thermodynamic state Generates information at the microscopic
Javier Junquera Statistical mechanics From the microscopic to the macroscopic level: the realm of statistical mechanics Computer simulations Thermodynamic state Generates information at the microscopic
The micro-properties of [hmpy+] [Tf 2 N-] Ionic liquid: a simulation. study. 1. Introduction
![The micro-properties of [hmpy+] [Tf 2 N-] Ionic liquid: a simulation. study. 1. Introduction](/thumbs/72/66941683.jpg "The micro-properties of [hmpy+] [Tf 2 N-] Ionic liquid: a simulation. study. 1. Introduction") ISBN 978-1-84626-081-0 Proceedings of the 2010 International Conference on Application of Mathematics and Physics Volume 1: Advances on Space Weather, Meteorology and Applied Physics Nanjing, P. R. China,
ISBN 978-1-84626-081-0 Proceedings of the 2010 International Conference on Application of Mathematics and Physics Volume 1: Advances on Space Weather, Meteorology and Applied Physics Nanjing, P. R. China,
Molecular Dynamics Simulations
 Molecular Dynamics Simulations Dr. Kasra Momeni www.knanosys.com Outline Long-range Interactions Ewald Sum Fast Multipole Method Spherically Truncated Coulombic Potential Speeding up Calculations SPaSM
Molecular Dynamics Simulations Dr. Kasra Momeni www.knanosys.com Outline Long-range Interactions Ewald Sum Fast Multipole Method Spherically Truncated Coulombic Potential Speeding up Calculations SPaSM
Using Molecular Dynamics to Compute Properties CHEM 430
 Using Molecular Dynamics to Compute Properties CHEM 43 Heat Capacity and Energy Fluctuations Running an MD Simulation Equilibration Phase Before data-collection and results can be analyzed the system
Using Molecular Dynamics to Compute Properties CHEM 43 Heat Capacity and Energy Fluctuations Running an MD Simulation Equilibration Phase Before data-collection and results can be analyzed the system
Multiscale Coarse-Graining of Ionic Liquids
 3564 J. Phys. Chem. B 2006, 110, 3564-3575 Multiscale Coarse-Graining of Ionic Liquids Yanting Wang, Sergei Izvekov, Tianying Yan, and Gregory A. Voth* Center for Biophysical Modeling and Simulation and
3564 J. Phys. Chem. B 2006, 110, 3564-3575 Multiscale Coarse-Graining of Ionic Liquids Yanting Wang, Sergei Izvekov, Tianying Yan, and Gregory A. Voth* Center for Biophysical Modeling and Simulation and
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Unit Cell-Level Thickness Control of Single-Crystalline Zinc Oxide Nanosheets Enabled by Electrical Double Layer Confinement
 Unit Cell-Level Thickness Control of Single-Crystalline Zinc Oxide Nanosheets Enabled by Electrical Double Layer Confinement Xin Yin, Yeqi Shi, Yanbing Wei, Yongho Joo, Padma Gopalan, Izabela Szlufarska,
Unit Cell-Level Thickness Control of Single-Crystalline Zinc Oxide Nanosheets Enabled by Electrical Double Layer Confinement Xin Yin, Yeqi Shi, Yanbing Wei, Yongho Joo, Padma Gopalan, Izabela Szlufarska,
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
7 To solve numerically the equation of motion, we use the velocity Verlet or leap frog algorithm. _ V i n = F i n m i (F.5) For time step, we approxim
 For time step, we approxim") 69 Appendix F Molecular Dynamics F. Introduction In this chapter, we deal with the theories and techniques used in molecular dynamics simulation. The fundamental dynamics equations of any system is the
69 Appendix F Molecular Dynamics F. Introduction In this chapter, we deal with the theories and techniques used in molecular dynamics simulation. The fundamental dynamics equations of any system is the
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
T6.2 Molecular Mechanics
 T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
Van Gunsteren et al. Angew. Chem. Int. Ed. 45, 4064 (2006)
") Van Gunsteren et al. Angew. Chem. Int. Ed. 45, 4064 (2006) Martini Workshop 2015 Coarse Graining Basics Alex de Vries Every word or concept, clear as it may seem to be, has only a limited range of applicability
Van Gunsteren et al. Angew. Chem. Int. Ed. 45, 4064 (2006) Martini Workshop 2015 Coarse Graining Basics Alex de Vries Every word or concept, clear as it may seem to be, has only a limited range of applicability
Supporting Information. for. Influence of Cononsolvency on the. Aggregation of Tertiary Butyl Alcohol in. Methanol-Water Mixtures
 Supporting Information for Influence of Cononsolvency on the Aggregation of Tertiary Butyl Alcohol in Methanol-Water Mixtures Kenji Mochizuki,, Shannon R. Pattenaude, and Dor Ben-Amotz Research Institute
Supporting Information for Influence of Cononsolvency on the Aggregation of Tertiary Butyl Alcohol in Methanol-Water Mixtures Kenji Mochizuki,, Shannon R. Pattenaude, and Dor Ben-Amotz Research Institute
Shear Properties and Wrinkling Behaviors of Finite Sized Graphene
 Shear Properties and Wrinkling Behaviors of Finite Sized Graphene Kyoungmin Min, Namjung Kim and Ravi Bhadauria May 10, 2010 Abstract In this project, we investigate the shear properties of finite sized
Shear Properties and Wrinkling Behaviors of Finite Sized Graphene Kyoungmin Min, Namjung Kim and Ravi Bhadauria May 10, 2010 Abstract In this project, we investigate the shear properties of finite sized
Energy functions and their relationship to molecular conformation. CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror
 Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Outline Energy functions for proteins (or biomolecular systems more generally)
Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Outline Energy functions for proteins (or biomolecular systems more generally)
Biomolecular modeling I
 2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
The Molecular Dynamics Method
 H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
Protein Structure Analysis
 BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
Molecular Mechanics. Yohann Moreau. November 26, 2015
 Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Universal Repulsive Contribution to the. Solvent-Induced Interaction Between Sizable, Curved Hydrophobes: Supporting Information
 Universal Repulsive Contribution to the Solvent-Induced Interaction Between Sizable, Curved Hydrophobes: Supporting Information B. Shadrack Jabes, Dusan Bratko, and Alenka Luzar Department of Chemistry,
Universal Repulsive Contribution to the Solvent-Induced Interaction Between Sizable, Curved Hydrophobes: Supporting Information B. Shadrack Jabes, Dusan Bratko, and Alenka Luzar Department of Chemistry,
Molecular Mechanics / ReaxFF
 Molecular Dynamics simulations Lecture 09: Molecular Mechanics / ReaxFF Dr. Olli Pakarinen University of Helsinki Fall 2012 Lecture notes based on notes by Dr. Jani Kotakoski, 2010 CONTENTS Molecular mechanics
Molecular Dynamics simulations Lecture 09: Molecular Mechanics / ReaxFF Dr. Olli Pakarinen University of Helsinki Fall 2012 Lecture notes based on notes by Dr. Jani Kotakoski, 2010 CONTENTS Molecular mechanics
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Dynamics Studies of Water Flow in Carbon Nanotubes
 Western University Scholarship@Western Electronic Thesis and Dissertation Repository September 2012 Molecular Dynamics Studies of Water Flow in Carbon Nanotubes Alexander D. Marshall The University of
Western University Scholarship@Western Electronic Thesis and Dissertation Repository September 2012 Molecular Dynamics Studies of Water Flow in Carbon Nanotubes Alexander D. Marshall The University of
Supporting Information
 Supporting Information ph-responsive self-assembly of polysaccharide through a rugged energy landscape Brian H. Morrow, Gregory F. Payne, and Jana Shen Department of Pharmaceutical Sciences, School of
Supporting Information ph-responsive self-assembly of polysaccharide through a rugged energy landscape Brian H. Morrow, Gregory F. Payne, and Jana Shen Department of Pharmaceutical Sciences, School of
Electronic excitations in conjugated. Many-Body Green's Functions. Behnaz Bagheri Varnousfaderani. Behnaz Bagheri Varnousfaderani
 Technische Universiteit Eindhoven University of Technology Electronic excitations in conjugated polymers Electronic from excitations QM/MM simulations in conjugated pol based from on Many-Body QM/MM simulations
Technische Universiteit Eindhoven University of Technology Electronic excitations in conjugated polymers Electronic from excitations QM/MM simulations in conjugated pol based from on Many-Body QM/MM simulations
Molecular Dynamics. A very brief introduction
 Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Supplementary Materials
 Supplementary Materials Atomistic Origin of Brittle Failure of Boron Carbide from Large Scale Reactive Dynamics Simulations; Suggestions toward Improved Ductility Qi An and William A. Goddard III * Materials
Supplementary Materials Atomistic Origin of Brittle Failure of Boron Carbide from Large Scale Reactive Dynamics Simulations; Suggestions toward Improved Ductility Qi An and William A. Goddard III * Materials
Advanced in silico drug design
 Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro. Karel Berka, Ph.D.
 4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
Energy functions and their relationship to molecular conformation. CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror
 Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Yesterday s Nobel Prize: single-particle cryoelectron microscopy 2 Outline Energy
Energy functions and their relationship to molecular conformation CS/CME/BioE/Biophys/BMI 279 Oct. 3 and 5, 2017 Ron Dror Yesterday s Nobel Prize: single-particle cryoelectron microscopy 2 Outline Energy
Exercise 2: Solvating the Structure Before you continue, follow these steps: Setting up Periodic Boundary Conditions
 Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Supporting information
 Electronic Supplementary Material ESI for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Supporting information Understanding three-body contributions to coarse-grained force
Electronic Supplementary Material ESI for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Supporting information Understanding three-body contributions to coarse-grained force
Ideal Gas Behavior. NC State University
 Chemistry 331 Lecture 6 Ideal Gas Behavior NC State University Macroscopic variables P, T Pressure is a force per unit area (P= F/A) The force arises from the change in momentum as particles hit an object
Chemistry 331 Lecture 6 Ideal Gas Behavior NC State University Macroscopic variables P, T Pressure is a force per unit area (P= F/A) The force arises from the change in momentum as particles hit an object
Molecular modeling for functional materials design in chemical engineering and more
 Molecular modeling for functional materials design in chemical engineering and more Dr inż. Łukasz Radosiński Group of Bioprocess and Biomedical Engineering Wroclaw University of Science and Technology
Molecular modeling for functional materials design in chemical engineering and more Dr inż. Łukasz Radosiński Group of Bioprocess and Biomedical Engineering Wroclaw University of Science and Technology
Dynamic force matching: Construction of dynamic coarse-grained models with realistic short time dynamics and accurate long time dynamics
 for resubmission Dynamic force matching: Construction of dynamic coarse-grained models with realistic short time dynamics and accurate long time dynamics Aram Davtyan, 1 Gregory A. Voth, 1 2, a) and Hans
for resubmission Dynamic force matching: Construction of dynamic coarse-grained models with realistic short time dynamics and accurate long time dynamics Aram Davtyan, 1 Gregory A. Voth, 1 2, a) and Hans
Molecular mechanics. classical description of molecules. Marcus Elstner and Tomáš Kubař. April 29, 2016
 classical description of molecules April 29, 2016 Chemical bond Conceptual and chemical basis quantum effect solution of the SR numerically expensive (only small molecules can be treated) approximations
classical description of molecules April 29, 2016 Chemical bond Conceptual and chemical basis quantum effect solution of the SR numerically expensive (only small molecules can be treated) approximations
Ab initio molecular dynamics. Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy. Bangalore, 04 September 2014
 Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Gear methods I + 1/18
 Gear methods I + 1/18 Predictor-corrector type: knowledge of history is used to predict an approximate solution, which is made more accurate in the following step we do not want (otherwise good) methods
Gear methods I + 1/18 Predictor-corrector type: knowledge of history is used to predict an approximate solution, which is made more accurate in the following step we do not want (otherwise good) methods
Citation for published version (APA): Hess, B. (2002). Stochastic concepts in molecular simulation Groningen: s.n.
: Hess, B. (2002). Stochastic concepts in molecular simulation Groningen: s.n.") University of Groningen Stochastic concepts in molecular simulation Hess, Berk IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check
University of Groningen Stochastic concepts in molecular simulation Hess, Berk IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check
Temperature and Pressure Controls
 Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Temperature and Pressure Controls
 Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D.
 3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
Protein Structure Analysis
 BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
The GROMOS Software for (Bio)Molecular Simulation
Molecular Simulation") The GROMOS Software for (Bio)Molecular Simulation Volume 2: Algorithms and Formulae for Modelling of Molecular Systems September 28, 2017 Contents Chapter 1. Introduction 2-1 Chapter 2. Basic choices
The GROMOS Software for (Bio)Molecular Simulation Volume 2: Algorithms and Formulae for Modelling of Molecular Systems September 28, 2017 Contents Chapter 1. Introduction 2-1 Chapter 2. Basic choices
k θ (θ θ 0 ) 2 angles r i j r i j
 2 angles r i j r i j") 1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model. John Dood Hope College
 Water Model. John Dood Hope College") Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model John Dood Hope College What are MD simulations? Model and predict the structure and dynamics of large macromolecules.
Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model John Dood Hope College What are MD simulations? Model and predict the structure and dynamics of large macromolecules.
c 2010 ANJAN V. RAGHUNATHAN
 c 2010 ANJAN V. RAGHUNATHAN MOLECULAR UNDERSTANDING OF OSMOSIS AND A MULTISCALE FRAMEWORK TO INVESTIGATE CONFINED FLUID PROPERTIES BY ANJAN V. RAGHUNATHAN DISSERTATION Submitted in partial fulfillment
c 2010 ANJAN V. RAGHUNATHAN MOLECULAR UNDERSTANDING OF OSMOSIS AND A MULTISCALE FRAMEWORK TO INVESTIGATE CONFINED FLUID PROPERTIES BY ANJAN V. RAGHUNATHAN DISSERTATION Submitted in partial fulfillment
Interface Resistance and Thermal Transport in Nano-Confined Liquids
 1 Interface Resistance and Thermal Transport in Nano-Confined Liquids Murat Barisik and Ali Beskok CONTENTS 1.1 Introduction...1 1.2 Onset of Continuum Behavior...2 1.3 Boundary Treatment Effects on Interface
1 Interface Resistance and Thermal Transport in Nano-Confined Liquids Murat Barisik and Ali Beskok CONTENTS 1.1 Introduction...1 1.2 Onset of Continuum Behavior...2 1.3 Boundary Treatment Effects on Interface
I690/B680 Structural Bioinformatics Spring Protein Structure Determination by NMR Spectroscopy
 I690/B680 Structural Bioinformatics Spring 2006 Protein Structure Determination by NMR Spectroscopy Suggested Reading (1) Van Holde, Johnson, Ho. Principles of Physical Biochemistry, 2 nd Ed., Prentice
I690/B680 Structural Bioinformatics Spring 2006 Protein Structure Determination by NMR Spectroscopy Suggested Reading (1) Van Holde, Johnson, Ho. Principles of Physical Biochemistry, 2 nd Ed., Prentice
Supporting Information
 Supporting Information Sánchez et al. 10.1073/pnas.1612893114 SI Materials and Methods Growth of Single Crystalline Ice. The single crystal ice boule growth method is based on withdrawing a single crystalline
Supporting Information Sánchez et al. 10.1073/pnas.1612893114 SI Materials and Methods Growth of Single Crystalline Ice. The single crystal ice boule growth method is based on withdrawing a single crystalline
Molecular Modeling of Matter
 Molecular Modeling of Matter Keith E. Gubbins Lecture 2 Atomistic Simulation. General Features. Molecular Dynamics Outline General features of atomistic simulation Brief description of MC and MD Features
Molecular Modeling of Matter Keith E. Gubbins Lecture 2 Atomistic Simulation. General Features. Molecular Dynamics Outline General features of atomistic simulation Brief description of MC and MD Features
Atoms, electrons and Solids
 Atoms, electrons and Solids Shell model of an atom negative electron orbiting a positive nucleus QM tells that to minimize total energy the electrons fill up shells. Each orbit in a shell has a specific
Atoms, electrons and Solids Shell model of an atom negative electron orbiting a positive nucleus QM tells that to minimize total energy the electrons fill up shells. Each orbit in a shell has a specific
Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia
 University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
Aspects of nonautonomous molecular dynamics
 Aspects of nonautonomous molecular dynamics IMA, University of Minnesota, Minneapolis January 28, 2007 Michel Cuendet Swiss Institute of Bioinformatics, Lausanne, Switzerland Introduction to the Jarzynski
Aspects of nonautonomous molecular dynamics IMA, University of Minnesota, Minneapolis January 28, 2007 Michel Cuendet Swiss Institute of Bioinformatics, Lausanne, Switzerland Introduction to the Jarzynski
Supporting Information for Solid-liquid Thermal Transport and its Relationship with Wettability and the Interfacial Liquid Structure
 Supporting Information for Solid-liquid Thermal Transport and its Relationship with Wettability and the Interfacial Liquid Structure Bladimir Ramos-Alvarado, Satish Kumar, and G. P. Peterson The George
Supporting Information for Solid-liquid Thermal Transport and its Relationship with Wettability and the Interfacial Liquid Structure Bladimir Ramos-Alvarado, Satish Kumar, and G. P. Peterson The George
Rate of Heating and Cooling
 Rate of Heating and Cooling 35 T [ o C] Example: Heating and cooling of Water E 30 Cooling S 25 Heating exponential decay 20 0 100 200 300 400 t [sec] Newton s Law of Cooling T S > T E : System S cools
Rate of Heating and Cooling 35 T [ o C] Example: Heating and cooling of Water E 30 Cooling S 25 Heating exponential decay 20 0 100 200 300 400 t [sec] Newton s Law of Cooling T S > T E : System S cools
Computer simulation methods (2) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
3. An Introduction to Molecular Mechanics
 3. An Introduction to Molecular Mechanics Introduction When you use Chem3D to draw molecules, the program assigns bond lengths and bond angles based on experimental data. The program does not contain real
3. An Introduction to Molecular Mechanics Introduction When you use Chem3D to draw molecules, the program assigns bond lengths and bond angles based on experimental data. The program does not contain real
From Atoms to Materials: Predictive Theory and Simulations
 From Atoms to Materials: Predictive Theory and Simulations Week 3 Lecture 4 Potentials for metals and semiconductors Ale Strachan strachan@purdue.edu School of Materials Engineering & Birck anotechnology
From Atoms to Materials: Predictive Theory and Simulations Week 3 Lecture 4 Potentials for metals and semiconductors Ale Strachan strachan@purdue.edu School of Materials Engineering & Birck anotechnology
Glass-Transition and Side-Chain Dynamics in Thin Films: Explaining. Dissimilar Free Surface Effects for Polystyrene and Poly(methyl methacrylate)
") Supporting Information for Glass-Transition and Side-Chain Dynamics in Thin Films: Explaining Dissimilar Free Surface Effects for Polystyrene and Poly(methyl methacrylate) David D. Hsu, Wenjie Xia, Jake
Supporting Information for Glass-Transition and Side-Chain Dynamics in Thin Films: Explaining Dissimilar Free Surface Effects for Polystyrene and Poly(methyl methacrylate) David D. Hsu, Wenjie Xia, Jake
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 If you throw up a stone, it is Physics. If you throw up a stone, it is Physics. If it lands on your head, it is
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 If you throw up a stone, it is Physics. If you throw up a stone, it is Physics. If it lands on your head, it is
Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012
 Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012 K. Kremer Max Planck Institute for Polymer Research, Mainz Overview Simulations, general considerations
Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012 K. Kremer Max Planck Institute for Polymer Research, Mainz Overview Simulations, general considerations
Routine access to millisecond timescale events with accelerated molecular dynamics
 Routine access to millisecond timescale events with accelerated molecular dynamics Levi C.T. Pierce, Romelia Salomon-Ferrer, Cesar Augusto F. de Oliveira #, J. Andrew McCammon #, Ross C. Walker * SUPPORTING
Routine access to millisecond timescale events with accelerated molecular dynamics Levi C.T. Pierce, Romelia Salomon-Ferrer, Cesar Augusto F. de Oliveira #, J. Andrew McCammon #, Ross C. Walker * SUPPORTING
MODELLING THE STRUCTURE OF. NANOPARTICLE EMBEDDING MATRICES : MOLECULAR DYNAMICS IN Li 2 B 4 O 7
 MODELLING THE STRUCTURE OF NANOPARTICLE EMBEDDING MATRICES : MOLECULAR DYNAMICS IN Li 2 B 4 O 7 Alain MARBEUF and Janis KLIAVA Centre de Physique Moléculaire Optique et Hertzienne UMR5798, Université Bordeaux
MODELLING THE STRUCTURE OF NANOPARTICLE EMBEDDING MATRICES : MOLECULAR DYNAMICS IN Li 2 B 4 O 7 Alain MARBEUF and Janis KLIAVA Centre de Physique Moléculaire Optique et Hertzienne UMR5798, Université Bordeaux