NWChem: Molecular Dynamics and QM/MM
|
|
|
- Jayson Wilkinson
- 6 years ago
- Views:
Transcription

1 NWChem: Molecular Dynamics and QM/MM
2 Molecular Dynamics Functionality Target systems: General features: biomolecules (proteins, DNA/RNA, biomembranes) energy evaluation (SP) energy minimization (EM) molecular dynamics simulation (MD) free energy evaluation (MSTP & MCTI) quantum molecular dynamics (QMD) hybrid molecular dynamics (QM/MM)
3 Force Fields Classical empirical force fields Bond-stretching, angle-bending, torsional, out-of-plane-bending, electrostatic, and van der Waals non-bonded interactions Electronic polarization First order Self-consistent induced fields Smooth particle-mesh Ewald electrostatics Effective pair potential MD only Self-guided MD
4 Simulation Integration Newton s Equations of Motion (leapfrog, Browne-Clark) Constant Temperature and Pressure (Berendsen weak coupling) Periodic Boundary Conditions (minimum image convention) Geometry Optimization (steepest descent, conjugate gradient) Twin-Range Verlet Neighbor Lists (cell index method) Constraints (SHAKE)
5 Free Energy Evaluation Single Step Thermodynamic Perturbation (SSTP) G = G 1 - G 0 = - RT ln < exp ( - H/RT)> 0 with H=H 1 -H 0 Multiple Step Thermodynamic Perturbation (MSTP) DG = S i ( G i+1 - G i ) = -S i RT ln < exp ( - H i /RT)> i with H i =H i+1 H i Multiconfiguration Thermodynamic Integration (MCTI) DG = S i ( G i+1 - G i ) = S i < H(λ) / λ > i Dλ i Single and dual topology Hamiltonians Double-wide sampling Separation-shifted scaling (SSS) Potentials of mean force over multiple processors Statistical error correlation analysis
6 Database Directories ffield_lvl ffield force field e.g. amber, charmm lvl 1, 2, 3, 4, 5, 6, 7, 8, or 9 Directories supplied with NWChem are named ffield_l where l is one of: s x q u t c standard parameters as published for the force field extensions as published in open literature contributed parameters by NWChem team user contributed parameters temporary current Defined in input file or ~/.nwchemrc ffield amber amber_1 /software/nwchem/share/data/amber/amber_s/ amber_2 /software/nwchem/share/data/amber/amber_x/ amber_3 /home/newton/data/amber/amber_q/ spce /software/nwchem/share/data/solvents/spce.rst
7 File Names system.ext system user defined molecular system name ext file type, e.g. pdb PDB file top topology file seq sequence file system_calc.ext system user defined molecular system name calc user defined identification for the calculation identification, e.g. em, md002, tia ext file type, e.g. out output file rst restart file qrs energy minimized restart file prp property file trj trajectory file gib free energy file
8 PREPARE Functionality and requirements frg sgm seq top pdb rst Functionality: Topology and restart file generation Coordinates from pdb formatted file or geometry input Fragment and segment file generation from coordinates Solvation Potential of mean force functions Topology modification for free energy and QM/MM calculations File format conversion, e.g. from rst to pdb Requirements PDB format, i.e. IUPAC atom names, residue names, etc. Automated atom typing based on force field typing rules Force field parameters from par file(s) Partial atomic charges from frg or sgm files
9 PREPARE Input Example PDB format, i.e. IUPAC atom names, residue names, etc. Automated atom typing based on force field typing rules Force field parameters from par file(s) Partial atomic charges from frg or sgm files memory noverify heap 1 mb stack 48 mb global 24 mb start crown basis "ao basis" print H library sto-3g C library sto-3g O library sto-3g Na library sto-3g end Define basis sets in case partial charges need to be calculated prepare system crown_em modify atom 7:Na set 3 type K end task prepare Read coordinates from crown.pdb Specify atom type change in topology Generate topology file crown.top Restart file crown_em.rst
10 MD Input Example md system crown_md data 1000 isotherm isobar record rest 1000 coord 100 prop 10 end task md dynamics task shell cp crown_md.rst crown_ti.rst Molecular dynamics input: NpT ensemble, 1000 steps Copy restart file. md system crown_ti equil 1000 data 2000 over 1000 step Free energy simulation isotherm MCTI: 21*( ) isobar NpT ensemble new forward 21 of 21 print step 100 stat 1000 record rest 1000 free 1 end task md thermodynamics
11 ANALYZE input memory heap 8 mb stack 64 mb global 24 mb start job analysis system system_calc reference system_calc.rst file crown_md?.trj 0 10 #? Is wild card, replaced by series 0-10 select _C? _O? # select subset of atoms essential # Perform essential dynamics analysis project 1 crown_vec1 Projection onto specified vector project 2 crown_vec2 end task analyze Files required: system.top ( molecular topology ) system_calc.rst ( coordinates )
12 Relative solvation G Input Example memory noverify heap 10 mb stack 32 mb global 32 mb start crown basis "ao basis" print H library sto-3g C library sto-3g O library sto-3g Na library sto-3g end prepare system crown_em modify atom 7:Na set 3 type K end task prepare md system crown_em sd 1000 noshake end task md optimize prepare read crown_em.qrs write crown_em.pdb solvate write crown_md.rst end task prepare Define basis sets Read coordinates from crown.pdb Specify atom type change in topology Generate topology file crown.top Restart file crown_em.rst Perform energy minimization Solvate the solute Generate crown_md.rst
13 Relative solvation G Input Example (continued) md system crown_md data 1000 isotherm trelax isobar record rest 1000 scoor 100 prop 10 end task md dynamics task shell cp crown_md.rst crown_ti.rst Molecular dynamics input: NpT ensemble, 1000 steps Copy restart file. md system crown_ti equil 1000 data 2000 over 1000 step Free energy simulation input: isotherm trelax MCTI: 21*( ) isobar NpT ensemble new forward 21 of 21 print step 100 stat 1000 record rest 100 free 1 end task md thermodynamics

 MM region, 80,000 atoms Cytochrome IFC3, Schewanella QM Region")
14 QM/MM approach Combines two different descriptions quantummechanical and classical The level theory changes based on a particular region Reactive regions quantum mechanical description (QM) Regions where no chemical changes occur (or important) are treated a the classical molecular mechanics level (MM) MM region, 80,000 atoms Cytochrome IFC3, Schewanella QM Region 109 atoms, DFT 14
15 Structure of QM/MM energy functional ( ) ( ) ( ) E r, R; ψ = E r, R; ψ + qm / mm qm mm E r, R All QM-dependencies are in the first term int ext [ rr, ; ψ] = [ rr, ; ψ] + [ rr, ; ρ] E E E Internal QM energy (theory dependent) qm qm qm Coulomb interactions with MM atoms I Z I R ρ I ( r ') r ' dr ' All other classical terms are in the second term Bonded, angle, dihedral Coulomb interactions Vdw interactions 15

 modules Generic")



16 QM/MM Interface in NWChem MM QM QM/MM Modular implementation sits on top of molecular mechanics (MM) and quantum (QM) modules Generic interface driven by function calls Manages data flow domain decomposition of coordinates in MM module replicated geometry data in QM module all data transfers happen in core Dispatches high level operations (e.g. optimization) 16
17 Summary of QM/MM Capabilities Ground and excited state properties Structural optimization Reaction Pathway Calculations Dynamical Simulations Statistical Sampling (free energies) 17
 250 200 150 100 basis * library \"Ahlrichs pvdz\" end qmmm link_atoms")
18 Example of QM/MM Single Point Calculation start capk charge 2 prepare source capk.pdb modify segment 358 quantum ignore update lists write capk.rst end task prepare md system capk cutoff 1.5 end QM region definition Active Site of capk kinase 75 atoms, 817 basis Scaling of QM/MM DFT calculation functions 300 Time (s) basis * library "Ahlrichs pvdz" end qmmm link_atoms hydrogen end task qmmm dft energy Hydrogen Link Atoms 50 Commence QM/MM DFT Calculation Processors 18 Valiev, M., et al.jpc B, (47): p

19 Example: QM/MM Excited State Calculations of cytosine base in DNA QM/MM coupled-cluster CR-EOMCCSD(T) calculations of two lowest excited states Protein environment has a significant influence on the excitation leading to a 0.4 ev stabilization of the ππ* excited state compared to gas phase M. Valiev, K. Kowalski, JCP, 125(21), (2006) 19
20 QM/MM Optimization Large system sizes ( ) makes direct optimizations impractical even with QM/MM approximation Key observations Most of degrees of freedom are in MM region The structure of far away MM regions has a little effect on the structure of QM region Small displacements of MM atoms affect little the electronic structure of QM region. Decouple optimization of QM and MM regions 20

21 Optimization Algorithm 1. QM Region Optimization a. MM region is fixed b. Typically steps c. Requires solution of Schrödinger Equation 2. MM Region Optimization a. QM region is fixed b steps c. QM region is represented either as effective point charges or static electron density distribution 3. Repeat the cycle until convergence 21

22 Example: Optimization of Zn-porphyrin in solution QM region Zn-porphyrin (37 atoms) DFT/B3LYP MM region 869 SPC/E waters 4.5 hours on 48 processors versus direct optimization would take ~ 2 days. qmmm region qm solvent maxiter method lbfgs sd ncycles 20 density espfit end task qmmm dft optimize Input File 22


23 Reaction Pathway II Nudged Elastic Band Method Pathway approximated discrete set of intermediate structures Beads represent different snapshots of reactive QM region along the pathway Forces on beads are calculated at the relaxed solvent configuration 23
24 Free Energy Differences W = AB 1 E A E B ln e β β ( ( ) ( )) A A B Accurate quantum mechanical description is a major challenge especially for high level methods ( energy evaluations) The solution Introduce intermediate less expensive representation(s) Redistribute sampling using thermodynamic cycles 24
 ZI ρ R r' I dr' = i, I Z R I I Qi r i")
25 QM/MM Representations Different QM/MM representations Computational Efficiency CC/MM DFT/MM MM/MM Example of MM/MM representation - QM atoms are replaced by effective point charges Q i reproducing correct field E qm Z IQi = r i, I R I Complexity and Accuracy i I ( r' ) ZI ρ R r' I dr' = i, I Z R I I Qi r i 25
26 Free Energy Ladder 1 E A E B WAB = ln e β β ( ( ) ( )) A A B Representation MM/MM DFT/MM E MM/MM (A) E DFT/MM (A) E MM/MM (B) E DFT/MM (B) A B Configuration DFT W AB = W MM DFT MM MM ( AA W BB )+ W AB Valiev et al JCP 127, (2007) 26
27 Free Energy Ladder 1 E A E B WAB = ln e β β ( ( ) ( )) A A B Representation MM/MM DFT/MM E MM/MM (A) E DFT/MM (A) E MM/MM (B) E DFT/MM (B) CC/MM E CC/MM (A) E CC/MM (B) A Configuration CC W AB = W DFT CC DFT DFT ( AA W BB )+ W MM DFT MM MM ( AA W BB )+ W AB B 27

q A + λq B Free Energy Perturbation = ESP W AB e i 1 ESP β Eλ i")
28 Calculation of MM/MM free energy E MM/MM (A) E MM/MM (B) Can use any of the methods developed for classical free energy calculations Transformation between A and B configurations r λ = ( 1 λ)r A + λr B Q λ = ( 1 λ)q A + λq B Free Energy Perturbation = ESP W AB e i 1 ESP β Eλ i λi+ 1 ln β λ i 28
 MM representation is closely tailored to DFT by point charge fitting Approximate by the energy difference Can utilize free energy perturbation approach by resampling")
29 Calculation of DFT->MM free energy E MM/MM (A) W DFT MM AA 1 β AA = ln β e DFT MM ( E ) MM / MM E DFT/MM (A) ( r R; ψ ) E ( r, Q ) DFT MM E AA = EDFT / MM A, A MM / MM A R; A Vertical change of transformation (fixed QM region) MM representation is closely tailored to DFT by point charge fitting Approximate by the energy difference Can utilize free energy perturbation approach by resampling MM/MM trajectory 29


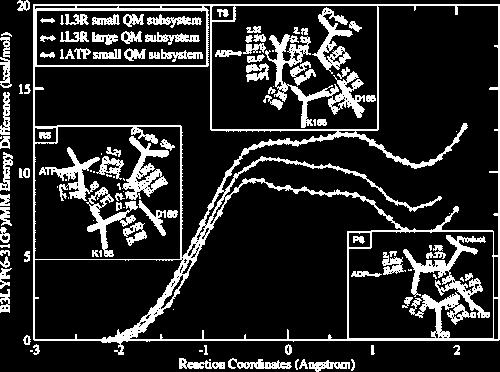
30 Example : Free energy barrier for the phosphorylation reaction in kinase protein ~15 kcal/mol reaction barrier 30 capk protein kinase catalyze the transfer of the g-phosphoryl Determination of the reaction pathway using NEB QM/MM ap Calculation of free energy using effective charge approximatio Valiev et al J. Phys Chem B. 111(47): (2007).


31 Importance of free energy (Cheng et al JACS, 127,1553,2005) PKA 31

32 32 Questions?
Basic introduction of NWChem software
 Basic introduction of NWChem software Background NWChem is part of the Molecular Science Software Suite Designed and developed to be a highly efficient and portable Massively Parallel computational chemistry
Basic introduction of NWChem software Background NWChem is part of the Molecular Science Software Suite Designed and developed to be a highly efficient and portable Massively Parallel computational chemistry
Hands-on : Model Potential Molecular Dynamics
 Hands-on : Model Potential Molecular Dynamics OUTLINE 0. DL_POLY code introduction 0.a Input files 1. THF solvent molecule 1.a Geometry optimization 1.b NVE/NVT dynamics 2. Liquid THF 2.a Equilibration
Hands-on : Model Potential Molecular Dynamics OUTLINE 0. DL_POLY code introduction 0.a Input files 1. THF solvent molecule 1.a Geometry optimization 1.b NVE/NVT dynamics 2. Liquid THF 2.a Equilibration
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Multiscale Materials Modeling
 Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Molecular Dynamics Simulations. Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia
 Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Basic introduction of NWChem software
 Basic introduction of NWChem software Background! NWChem is part of the Molecular Science Software Suite! Designed and developed to be a highly efficient and portable Massively Parallel computational chemistry
Basic introduction of NWChem software Background! NWChem is part of the Molecular Science Software Suite! Designed and developed to be a highly efficient and portable Massively Parallel computational chemistry
Fragmentation methods
 Fragmentation methods Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method
Fragmentation methods Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
Advanced in silico drug design
 Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Introduction to Classical Molecular Dynamics. Giovanni Chillemi HPC department, CINECA
 Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
Protein Structure Analysis
 BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
TECHNIQUES TO LOCATE A TRANSITION STATE
 32 III. TECHNIQUES TO LOCATE A TRANSITION STATE In addition to the location of minima, the calculation of transition states and reaction pathways is an interesting task for Quantum Chemistry (QC). The
32 III. TECHNIQUES TO LOCATE A TRANSITION STATE In addition to the location of minima, the calculation of transition states and reaction pathways is an interesting task for Quantum Chemistry (QC). The
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Free Energy Simulation Methods
 Free Energy Simulation Methods Free energy simulation methods Many methods have been developed to compute (relative) free energies on the basis of statistical mechanics Free energy perturbation Thermodynamic
Free Energy Simulation Methods Free energy simulation methods Many methods have been developed to compute (relative) free energies on the basis of statistical mechanics Free energy perturbation Thermodynamic
Computational Chemistry - MD Simulations
 Computational Chemistry - MD Simulations P. Ojeda-May pedro.ojeda-may@umu.se Department of Chemistry/HPC2N, Umeå University, 901 87, Sweden. May 2, 2017 Table of contents 1 Basics on MD simulations Accelerated
Computational Chemistry - MD Simulations P. Ojeda-May pedro.ojeda-may@umu.se Department of Chemistry/HPC2N, Umeå University, 901 87, Sweden. May 2, 2017 Table of contents 1 Basics on MD simulations Accelerated
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 If you throw up a stone, it is Physics. If you throw up a stone, it is Physics. If it lands on your head, it is
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 If you throw up a stone, it is Physics. If you throw up a stone, it is Physics. If it lands on your head, it is
Charge equilibration
 Charge equilibration Taylor expansion of energy of atom A @E E A (Q) =E A0 + Q A + 1 @Q A 0 2 Q2 A @ 2 E @Q 2 A 0 +... The corresponding energy of cation/anion and neutral atom E A (+1) = E A0 + @E @Q
Charge equilibration Taylor expansion of energy of atom A @E E A (Q) =E A0 + Q A + 1 @Q A 0 2 Q2 A @ 2 E @Q 2 A 0 +... The corresponding energy of cation/anion and neutral atom E A (+1) = E A0 + @E @Q
Hyeyoung Shin a, Tod A. Pascal ab, William A. Goddard III abc*, and Hyungjun Kim a* Korea
 The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
Subject of the Lecture:
 Subject of the Lecture: Conceptual basis for the development of force fields. Implementation/validation Water - a worked example Extensions - combining molecular mechanics and quantum mechanics (QM/MM)
Subject of the Lecture: Conceptual basis for the development of force fields. Implementation/validation Water - a worked example Extensions - combining molecular mechanics and quantum mechanics (QM/MM)
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Development of Molecular Dynamics Simulation System for Large-Scale Supra-Biomolecules, PABIOS (PArallel BIOmolecular Simulator)
") Development of Molecular Dynamics Simulation System for Large-Scale Supra-Biomolecules, PABIOS (PArallel BIOmolecular Simulator) Group Representative Hisashi Ishida Japan Atomic Energy Research Institute
Development of Molecular Dynamics Simulation System for Large-Scale Supra-Biomolecules, PABIOS (PArallel BIOmolecular Simulator) Group Representative Hisashi Ishida Japan Atomic Energy Research Institute
Molecular Dynamics. A very brief introduction
 Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Biomolecular modeling I
 2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
Bridging Scales Through Wavefunction Analysis
 Bridging Scales Through Wavefunction Analysis Felix Plasser Institute for Theoretical Chemistry, University of Vienna Excited States Bridging Scales Marseille, November 7 10, 2016 F. Plasser Wavefunction
Bridging Scales Through Wavefunction Analysis Felix Plasser Institute for Theoretical Chemistry, University of Vienna Excited States Bridging Scales Marseille, November 7 10, 2016 F. Plasser Wavefunction
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Free energy simulations
 Free energy simulations Marcus Elstner and Tomáš Kubař January 14, 2013 Motivation a physical quantity that is of most interest in chemistry? free energies Helmholtz F or Gibbs G holy grail of computational
Free energy simulations Marcus Elstner and Tomáš Kubař January 14, 2013 Motivation a physical quantity that is of most interest in chemistry? free energies Helmholtz F or Gibbs G holy grail of computational
The Molecular Dynamics Method
 H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
Free energy calculations and the potential of mean force
 Free energy calculations and the potential of mean force IMA Workshop on Classical and Quantum Approaches in Molecular Modeling Mark Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Science
Free energy calculations and the potential of mean force IMA Workshop on Classical and Quantum Approaches in Molecular Modeling Mark Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Science
Geometry Optimisation
 Geometry Optimisation Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, UK http://www.cmt.york.ac.uk/cmd http://www.castep.org Motivation Overview of Talk Background
Geometry Optimisation Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, UK http://www.cmt.york.ac.uk/cmd http://www.castep.org Motivation Overview of Talk Background
Computational Chemistry. An Introduction to Molecular Dynamic Simulations
 Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Biomolecules are dynamic no single structure is a perfect model
 Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
NTChem. ntprep. ntprep
 NTChem NTChem.inp ) bash ) csh RCCS)) ntprep ntprep /opt/aics/ntchem/scripts RCCS) ccpg: /local/apl/pg/ntchem/scripts ccuv: /local/apl/uv/ntchem/scripts Focus: /home1/share/ntchem/ntchem2013.4.0/scripts
NTChem NTChem.inp ) bash ) csh RCCS)) ntprep ntprep /opt/aics/ntchem/scripts RCCS) ccpg: /local/apl/pg/ntchem/scripts ccuv: /local/apl/uv/ntchem/scripts Focus: /home1/share/ntchem/ntchem2013.4.0/scripts
Towards accurate calculations of Zn 2+ binding free energies in zinc finger proteins
 Towards accurate calculations of Zn 2+ binding free energies in zinc finger proteins UNDERGRADUATE HONORS RESEARCH THESIS Presented in Partial Fulfillment of the Requirements for the Bachelor of Science
Towards accurate calculations of Zn 2+ binding free energies in zinc finger proteins UNDERGRADUATE HONORS RESEARCH THESIS Presented in Partial Fulfillment of the Requirements for the Bachelor of Science
Methods of Computer Simulation. Molecular Dynamics and Monte Carlo
 Molecular Dynamics Time is of the essence in biological processes therefore how do we understand time-dependent processes at the molecular level? How do we do this experimentally? How do we do this computationally?
Molecular Dynamics Time is of the essence in biological processes therefore how do we understand time-dependent processes at the molecular level? How do we do this experimentally? How do we do this computationally?
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
Multi-scale approaches in description and design of enzymes
 Multi-scale approaches in description and design of enzymes Anastassia Alexandrova and Manuel Sparta UCLA & CNSI Catalysis: it is all about the barrier The inside-out protocol: Big Aim: development of
Multi-scale approaches in description and design of enzymes Anastassia Alexandrova and Manuel Sparta UCLA & CNSI Catalysis: it is all about the barrier The inside-out protocol: Big Aim: development of
Gromacs Workshop Spring CSC
 Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Semi Empirical Force Fields and Their Limitations. Potential Energy Surface (PES)
") Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model. John Dood Hope College
 Water Model. John Dood Hope College") Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model John Dood Hope College What are MD simulations? Model and predict the structure and dynamics of large macromolecules.
Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model John Dood Hope College What are MD simulations? Model and predict the structure and dynamics of large macromolecules.
Alchemical free energy calculations in OpenMM
 Alchemical free energy calculations in OpenMM Lee-Ping Wang Stanford Department of Chemistry OpenMM Workshop, Stanford University September 7, 2012 Special thanks to: John Chodera, Morgan Lawrenz Outline
Alchemical free energy calculations in OpenMM Lee-Ping Wang Stanford Department of Chemistry OpenMM Workshop, Stanford University September 7, 2012 Special thanks to: John Chodera, Morgan Lawrenz Outline
Development and Application of Combined Quantum Mechanical and Molecular Mechanical Methods
 University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Student Research Projects, Dissertations, and Theses - Chemistry Department Chemistry, Department of Fall 12-2014 Development
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Student Research Projects, Dissertations, and Theses - Chemistry Department Chemistry, Department of Fall 12-2014 Development
Integrated Tools for Computational Chemical Dynamics
 Integrated Tools for Computational Chemical Dynamics Research Tools Design Consortium Department of Chemistry, UM Chemical & Materials Sciences Div. and Environmental Molecular Sci. Lab Participants U
Integrated Tools for Computational Chemical Dynamics Research Tools Design Consortium Department of Chemistry, UM Chemical & Materials Sciences Div. and Environmental Molecular Sci. Lab Participants U
Density Functional Theory: from theory to Applications
 Density Functional Theory: from theory to Applications Uni Mainz January 17, 2011 CP-PAW/COSMO Interface Mixed quantum/ classical molecular dynamics (QM/MM) Quantum mechanics is computationally expensive
Density Functional Theory: from theory to Applications Uni Mainz January 17, 2011 CP-PAW/COSMO Interface Mixed quantum/ classical molecular dynamics (QM/MM) Quantum mechanics is computationally expensive
Multi-reference Density Functional Theory. COLUMBUS Workshop Argonne National Laboratory 15 August 2005
 Multi-reference Density Functional Theory COLUMBUS Workshop Argonne National Laboratory 15 August 2005 Capt Eric V. Beck Air Force Institute of Technology Department of Engineering Physics 2950 Hobson
Multi-reference Density Functional Theory COLUMBUS Workshop Argonne National Laboratory 15 August 2005 Capt Eric V. Beck Air Force Institute of Technology Department of Engineering Physics 2950 Hobson
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular mechanics. classical description of molecules. Marcus Elstner and Tomáš Kubař. April 29, 2016
 classical description of molecules April 29, 2016 Chemical bond Conceptual and chemical basis quantum effect solution of the SR numerically expensive (only small molecules can be treated) approximations
classical description of molecules April 29, 2016 Chemical bond Conceptual and chemical basis quantum effect solution of the SR numerically expensive (only small molecules can be treated) approximations
22 Path Optimisation Methods
 22 Path Optimisation Methods 204 22 Path Optimisation Methods Many interesting chemical and physical processes involve transitions from one state to another. Typical examples are migration paths for defects
22 Path Optimisation Methods 204 22 Path Optimisation Methods Many interesting chemical and physical processes involve transitions from one state to another. Typical examples are migration paths for defects
Molecular Simulation II. Classical Mechanical Treatment
 Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Biomolecular modeling I
 2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
What is Classical Molecular Dynamics?
 What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
Molecular Mechanics. Yohann Moreau. November 26, 2015
 Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Free energy calculations
 Free energy calculations Berk Hess May 5, 2017 Why do free energy calculations? The free energy G gives the population of states: ( ) P 1 G = exp, G = G 2 G 1 P 2 k B T Since we mostly simulate in the
Free energy calculations Berk Hess May 5, 2017 Why do free energy calculations? The free energy G gives the population of states: ( ) P 1 G = exp, G = G 2 G 1 P 2 k B T Since we mostly simulate in the
Computer simulation methods (2) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
CHEM 498Q / CHEM 630Q: Molecular Modeling of Proteins TUTORIAL #3a: Empirical force fields
 Department of Chemistry and Biochemistry, Concordia University! page 1 of 6 CHEM 498Q / CHEM 630Q: Molecular Modeling of Proteins TUTORIAL #3a: Empirical force fields INTRODUCTION The goal of this tutorial
Department of Chemistry and Biochemistry, Concordia University! page 1 of 6 CHEM 498Q / CHEM 630Q: Molecular Modeling of Proteins TUTORIAL #3a: Empirical force fields INTRODUCTION The goal of this tutorial
Molecular simulation and structure prediction using CHARMM and the MMTSB Tool Set Free Energy Methods
 Molecular simulation and structure prediction using CHARMM and the MMTSB Tool Set Free Energy Methods Charles L. Brooks III MMTSB/CTBP 2006 Summer Workshop CHARMM Simulations The flow of data and information
Molecular simulation and structure prediction using CHARMM and the MMTSB Tool Set Free Energy Methods Charles L. Brooks III MMTSB/CTBP 2006 Summer Workshop CHARMM Simulations The flow of data and information
APBS electrostatics in VMD - Software. APBS! >!Examples! >!Visualization! >! Contents
 Software Search this site Home Announcements An update on mailing lists APBS 1.2.0 released APBS 1.2.1 released APBS 1.3 released New APBS 1.3 Windows Installer PDB2PQR 1.7.1 released PDB2PQR 1.8 released
Software Search this site Home Announcements An update on mailing lists APBS 1.2.0 released APBS 1.2.1 released APBS 1.3 released New APBS 1.3 Windows Installer PDB2PQR 1.7.1 released PDB2PQR 1.8 released
Applications of Molecular Dynamics
 June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
Analysis of the simulation
 Analysis of the simulation Marcus Elstner and Tomáš Kubař January 7, 2014 Thermodynamic properties time averages of thermodynamic quantites correspond to ensemble averages (ergodic theorem) some quantities
Analysis of the simulation Marcus Elstner and Tomáš Kubař January 7, 2014 Thermodynamic properties time averages of thermodynamic quantites correspond to ensemble averages (ergodic theorem) some quantities
Phase Equilibria and Molecular Solutions Jan G. Korvink and Evgenii Rudnyi IMTEK Albert Ludwig University Freiburg, Germany
 Phase Equilibria and Molecular Solutions Jan G. Korvink and Evgenii Rudnyi IMTEK Albert Ludwig University Freiburg, Germany Preliminaries Learning Goals Phase Equilibria Phase diagrams and classical thermodynamics
Phase Equilibria and Molecular Solutions Jan G. Korvink and Evgenii Rudnyi IMTEK Albert Ludwig University Freiburg, Germany Preliminaries Learning Goals Phase Equilibria Phase diagrams and classical thermodynamics
T6.2 Molecular Mechanics
 T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
Jack Smith. Center for Environmental, Geotechnical and Applied Science. Marshall University
 Jack Smith Center for Environmental, Geotechnical and Applied Science Marshall University -- Division of Science and Research WV Higher Education Policy Commission WVU HPC Summer Institute June 20, 2014
Jack Smith Center for Environmental, Geotechnical and Applied Science Marshall University -- Division of Science and Research WV Higher Education Policy Commission WVU HPC Summer Institute June 20, 2014
Tools for QM studies of large systems
 Tools for QM studies of large systems Automated, hessian-free saddle point search & characterization QM/MM implementation for zeolites Shaama Mallikarjun Sharada Advisors: Prof. Alexis T Bell, Prof. Martin
Tools for QM studies of large systems Automated, hessian-free saddle point search & characterization QM/MM implementation for zeolites Shaama Mallikarjun Sharada Advisors: Prof. Alexis T Bell, Prof. Martin
Using Web-Based Computations in Organic Chemistry
 10/30/2017 1 Using Web-Based Computations in Organic Chemistry John Keller UAF Department of Chemistry & Biochemistry The UAF WebMO site Practical aspects of computational chemistry theory and nomenclature
10/30/2017 1 Using Web-Based Computations in Organic Chemistry John Keller UAF Department of Chemistry & Biochemistry The UAF WebMO site Practical aspects of computational chemistry theory and nomenclature
Protein Structure Analysis
 BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
BINF 731 Protein Modeling Methods Protein Structure Analysis Iosif Vaisman Ab initio methods: solution of a protein folding problem search in conformational space Energy-based methods: energy minimization
Free energy calculations with alchemlyb
 Free energy calculations with alchemlyb Oliver Beckstein Arizona State University SPIDAL Teleconference 2019-02-01 Binding free energy measure of how strong a protein P and a ligand X stick together key
Free energy calculations with alchemlyb Oliver Beckstein Arizona State University SPIDAL Teleconference 2019-02-01 Binding free energy measure of how strong a protein P and a ligand X stick together key
2008 Biowerkzeug Ltd.
 2008 Biowerkzeug Ltd. 1 Contents Summary...3 1 Simulation...4 1.1 Setup...4 1.2 Output...4 2 Settings...5 3 Analysis...9 3.1 Setup...9 3.2 Input options...9 3.3 Descriptions...10 Please note that we cannot
2008 Biowerkzeug Ltd. 1 Contents Summary...3 1 Simulation...4 1.1 Setup...4 1.2 Output...4 2 Settings...5 3 Analysis...9 3.1 Setup...9 3.2 Input options...9 3.3 Descriptions...10 Please note that we cannot
Intermolecular Forces in Density Functional Theory
 Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations
 Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations 1 / 30
 Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Prediction of spectroscopic parameters for bio-organic and bio-inorganic intermediates in complex systems
 Prediction of spectroscopic parameters for bio-organic and bio-inorganic intermediates in complex systems Erik Donovan Hedegård Department of Physics, Chemistry and Pharmacy University of Southern Denmark
Prediction of spectroscopic parameters for bio-organic and bio-inorganic intermediates in complex systems Erik Donovan Hedegård Department of Physics, Chemistry and Pharmacy University of Southern Denmark
Reactive Empirical Force Fields
 Reactive Empirical Force Fields Jason Quenneville jasonq@lanl.gov X-1: Solid Mechanics, EOS and Materials Properties Applied Physics Division Los Alamos National Laboratory Timothy C. Germann, Los Alamos
Reactive Empirical Force Fields Jason Quenneville jasonq@lanl.gov X-1: Solid Mechanics, EOS and Materials Properties Applied Physics Division Los Alamos National Laboratory Timothy C. Germann, Los Alamos
Parameterization of a reactive force field using a Monte Carlo algorithm
 Parameterization of a reactive force field using a Monte Carlo algorithm Eldhose Iype (e.iype@tue.nl) November 19, 2015 Where innovation starts Thermochemical energy storage 2/1 MgSO 4.xH 2 O+Q MgSO 4
Parameterization of a reactive force field using a Monte Carlo algorithm Eldhose Iype (e.iype@tue.nl) November 19, 2015 Where innovation starts Thermochemical energy storage 2/1 MgSO 4.xH 2 O+Q MgSO 4
Molecular Mechanics / ReaxFF
 Molecular Dynamics simulations Lecture 09: Molecular Mechanics / ReaxFF Dr. Olli Pakarinen University of Helsinki Fall 2012 Lecture notes based on notes by Dr. Jani Kotakoski, 2010 CONTENTS Molecular mechanics
Molecular Dynamics simulations Lecture 09: Molecular Mechanics / ReaxFF Dr. Olli Pakarinen University of Helsinki Fall 2012 Lecture notes based on notes by Dr. Jani Kotakoski, 2010 CONTENTS Molecular mechanics
Methods and Code Integration. Ryan M. Olson University of Minnesota
 Methods and Code Integration Ryan M. Olson University of Minnesota Overview Methods Electrostatically Embedded Many-Body Method Adaptive Partitioning Configurational-Biased Grand Canonical Monte Carlo
Methods and Code Integration Ryan M. Olson University of Minnesota Overview Methods Electrostatically Embedded Many-Body Method Adaptive Partitioning Configurational-Biased Grand Canonical Monte Carlo
Opportunities from Accurate and Efficient Density Functional Theory Calculations for Large Systems
 Seminar CENTRE FOR PREDICTIVE MODELLING, WARWICK Opportunities from Accurate and Efficient Density Functional Theory Calculations for Large Systems Luigi Genovese L_Sim CEA Grenoble October 30, 2017 http://bigdft.org
Seminar CENTRE FOR PREDICTIVE MODELLING, WARWICK Opportunities from Accurate and Efficient Density Functional Theory Calculations for Large Systems Luigi Genovese L_Sim CEA Grenoble October 30, 2017 http://bigdft.org
Efficient Parallelization of Molecular Dynamics Simulations on Hybrid CPU/GPU Supercoputers
 Efficient Parallelization of Molecular Dynamics Simulations on Hybrid CPU/GPU Supercoputers Jaewoon Jung (RIKEN, RIKEN AICS) Yuji Sugita (RIKEN, RIKEN AICS, RIKEN QBiC, RIKEN ithes) Molecular Dynamics
Efficient Parallelization of Molecular Dynamics Simulations on Hybrid CPU/GPU Supercoputers Jaewoon Jung (RIKEN, RIKEN AICS) Yuji Sugita (RIKEN, RIKEN AICS, RIKEN QBiC, RIKEN ithes) Molecular Dynamics
Electronic excitations in conjugated. Many-Body Green's Functions. Behnaz Bagheri Varnousfaderani. Behnaz Bagheri Varnousfaderani
 Technische Universiteit Eindhoven University of Technology Electronic excitations in conjugated polymers Electronic from excitations QM/MM simulations in conjugated pol based from on Many-Body QM/MM simulations
Technische Universiteit Eindhoven University of Technology Electronic excitations in conjugated polymers Electronic from excitations QM/MM simulations in conjugated pol based from on Many-Body QM/MM simulations
Design of Efficient Catalysts with Double Transition Metal. Atoms on C 2 N Layer
 Supporting Information Design of Efficient Catalysts with Double Transition Metal Atoms on C 2 N Layer Xiyu Li, 1, Wenhui Zhong, 2, Peng Cui, 1 Jun Li, 1 Jun Jiang 1, * 1 Hefei National Laboratory for
Supporting Information Design of Efficient Catalysts with Double Transition Metal Atoms on C 2 N Layer Xiyu Li, 1, Wenhui Zhong, 2, Peng Cui, 1 Jun Li, 1 Jun Jiang 1, * 1 Hefei National Laboratory for
Ab initio molecular dynamics. Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy. Bangalore, 04 September 2014
 Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Oxygen Binding in Hemocyanin
 Supporting Information for Quantum Mechanics/Molecular Mechanics Study of Oxygen Binding in Hemocyanin Toru Saito and Walter Thiel* Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470
Supporting Information for Quantum Mechanics/Molecular Mechanics Study of Oxygen Binding in Hemocyanin Toru Saito and Walter Thiel* Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470
Ab initio Berechungen für Datenbanken
 J Ab initio Berechungen für Datenbanken Jörg Neugebauer University of Paderborn Lehrstuhl Computational Materials Science Computational Materials Science Group CMS Group Scaling Problem in Modeling length
J Ab initio Berechungen für Datenbanken Jörg Neugebauer University of Paderborn Lehrstuhl Computational Materials Science Computational Materials Science Group CMS Group Scaling Problem in Modeling length