Fragmentation methods
|
|
|
- Maude Butler
- 5 years ago
- Views:
Transcription

1 Fragmentation methods
2 Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method would scale linearly with respect to the system size. This is the goal of fragmentation methods. *(where N is a measure of system size such as the number of basis functions)

 methods have become popular in recent years,")


3 Fragmentation Methods Biomolecules contain hundreds or thousands of atoms, making accurate quantum calculations either very difficult or impossible Quantum Mechanics/Molecular Mechanics (QM/MM) methods have become popular in recent years, however, As system size grows the QM region can get unwieldy The energy contribution from the environment becomes too large to obtain reasonable accuracy from molecular mechanics Fragmentation methods offer a unique solution to accurate calculations on large molecules
4 Background & Motivations Fragmentation methods can be divided into two groups, one-step and two-step One-step methods obtain the energy and properties directly from fragment calculations. Methods included are: X-pol, FMO, KEM, MFCC, SFM (SMF) and MTA Two-step methods first compute some total property, for example the density, from fragment calculations. Other properties such as the total energy are then evaluated from the total property obtained in the first step. Examples of two-step methods are: X-Pol-X, MFCC-DM and DC M.S. Gordon, D.G. Fedorov, S.R Pruitt, and L.V. Slipchenko, Fragmentation Methods: A Route to Accurate Calculations on Large Systems, Chem. Rev, 112, 632 (2012)
5 Division of methods can be further refined into one-body, many-body and conglomerate methods. one-body methods do not perform any higher order QM many-body calculations many-body methods include pairs, triples etc QM corrections to the one-body methods conglomerate methods use other means to obtain many-body corrections (SFM/SMF)
6 The Fragment Molecular Orbital (FMO) Method: Kitaura et al.: CPL, 313, 701 (1999) Exchange and self-consistency are local in most molecules Treat non-local parts using just the Coulomb operator, thereby ignoring exchange monomer electron density being calculated = monomer Perform the molecular calculations individually in the rigorous Coulomb field of the whole system Improved by explicit many-body corrections for pairs and triples (dimers & trimers) The Coulomb bath allows for fragmentation without hydrogen capping Coulomb bath of the full system being taken into account


")

7 The Fragment Molecular Orbital Method Bonds are fractioned electrostatically Electrons are assigned heterolytically FMO fragmentation should be conducted based upon chemical knowledge (not a formal mathematical exercise ) Hydrogen bonding is accounted for by explicit dimer (fragment pair) calculations Dimer & trimer (fragment triple) calculations allow for other quantum effects to be taken into account


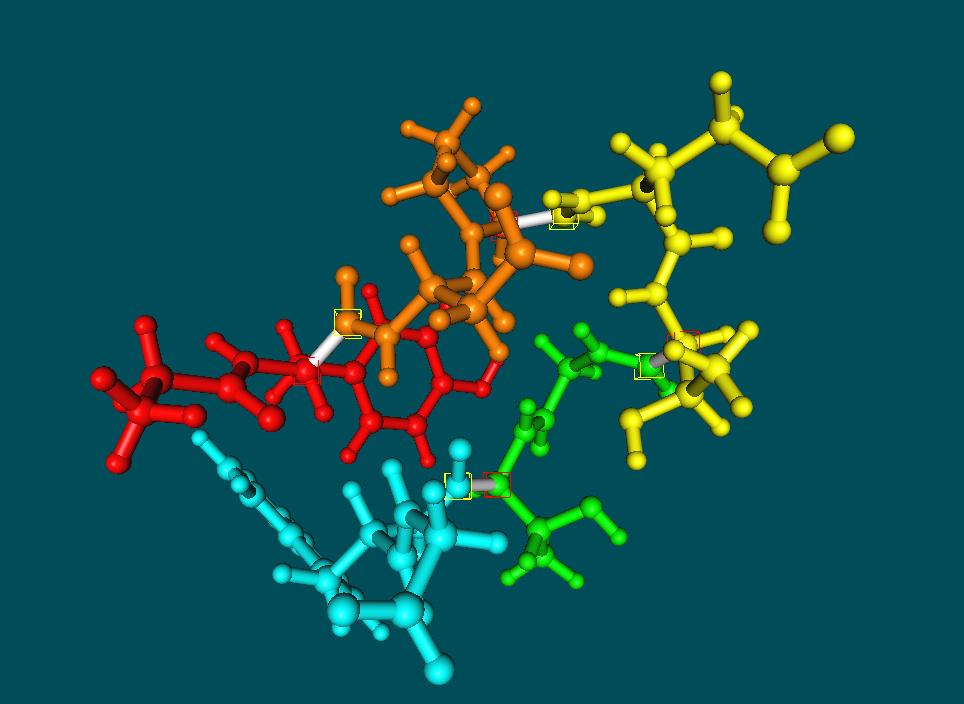
8 The Fragment Molecular Orbital Method: Fragmentation Scheme H H H H H H C C C OH For covalently bonded molecules, we divide the fragment into pieces so as not to destroy bond electron pairs. H In molecular clusters, fragmentation is easier, requiring no covalent bond breaking. We can have one molecule per fragment, two molecules per fragment etc
9 The Fragment Molecular Orbital Method - Basic Methodology The total energy of the system can be written as Where the monomer (I), dimer (IJ) and trimer (IJK) energies are obtained using the SCF method with modified Fock operators
10 The Fragment Molecular Orbital Method 1. Divide molecule into fragments and assign electrons to these fragments 2. Calculate initial electron density distribution of the fragments in the Coulomb bath of the full system 3. Construct the individual fragment Fock operators using the densities calculated in 2 and solve for the fragment energies 4. Determine if the density has converged for all the fragments. If not, go back to step 3 5. Construct Hamiltonians for each dimer (trimer) calculation using the converged monomer densities from steps Calculate total energy and electron density
 electron density being calculated in the presence of the converged ESP")
11 Steps 1-3: Coulomb bath of the full system being taken into account monomer electron density being calculated = monomer Step 4: Steps 1 through 3 are looped until the density of the full system converges to some predetermined threshold Steps 5-6: converged Coulomb bath of the full system from monomer SCF dimer (trimer) electron density being calculated in the presence of the converged ESP each dimer (trimer) calculation is performed once
12 The Fragment Molecular Orbital Method The number of dimer calculations increases as n choose 2 where n is the number of fragments. # of dimers = n! 2!(n 2)! 7 fragments = 21 dimers 8 fragments = 28 dimers 16 fragments = 120 dimers 32 fragments = 496 dimers 64 fragments = 2016 dimers 128 fragments = 8128 dimers The total number of dimer calculations increases rapidly! Two solutions: Approximations and parallelization
% R I,J User defined cut-off value Rcut R I,J R cut R I,J >R cut E FMO2 = E I + ΔE IJ + ΔE IJ I I>J I>J")
13 The Fragment Molecular Orbital Method: Approximations # % r i r ' j % R I,J = min $ ( i I, j J r vdw vdw &% i + r j )% R I,J User defined cut-off value Rcut R I,J R cut R I,J >R cut E FMO2 = E I + ΔE IJ + ΔE IJ I I>J I>J sep
14 The Fragment Molecular Orbital Method: Approximations Parallelization The Generalized Distributed Data Interface (GDDI) GDDI allows for massively parallel calculations on clusters of computers or supercomputers After the molecule is divided into fragments, each fragment is sent to a group which is composed of more than one processor or SMP enclosure Each fragment is then run in parallel in each group This provides two levels of parallelization, greatly speeding up the calculation


15 The Fragment Molecular Orbital Method: Parallelization Group Master Group #1 Group #2 Group #3 CPU CPU CPU CPU CPU CPU CPU CPU CPU CPU CPU CPU
16 The Fragment Molecular Orbital Method FMO has all of the following wavefunction types implemented RHF, ROHF, DFT/TDDFT, MP2, CC, CIS and MCSCF (all of which support FMO3 except MCSCF) FMO also has a multilayer implementation allowing you to specify different levels of electron correlation or basis sets in different layers FMO is also interfaced with PCM and EFP for solvent effects RUNTYPs available are ENERGY, GRADIENT, OPTIMIZE and GLOBOP
17 The Fragment Molecular Orbital Method - Examples 32-WATER CLUSTERS
18 The Fragment Molecular Orbital Method - Examples 32-WATER CLUSTERS
 to enter and form")
19 The Fragment Molecular Orbital Method - Examples Heterogeneous Catalysis on MSN Gatekeeper groups selectively allow reactants A (not B ) to enter and form product P


20 1770 atoms Mesoporous Silica Nanoparticle (MSN)
21 Diffusion Through MSN Pore FMO/HF FMO/HF-D
22 Path A FMO/HF FMO/HF-D
23 Path B
24 Path C
25 The Fragment Molecular Orbital Method - Examples Hydrated cisplatin-dna complex. FMO2-MP2 calculations were performed using the MCP model Sodium ions and water molecules were relaxed with the Amber99 force field This calculation included: 3596 atoms ~997 fragments Chemical Physics Letters 427 (2006)
 optimized structure (yellow) MP2 optimization took 105 geometry steps and ~63 hours FMO2-MP2")

26 The Fragment Molecular Orbital Method - Examples: Ionic Liquid FMO2-MP2/6-31G(d) optimized* octamer structure (red) overlaid with ab initio MP2/6-31G(d) optimized structure (yellow) MP2 optimization took 105 geometry steps and ~63 hours FMO2-MP2 optimization took 94 geometry steps and ~5.5 hours *Optimizations performed on 256 CPUs of an IBM Power6 cluster with 32 CPUs per node and 2 GB of RAM per CPU. GDDI was used for FMO2 calculation with one node per GDDI group. Error = kcal/mol RMSD = 0.04 angstroms
 Wall time (min) 1103 1533 566")

27 The Fragment Molecular Orbital Method - Examples 2VDA protein from the PDB database Cell 131 (2007) # Groups # CPUs* CPU time * (min) Wall time (min) * 4.7 GHz IBM Power6 processors with 2GB of RAM per CPU and 32 CPUs per node FMO2-MP2 single point energy calculation 6-31G(d) basis set 855 fragments (one residue per fragment) atoms
 energy calculation - 131,432 basis functions - 16,384 nodes (262,144 cores) - ~10 hours - ~72% of")
28 The Fragment Molecular Orbital Method - Examples amino acid residues water molecules - one residue or water per fragment - FMO2-MP2/6-311G(d,p) energy calculation - 131,432 basis functions - 16,384 nodes (262,144 cores) - ~10 hours - ~72% of Blue Waters
29
30 FMO2-MP2/aug-cc-pVDZ gradients: BG/P: ANL (Graham Fletcher)! G.D. Fletcher, D.G. Fedorov, S.R. Pruitt, T.L. Windus and M.S. Gordon, J. Chem. Theor. Comp., 8, 75 (2012).
31
32 The Fragment Molecular Orbital Method - Memory Requirements CCSD(T)/aug-cc-pVQZ single point energy calculation on a cluster of six water molecules 64 cores 8 cores/node 64 cores 1 core/node CCSD(T) 80 GB RAM per node 32 GB RAM per node FMO3-CC 12 GB RAM per node 4 GB RAM per node *One water molecule per FMO fragment CCSD(T)/aug-cc-pVQZ single point energy calculation on a cluster of six argon atoms 64 cores 8 cores/node 64 cores 1 core/node CCSD(T) 36 GB RAM per node 19 GB RAM per node FMO2-CC 9 GB RAM per node 4 GB RAM per node *Two argon atoms per FMO fragment (total of three fragments)
33
34 The Effective Fragment Potential Method Originally developed to describe aqueous solvent effects on molecules of biological interest. The EFP method is an ab initio based model potential for the evaluation of intermolecular forces. Each molecule is represented as a fragment of fixed geometry with a set of parameters derived from ab initio calculations. In the original implementation (EFP1 - only water) the interaction energy between fragments consists of three terms: Coulomb, polarization and exchange repulsion. E EFP1 = E Coul + E pol + E exrep E EFP2 = E Coul + E pol + E disp + E exrep + E ct
35 The Effective Fragment Molecular Orbital Method The EFMO method was developed to integrate the FMO and EFP methods in an effort to provide a generally applicable, accurate and efficient approach to large molecular systems. (FMO fragmentation) + (EFP interactions) + (other modifications) = EFMO R I,J R cut R I,J >R cut E FMO2 = E I + ΔE IJ + ΔE IJ I I>J I>J sep E EFP2 = E Coul + E pol + E disp + E exrep + E ct R I,J R cut E EFMO = E 0 I + (ΔE 0 IJ E pol Coul IJ ) + E IJ I I > J R I,J > R cut I > J pol + E tot
36 The Effective Fragment Molecular Orbital Method R I,J R cut E EFMO = E 0 I + (ΔE 0 IJ E pol Coul IJ ) + E IJ I I > J R I,J > R cut I > J pol + E tot The use of isolated fragment energies eliminates the need to calculate the ESP used in standard FMO method calculations. The many-body interaction energy formerly computed using the ESP is replaced by the total EFP polarization energy. However, the lack of dispersion limits the EFMO method to HF and DFT calculations. Additional computational savings could be achieved by reducing the value of Rcut
37 The Effective Fragment Molecular Orbital Method: Fully Integrated The original formulation of the total EFMO energy was fully integrated to include all five components of the EFP energy. R I,J R cut E EFMO = E 0 I + (ΔE 0 IJ E pol Coul IJ ) + E IJ I I > J E EFP2 = E Coul + E pol + E disp + E exrep + E ct R I,J R cut R I,J > R cut I > J pol + E tot E EFMO = E 0 I + (ΔE 0 IJ E pol IJ ) + (E Coul disp IJ + E IJ I I > J R I,J > R cut I > J + E exrep IJ + E ct pol IJ ) + E tot By including all intermolecular interactions, the user defined cut-off value Rcut can be reduced to neglect additional QM dimers. The reduction in QM dimers lowers the computational requirements of FIEFMO calculations relative to standard FMO method calculations.
38 The Effective Fragment Molecular Orbital Method Average total number of separated and QM dimers 8 waters 16 waters 32 waters 64 waters Rcut separated QM separated QM separated QM separated QM
39 The Effective Fragment Molecular Orbital Method G(d,p) Average Signed Errors (kcal/mol) G(3df,2p) Rcut FMO2 FIEFMO FMO2 FIEFMO 8 water molecules water molecules Error = n i=1 (E i X E i MP2 ) n
40 The Effective Fragment Molecular Orbital Method Binding Energy per Water Molecule (kcal/mol) G(d, d,p) G(3df,2p) Rcut FMO2 FIEFMO MP2 FMO2 FIEFMO MP2 8 water molecules water molecules




41 The Effective Fragment Molecular Orbital Method Energy contributions of each intermolecular interaction for all dimer interactions in a cluster of 8 water molecules and 2 benzene molecules. 1 Interaction Energy (kcal/mol)
42 The Effective Fragment Molecular Orbital Method Timings performed on 10 nodes containing six 2.67 GHz Xeon X5650 cores per node with 24 GB of RAM MP2/ G(3df,2p) FIEFMO FMO2 FMO3 MP2 Rcut wall time error wall time error wall time error wall time 8 Water Molecules + 2 Benzene Molecules Water Molecules + 8 Methanol Molecules Times are in seconds, and errors are in kcal/mol
43 The Fragment Molecular Orbital Method Important GAMESS modules $FMO $GDDI: Specifies parallel run $FMOPRP: Sets up convergers, properties $FMOXYZ: Similar to $DATA $FMOBND: Bond detachment description $OPTFMO: Geometry optimization driver $FMOHYB ($FMOLMO): Fragmentation
44 The Fragment Molecular Orbital Method Example Input
45
46
47
48
49
Geometry Optimizations of Open-Shell Systems with the Fragment Molecular Orbital Method
 Chemistry Publications Chemistry 4-2012 Geometry Optimizations of Open-Shell Systems with the Fragment Molecular Orbital Method Spencer Pruitt Iowa State University Dmitri G. Fedorov National Institute
Chemistry Publications Chemistry 4-2012 Geometry Optimizations of Open-Shell Systems with the Fragment Molecular Orbital Method Spencer Pruitt Iowa State University Dmitri G. Fedorov National Institute
METHODS FOR TREATING SOLVENT EFFECTS AND INTERMOLECULAR FORCES. Mark S. Gordon Iowa State University Ames Laboratory
 METHODS FOR TREATING SOLVENT EFFECTS AND INTERMOLECULAR FORCES Mark S. Gordon Iowa State University Ames Laboratory OUTLINE Solvation Methods Explicit vs. implicit methods Explicit Methods TIP3P, TIP4P
METHODS FOR TREATING SOLVENT EFFECTS AND INTERMOLECULAR FORCES Mark S. Gordon Iowa State University Ames Laboratory OUTLINE Solvation Methods Explicit vs. implicit methods Explicit Methods TIP3P, TIP4P
Lec20 Fri 3mar17
 564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs
 Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs Founded in 1993, Q-Chem strives to bring its customers state-ofthe-art methods and algorithms for performing quantum chemistry calculations. Cutting-edge
Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs Founded in 1993, Q-Chem strives to bring its customers state-ofthe-art methods and algorithms for performing quantum chemistry calculations. Cutting-edge
AN INTRODUCTION TO QUANTUM CHEMISTRY. Mark S. Gordon Iowa State University
 AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
Basic introduction of NWChem software
 Basic introduction of NWChem software Background NWChem is part of the Molecular Science Software Suite Designed and developed to be a highly efficient and portable Massively Parallel computational chemistry
Basic introduction of NWChem software Background NWChem is part of the Molecular Science Software Suite Designed and developed to be a highly efficient and portable Massively Parallel computational chemistry
Computational chemistry with GAMESS: a very brief overview with examples
 Computational chemistry with GAMESS: a very brief overview with examples PHY-6120 Molecular Physics (Spring 2015), UConn Phys. Dept. Feb 17 th 2015 H = ħ2 2μ i Intro: V(R) for diatomic molecules + k Z
Computational chemistry with GAMESS: a very brief overview with examples PHY-6120 Molecular Physics (Spring 2015), UConn Phys. Dept. Feb 17 th 2015 H = ħ2 2μ i Intro: V(R) for diatomic molecules + k Z
Chemistry 4560/5560 Molecular Modeling Fall 2014
 Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Computational Modeling of Protein-Ligand Interactions
 Computational Modeling of Protein-Ligand Interactions Steven R. Gwaltney Department of Chemistry Mississippi State University Mississippi State, MS 39762 Auguste Comte, 1830 Every attempt to refer chemical
Computational Modeling of Protein-Ligand Interactions Steven R. Gwaltney Department of Chemistry Mississippi State University Mississippi State, MS 39762 Auguste Comte, 1830 Every attempt to refer chemical
----- Ver October 24, 2014 Bug about reading MOPAC2012 Ver.14 calculations of 1 atom and 2 atoms molecule was fixed.
 ***** Facio's Release History ***** ----- Ver.18.8.2 ----- October 24, 2014 Bug about reading MOPAC2012 Ver.14 calculations of 1 atom and 2 atoms molecule was fixed. ----- Ver.18.8.1 ----- August 14, 2014
***** Facio's Release History ***** ----- Ver.18.8.2 ----- October 24, 2014 Bug about reading MOPAC2012 Ver.14 calculations of 1 atom and 2 atoms molecule was fixed. ----- Ver.18.8.1 ----- August 14, 2014
Computational Chemistry. An Introduction to Molecular Dynamic Simulations
 Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational and spectroscopic investigation of 7-azaindole: Solvation and intermolecular interactions
 Computational and spectroscopic investigation of 7-azaindole: Solvation and intermolecular interactions Michael Kamrath, Krista Cruse, Nathan Erickson, Molly Beernink Abstract We report results of an experimental
Computational and spectroscopic investigation of 7-azaindole: Solvation and intermolecular interactions Michael Kamrath, Krista Cruse, Nathan Erickson, Molly Beernink Abstract We report results of an experimental
Electron Correlation Methods
 Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Beyond the Hartree-Fock Approximation: Configuration Interaction
 Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Beyond the Hartree-Fock Approximation: Configuration Interaction The Hartree-Fock (HF) method uses a single determinant (single electronic configuration) description of the electronic wavefunction. For
Electron Correlation
 Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
Non-covalent force fields computed ab initio
 Non-covalent force fields computed ab initio Supermolecule calculations Symmetry-adapted perturbation theory (SAPT) Supermolecule calculations Requirements: E = E AB E A E B. Include electron correlation,
Non-covalent force fields computed ab initio Supermolecule calculations Symmetry-adapted perturbation theory (SAPT) Supermolecule calculations Requirements: E = E AB E A E B. Include electron correlation,
Benzene Dimer: dispersion forces and electronic correlation
 Benzene Dimer: dispersion forces and electronic correlation Introduction The Benzene dimer is an ideal example of a system bound by π-π interaction, which is in several cases present in many biologically
Benzene Dimer: dispersion forces and electronic correlation Introduction The Benzene dimer is an ideal example of a system bound by π-π interaction, which is in several cases present in many biologically
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
PLEASE SCROLL DOWN FOR ARTICLE
 This article was downloaded by: [Purdue University] On: 30 March 009 Access details: Access Details: [subscription number 907055043] Publisher Taylor & Francis Informa Ltd Registered in England and Wales
This article was downloaded by: [Purdue University] On: 30 March 009 Access details: Access Details: [subscription number 907055043] Publisher Taylor & Francis Informa Ltd Registered in England and Wales
Energy Gradients in Combined Fragment Molecular Orbital and Polarizable Continuum Model (FMO/PCM) Calculation
 Calculation") University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Hui Li Publications Published Research - Department of Chemistry 2009 Energy Gradients in Combined Fragment Molecular Orbital
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Hui Li Publications Published Research - Department of Chemistry 2009 Energy Gradients in Combined Fragment Molecular Orbital
Q-Chem Workshop. Doubletree Hotel 2085 S. Harbor Boulevard Anaheim, CA March 26, Schedule
 Q-Chem Workshop Doubletree Hotel 2085 S. Harbor Boulevard Anaheim, CA 92802 March 26, 2011 1 8:30 Schedule Welcome remarks, Prof. Peter Gill, Australian National Univ and President of Q-Chem 8:45-9:15
Q-Chem Workshop Doubletree Hotel 2085 S. Harbor Boulevard Anaheim, CA 92802 March 26, 2011 1 8:30 Schedule Welcome remarks, Prof. Peter Gill, Australian National Univ and President of Q-Chem 8:45-9:15
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
NWChem: Coupled Cluster Method (Tensor Contraction Engine)
") NWChem: Coupled Cluster Method (Tensor Contraction Engine) Why CC is important?! Correlation effects are important!! CC is size-extensive theory: can be used to describe dissociation processes.! Higher-order
NWChem: Coupled Cluster Method (Tensor Contraction Engine) Why CC is important?! Correlation effects are important!! CC is size-extensive theory: can be used to describe dissociation processes.! Higher-order
Multiscale Materials Modeling
 Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Electronic structure theory: Fundamentals to frontiers. VI. Analysis and more.
 Electronic structure theory: Fundamentals to frontiers. VI. Analysis and more. MARTIN HEAD-GORDON Department of Chemistry, University of California, Berkeley, and, Chemical Sciences Division, Lawrence
Electronic structure theory: Fundamentals to frontiers. VI. Analysis and more. MARTIN HEAD-GORDON Department of Chemistry, University of California, Berkeley, and, Chemical Sciences Division, Lawrence
Alanine: Then There Was Water
 Chemistry Publications Chemistry 6-2009 Alanine: Then There Was Water Jonathan M. Mullin Iowa State University Mark S. Gordon Iowa State University, mgordon@iastate.edu Follow this and additional works
Chemistry Publications Chemistry 6-2009 Alanine: Then There Was Water Jonathan M. Mullin Iowa State University Mark S. Gordon Iowa State University, mgordon@iastate.edu Follow this and additional works
Chemistry 334 Part 2: Computational Quantum Chemistry
 Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
One-sided Communication Implementation in FMO Method
 One-sided Communication mplementation in FMO Method J. Maki, Y. nadomi, T. Takami, R. Susukita, H. Honda, J. Ooba, T. obayashi, R. Nogita,. noue and M. Aoyagi Computing and Communications Center, yushu
One-sided Communication mplementation in FMO Method J. Maki, Y. nadomi, T. Takami, R. Susukita, H. Honda, J. Ooba, T. obayashi, R. Nogita,. noue and M. Aoyagi Computing and Communications Center, yushu
Electric properties of molecules
 Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Feet on the potential energy surface, head in the π clouds
 Graduate Theses and Dissertations Iowa State University Capstones, Theses and Dissertations 2011 Feet on the potential energy surface, head in the π clouds Quentin Anthony Smith Iowa State University Follow
Graduate Theses and Dissertations Iowa State University Capstones, Theses and Dissertations 2011 Feet on the potential energy surface, head in the π clouds Quentin Anthony Smith Iowa State University Follow
The Atmospheric Significance of Water. Clusters and Ozone-water Complexes.
 The Atmospheric Significance of Water Clusters and Ozone-water Complexes. Josep M. Anglada, a,* Gerald J. Hoffman, b Lyudmila V. Slipchenko, c Marilia M.Costa, d Manuel F. Ruiz-López, d and Joseph S. Francisco.
The Atmospheric Significance of Water Clusters and Ozone-water Complexes. Josep M. Anglada, a,* Gerald J. Hoffman, b Lyudmila V. Slipchenko, c Marilia M.Costa, d Manuel F. Ruiz-López, d and Joseph S. Francisco.
Q-Chem 4.0: Expanding the Frontiers. Jing Kong Q-Chem Inc. Pittsburgh, PA
 Q-Chem 4.0: Expanding the Frontiers Jing Kong Q-Chem Inc. Pittsburgh, PA Q-Chem: Profile Q-Chem is a high performance quantum chemistry program; Contributed by best quantum chemists from 40 universities
Q-Chem 4.0: Expanding the Frontiers Jing Kong Q-Chem Inc. Pittsburgh, PA Q-Chem: Profile Q-Chem is a high performance quantum chemistry program; Contributed by best quantum chemists from 40 universities
Using Web-Based Computations in Organic Chemistry
 10/30/2017 1 Using Web-Based Computations in Organic Chemistry John Keller UAF Department of Chemistry & Biochemistry The UAF WebMO site Practical aspects of computational chemistry theory and nomenclature
10/30/2017 1 Using Web-Based Computations in Organic Chemistry John Keller UAF Department of Chemistry & Biochemistry The UAF WebMO site Practical aspects of computational chemistry theory and nomenclature
Jack Smith. Center for Environmental, Geotechnical and Applied Science. Marshall University
 Jack Smith Center for Environmental, Geotechnical and Applied Science Marshall University -- Division of Science and Research WV Higher Education Policy Commission WVU HPC Summer Institute June 20, 2014
Jack Smith Center for Environmental, Geotechnical and Applied Science Marshall University -- Division of Science and Research WV Higher Education Policy Commission WVU HPC Summer Institute June 20, 2014
Dimer Dissociation of a Photoreceptor Protein from QM/MM and MD Simulations
 Dimer Dissociation of a Photoreceptor Protein from QM/MM and MD Simulations IMA University of Minnesota Minneapolis, MN, July 20, 2015 Haisheng Ren Advisor: Prof. Jiali Gao Department of Chemistry, University
Dimer Dissociation of a Photoreceptor Protein from QM/MM and MD Simulations IMA University of Minnesota Minneapolis, MN, July 20, 2015 Haisheng Ren Advisor: Prof. Jiali Gao Department of Chemistry, University
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D.
 3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
A needed response: Fragment molecular orbital analytic gradients
 Graduate Theses and Dissertations Graduate College 2014 A needed response: Fragment molecular orbital analytic gradients Kurt Ryan Brorsen Iowa State University Follow this and additional works at: http://lib.dr.iastate.edu/etd
Graduate Theses and Dissertations Graduate College 2014 A needed response: Fragment molecular orbital analytic gradients Kurt Ryan Brorsen Iowa State University Follow this and additional works at: http://lib.dr.iastate.edu/etd
Intro to ab initio methods
 Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Performance of Hartree-Fock and Correlated Methods
 Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
Chemistry 460 Fall 2017 Dr. Jean M. Standard December 4, 2017 Performance of Hartree-Fock and Correlated Methods Hartree-Fock Methods Hartree-Fock methods generally yield optimized geomtries and molecular
2~:J~ -ryej- r- 2 Jr. A - f3. sr(djk nv~tor rn~ +~ rvjs (::-CJ) ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.
 ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.") .~, ~ I, sr(djk nv~tor rn~ +~ rvjs (::-CJ) ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.)1e'" 21t-ol Je C'...-------- lj-vi, J? Jr Jr \Ji 2~:J~ -ryej- r- 2 Jr A - f3 c _,~,= ~,.,w._..._.
.~, ~ I, sr(djk nv~tor rn~ +~ rvjs (::-CJ) ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.)1e'" 21t-ol Je C'...-------- lj-vi, J? Jr Jr \Ji 2~:J~ -ryej- r- 2 Jr A - f3 c _,~,= ~,.,w._..._.
Towards gas-phase accuracy for condensed phase problems
 Towards gas-phase accuracy for condensed phase problems Fred Manby Centre for Computational Chemistry, School of Chemistry University of Bristol STC 2006: Quantum Chemistry Methods and Applications Erkner,
Towards gas-phase accuracy for condensed phase problems Fred Manby Centre for Computational Chemistry, School of Chemistry University of Bristol STC 2006: Quantum Chemistry Methods and Applications Erkner,
Water Benzene Interactions: An Effective Fragment Potential and Correlated Quantum Chemistry Study
 Chemistry Publications Chemistry 2009 Water Benzene Interactions: An Effective Fragment Potential and Correlated Quantum Chemistry Study Lyudmila V. Slipchenko Iowa State University Mark S. Gordon Iowa
Chemistry Publications Chemistry 2009 Water Benzene Interactions: An Effective Fragment Potential and Correlated Quantum Chemistry Study Lyudmila V. Slipchenko Iowa State University Mark S. Gordon Iowa
计算物理作业二. Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit.
 计算物理作业二 Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit. In this exercise, basis indicates one of the following basis sets: STO-3G, cc-pvdz, cc-pvtz, cc-pvqz
计算物理作业二 Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit. In this exercise, basis indicates one of the following basis sets: STO-3G, cc-pvdz, cc-pvtz, cc-pvqz
Development and Application of Combined Quantum Mechanical and Molecular Mechanical Methods
 University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Student Research Projects, Dissertations, and Theses - Chemistry Department Chemistry, Department of Fall 12-2014 Development
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Student Research Projects, Dissertations, and Theses - Chemistry Department Chemistry, Department of Fall 12-2014 Development
Advanced in silico drug design
 Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Introduction to Computational Chemistry: Theory
 Introduction to Computational Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC andrew.gilbert@anu.edu.au 3023 Course Lectures Introduction Hartree Fock Theory Basis Sets Lecture 1 1 Introduction
Introduction to Computational Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC andrew.gilbert@anu.edu.au 3023 Course Lectures Introduction Hartree Fock Theory Basis Sets Lecture 1 1 Introduction
Advanced Quantum Chemistry III: Part 6
 Advanced Quantum Chemistry III: Part 6 Norio Yoshida Kyushu University Last updated 16-1-6 2015 Winter Term 1 Quantum Chemistry for Condensed Phase Liquid phase Solid phase Biological systems 2 Divide
Advanced Quantum Chemistry III: Part 6 Norio Yoshida Kyushu University Last updated 16-1-6 2015 Winter Term 1 Quantum Chemistry for Condensed Phase Liquid phase Solid phase Biological systems 2 Divide
QMC dissociation energy of the water dimer: Time step errors and backflow calculations
 QMC dissociation energy of the water dimer: Time step errors and backflow calculations Idoia G. de Gurtubay and Richard J. Needs TCM group. Cavendish Laboratory University of Cambridge Idoia G. de Gurtubay.
QMC dissociation energy of the water dimer: Time step errors and backflow calculations Idoia G. de Gurtubay and Richard J. Needs TCM group. Cavendish Laboratory University of Cambridge Idoia G. de Gurtubay.
Anion-π and π-π cooperative interactions
 Chapter 5 Anion-π and π-π cooperative interactions 5.1 Introduction The design of selective receptors of anionic species is a very active area of research within supramolecular chemistry due to the potential
Chapter 5 Anion-π and π-π cooperative interactions 5.1 Introduction The design of selective receptors of anionic species is a very active area of research within supramolecular chemistry due to the potential
Uptake of OH radical to aqueous aerosol: a computational study
 Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Density Functional Theory
 Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Basic introduction of NWChem software
 Basic introduction of NWChem software Background! NWChem is part of the Molecular Science Software Suite! Designed and developed to be a highly efficient and portable Massively Parallel computational chemistry
Basic introduction of NWChem software Background! NWChem is part of the Molecular Science Software Suite! Designed and developed to be a highly efficient and portable Massively Parallel computational chemistry
G1-3 These methods are based on ab initio molecular orbital calculations. Electron correlation is calculated using MP2 or MP4 and QCI.
 23. High Accuracy Energy Methods 23.1 Gaussian-n Methods The method keywords G1, G2, G2MP2, G3, G3MP2, G3B3, G3MP2B3, G4, and G4MP2 perform high accuracy complex energy computations in Gaussian. Essentially,
23. High Accuracy Energy Methods 23.1 Gaussian-n Methods The method keywords G1, G2, G2MP2, G3, G3MP2, G3B3, G3MP2B3, G4, and G4MP2 perform high accuracy complex energy computations in Gaussian. Essentially,
Intermolecular Forces in Density Functional Theory
 Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
Lecture 4: methods and terminology, part II
 So theory guys have got it made in rooms free of pollution. Instead of problems with the reflux, they have only solutions... In other words, experimentalists will likely die of cancer From working hard,
So theory guys have got it made in rooms free of pollution. Instead of problems with the reflux, they have only solutions... In other words, experimentalists will likely die of cancer From working hard,
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations
 Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Dispersion Correction Derived from First Principles for Density Functional Theory and Hartree Fock Theory
 Chemistry Publications Chemistry 2-2015 Dispersion Correction Derived from First Principles for Density Functional Theory and Hartree Fock Theory Emilie B. Guidez Iowa State University, eguidez@ameslab.gov
Chemistry Publications Chemistry 2-2015 Dispersion Correction Derived from First Principles for Density Functional Theory and Hartree Fock Theory Emilie B. Guidez Iowa State University, eguidez@ameslab.gov
Electronic quantum effect on hydrogen bond geometry in. water dimer
 Electronic quantum effect on hydrogen bond geometry in water dimer Danhui Li 1,2, Zhiyuan Zhang 1,2 Wanrun Jiang 1,2 Depeng Zhang 1,2 Yu Zhu 1,2 and Zhigang Wang 1,2* 1 Institute of Atomic and Molecular
Electronic quantum effect on hydrogen bond geometry in water dimer Danhui Li 1,2, Zhiyuan Zhang 1,2 Wanrun Jiang 1,2 Depeng Zhang 1,2 Yu Zhu 1,2 and Zhigang Wang 1,2* 1 Institute of Atomic and Molecular
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro. Karel Berka, Ph.D.
 4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
Introduction to Computational Quantum Chemistry: Theory
 Introduction to Computational Quantum Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC 3108 Course Lectures 2007 Introduction Hartree Fock Theory Configuration Interaction Lectures 1 Introduction
Introduction to Computational Quantum Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC 3108 Course Lectures 2007 Introduction Hartree Fock Theory Configuration Interaction Lectures 1 Introduction
Advanced Quantum Chemistry III: Part 6
 Advanced Quantum Chemistry III: Part 6 Norio Yoshida Kyushu University Last updated 14-1-15 2013 Winter Term 1 Quantum Chemistry for Condensed Phase Liquid phase Solid phase Biological systems 2 Divide
Advanced Quantum Chemistry III: Part 6 Norio Yoshida Kyushu University Last updated 14-1-15 2013 Winter Term 1 Quantum Chemistry for Condensed Phase Liquid phase Solid phase Biological systems 2 Divide
Why Is Molecular Interaction Important in Our Life
 Why Is Molecular Interaction Important in ur Life QuLiS and Graduate School of Science iroshima University http://www.nabit.hiroshima-u.ac.jp/iwatasue/indexe.htm Suehiro Iwata Sept. 29, 2007 Department
Why Is Molecular Interaction Important in ur Life QuLiS and Graduate School of Science iroshima University http://www.nabit.hiroshima-u.ac.jp/iwatasue/indexe.htm Suehiro Iwata Sept. 29, 2007 Department
QUANTUM CHEMISTRY PROJECT 3: ATOMIC AND MOLECULAR STRUCTURE
 Chemistry 460 Fall 2017 Dr. Jean M. Standard November 1, 2017 QUANTUM CHEMISTRY PROJECT 3: ATOMIC AND MOLECULAR STRUCTURE OUTLINE In this project, you will carry out quantum mechanical calculations of
Chemistry 460 Fall 2017 Dr. Jean M. Standard November 1, 2017 QUANTUM CHEMISTRY PROJECT 3: ATOMIC AND MOLECULAR STRUCTURE OUTLINE In this project, you will carry out quantum mechanical calculations of
Quantum Chemical Calculations by Parallel Computer from Commodity PC Components
 Nonlinear Analysis: Modelling and Control, 2007, Vol. 12, No. 4, 461 468 Quantum Chemical Calculations by Parallel Computer from Commodity PC Components S. Bekešienė 1, S. Sėrikovienė 2 1 Institute of
Nonlinear Analysis: Modelling and Control, 2007, Vol. 12, No. 4, 461 468 Quantum Chemical Calculations by Parallel Computer from Commodity PC Components S. Bekešienė 1, S. Sėrikovienė 2 1 Institute of
Hydrogen and Halogen Bonds are Ruled by the Same Mechanisms S.J.Grabowski
 1 Hydrogen and Halogen Bonds are Ruled by the Same Mechanisms S.J.Grabowski Electronic supplementary information All results presented were obtained at MP2/6-311++G(d,p) level, QTAIM was applied to calculate
1 Hydrogen and Halogen Bonds are Ruled by the Same Mechanisms S.J.Grabowski Electronic supplementary information All results presented were obtained at MP2/6-311++G(d,p) level, QTAIM was applied to calculate
Same idea for polyatomics, keep track of identical atom e.g. NH 3 consider only valence electrons F(2s,2p) H(1s)
 H(1s)") XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
Solution of the Electronic Schrödinger Equation. Using Basis Sets to Solve the Electronic Schrödinger Equation with Electron Correlation
 Solution of the Electronic Schrödinger Equation Using Basis Sets to Solve the Electronic Schrödinger Equation with Electron Correlation Errors in HF Predictions: Binding Energies D e (kcal/mol) HF Expt
Solution of the Electronic Schrödinger Equation Using Basis Sets to Solve the Electronic Schrödinger Equation with Electron Correlation Errors in HF Predictions: Binding Energies D e (kcal/mol) HF Expt
Electronic Supplementary Information (ESI): First Principles Study of Photo-oxidation Degradation Mechanisms in P3HT for Organic Solar Cells
: First Principles Study of Photo-oxidation Degradation Mechanisms in P3HT for Organic Solar Cells") Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is The Royal Society of Chemistry 2014 Electronic Supplementary Information (ESI): First Principles Study of
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is The Royal Society of Chemistry 2014 Electronic Supplementary Information (ESI): First Principles Study of
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Chemistry Publications
 Chemistry Publications Chemistry 3-1998 An Approximate Formula for the Intermolecular Pauli Repulsion Between Closed Shell Molecules. II. Application to the Effective Fragment Potential Method Jan H. Jensen
Chemistry Publications Chemistry 3-1998 An Approximate Formula for the Intermolecular Pauli Repulsion Between Closed Shell Molecules. II. Application to the Effective Fragment Potential Method Jan H. Jensen
Modeling Chemical Reactions in Aqueous Solutions
 University of Arkansas, Fayetteville ScholarWorks@UARK Theses and Dissertations 8-2013 Modeling Chemical Reactions in Aqueous Solutions Osman Uner University of Arkansas, Fayetteville Follow this and additional
University of Arkansas, Fayetteville ScholarWorks@UARK Theses and Dissertations 8-2013 Modeling Chemical Reactions in Aqueous Solutions Osman Uner University of Arkansas, Fayetteville Follow this and additional
CLUSTER-BASED APPROACHES TO SOLVATION. Mark S. Gordon Iowa State University Ames Laboratory
 CLUSTER-BASED APPROACHES TO SOLVATION Mark S. Gordon Iowa State University Ames Laboratory 1 OUTLINE Solvation Methods Explicit vs. implicit methods Explicit Methods TIP3P, TIP4P SPC, SPC/E EFP Method
CLUSTER-BASED APPROACHES TO SOLVATION Mark S. Gordon Iowa State University Ames Laboratory 1 OUTLINE Solvation Methods Explicit vs. implicit methods Explicit Methods TIP3P, TIP4P SPC, SPC/E EFP Method
NWChem: Coupled Cluster Method (Tensor Contraction Engine)
") NWChem: Coupled Cluster Method (ensor Contraction Engine) What we want to solve H Ψ = E Ψ Many Particle Systems Molecular/Atomic Physics, Quantum Chemistry (electronic Schrödinger equations) Solid State
NWChem: Coupled Cluster Method (ensor Contraction Engine) What we want to solve H Ψ = E Ψ Many Particle Systems Molecular/Atomic Physics, Quantum Chemistry (electronic Schrödinger equations) Solid State
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
Bridging Scales Through Wavefunction Analysis
 Bridging Scales Through Wavefunction Analysis Felix Plasser Institute for Theoretical Chemistry, University of Vienna Excited States Bridging Scales Marseille, November 7 10, 2016 F. Plasser Wavefunction
Bridging Scales Through Wavefunction Analysis Felix Plasser Institute for Theoretical Chemistry, University of Vienna Excited States Bridging Scales Marseille, November 7 10, 2016 F. Plasser Wavefunction
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
The Effective Fragment Potential: A General Method for Predicting Intermolecular Interactions
 CHAPTER 10 The Effective Fragment Potential: A General Method for Predicting Intermolecular Interactions Mark S. Gordon *, Lyudmilla Slipchenko *,HuiLi ** and Jan H. Jensen *** Contents 1. Introduction
CHAPTER 10 The Effective Fragment Potential: A General Method for Predicting Intermolecular Interactions Mark S. Gordon *, Lyudmilla Slipchenko *,HuiLi ** and Jan H. Jensen *** Contents 1. Introduction
( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r)
Ψ el ( r;r) = E el ( R)Ψ el ( r;r)") Born Oppenheimer Approximation: Ĥ el ( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r) For a molecule with N electrons and M nuclei: Ĥ el What is E el (R)? s* potential surface Reaction Barrier Unstable intermediate
Born Oppenheimer Approximation: Ĥ el ( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r) For a molecule with N electrons and M nuclei: Ĥ el What is E el (R)? s* potential surface Reaction Barrier Unstable intermediate
Electron Correlation - Methods beyond Hartree-Fock
 Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Gustavus Adolphus College. Lab #5: Computational Chemistry
 CHE 372 Gustavus Adolphus College Lab #5: Computational Chemistry Introduction In this investigation we will apply the techniques of computational chemistry to several of the molecular systems that we
CHE 372 Gustavus Adolphus College Lab #5: Computational Chemistry Introduction In this investigation we will apply the techniques of computational chemistry to several of the molecular systems that we
QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C
 Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
Lec20 Wed 1mar17 update 3mar 10am
 564-17 Lec20 Wed 1mar17 update 3mar 10am Figure 15.2 Shows that increasing the diversity of the basis set lowers The HF-SCF energy considerably, but comes nowhere near the exact experimental energy, regardless
564-17 Lec20 Wed 1mar17 update 3mar 10am Figure 15.2 Shows that increasing the diversity of the basis set lowers The HF-SCF energy considerably, but comes nowhere near the exact experimental energy, regardless
Atom-molecule molecule collisions in spin-polarized polarized alkalis: potential energy surfaces and quantum dynamics
 Atom-molecule molecule collisions in spin-polarized polarized alkalis: potential energy surfaces and quantum dynamics Pavel Soldán, Marko T. Cvitaš and Jeremy M. Hutson University of Durham with Jean-Michel
Atom-molecule molecule collisions in spin-polarized polarized alkalis: potential energy surfaces and quantum dynamics Pavel Soldán, Marko T. Cvitaš and Jeremy M. Hutson University of Durham with Jean-Michel
Molecular mechanics. classical description of molecules. Marcus Elstner and Tomáš Kubař. April 29, 2016
 classical description of molecules April 29, 2016 Chemical bond Conceptual and chemical basis quantum effect solution of the SR numerically expensive (only small molecules can be treated) approximations
classical description of molecules April 29, 2016 Chemical bond Conceptual and chemical basis quantum effect solution of the SR numerically expensive (only small molecules can be treated) approximations
List of Figures Page Figure No. Figure Caption No. Figure 1.1.
 List of Figures Figure No. Figure Caption Page No. Figure 1.1. Cation- interactions and their modulations. 4 Figure 1.2. Three conformations of benzene dimer, S is not a minimum on the potential energy
List of Figures Figure No. Figure Caption Page No. Figure 1.1. Cation- interactions and their modulations. 4 Figure 1.2. Three conformations of benzene dimer, S is not a minimum on the potential energy
Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule. Vesa Hänninen
 Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the electronic energy
Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the electronic energy
IFM Chemistry Computational Chemistry 2010, 7.5 hp LAB2. Computer laboratory exercise 1 (LAB2): Quantum chemical calculations
: Quantum chemical calculations") Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Coupled-Cluster Perturbative Triples for Bond Breaking
 Coupled-Cluster Perturbative Triples for Bond Breaking Andrew G. Taube and Rodney J. Bartlett Quantum Theory Project University of Florida INT CC Meeting Seattle July 8, 2008 Why does chemistry need triples?
Coupled-Cluster Perturbative Triples for Bond Breaking Andrew G. Taube and Rodney J. Bartlett Quantum Theory Project University of Florida INT CC Meeting Seattle July 8, 2008 Why does chemistry need triples?
Atomic and molecular interaction forces in biology
 Atomic and molecular interaction forces in biology 1 Outline Types of interactions relevant to biology Van der Waals interactions H-bond interactions Some properties of water Hydrophobic effect 2 Types
Atomic and molecular interaction forces in biology 1 Outline Types of interactions relevant to biology Van der Waals interactions H-bond interactions Some properties of water Hydrophobic effect 2 Types
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
Biomolecular modeling I
 2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
Fixed-Node quantum Monte Carlo for Chemistry
 Fixed-Node quantum Monte Carlo for Chemistry Michel Caffarel Lab. Physique et Chimie Quantiques, CNRS-IRSAMC, Université de Toulouse e-mail : caffarel@irsamc.ups-tlse.fr. p.1/29 The N-body problem of Chemistry
Fixed-Node quantum Monte Carlo for Chemistry Michel Caffarel Lab. Physique et Chimie Quantiques, CNRS-IRSAMC, Université de Toulouse e-mail : caffarel@irsamc.ups-tlse.fr. p.1/29 The N-body problem of Chemistry
Study of Ozone in Tribhuvan University, Kathmandu, Nepal. Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal
 Study of Ozone in Tribhuvan University, Kathmandu, Nepal Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal 1 Country of the Mt Everest 2 View of the Mt Everest 3 4 5
Study of Ozone in Tribhuvan University, Kathmandu, Nepal Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal 1 Country of the Mt Everest 2 View of the Mt Everest 3 4 5
Interfacing Q-Chem and CHARMM to Perform QM/MM Reaction Path Calculations*
 Interfacing Q-Chem and CHARMM to Perform QM/MM Reaction Path Calculations* H. LEE WOODCOCK III, 1 MILAN HODO S CEK, 2 ANDREW T. B. GILBERT, 3 PETER M. W. GILL, 3 HENRY F. SCHAEFER III, 4 BERNARD R. BROOKS
Interfacing Q-Chem and CHARMM to Perform QM/MM Reaction Path Calculations* H. LEE WOODCOCK III, 1 MILAN HODO S CEK, 2 ANDREW T. B. GILBERT, 3 PETER M. W. GILL, 3 HENRY F. SCHAEFER III, 4 BERNARD R. BROOKS
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
ab initio Electronic Structure Calculations
 ab initio Electronic Structure Calculations New scalability frontiers using the BG/L Supercomputer C. Bekas, A. Curioni and W. Andreoni IBM, Zurich Research Laboratory Rueschlikon 8803, Switzerland ab
ab initio Electronic Structure Calculations New scalability frontiers using the BG/L Supercomputer C. Bekas, A. Curioni and W. Andreoni IBM, Zurich Research Laboratory Rueschlikon 8803, Switzerland ab
Report on Atomistic Modeling of Bonding in Carbon-Based Nanostructures
 Report on Atomistic Modeling of Bonding in Carbon-Based Nanostructures Timothy Stillings Department of Physics, Astronomy and Materials Science Missouri State University Advisor: Ridwan Sakidja Abstract
Report on Atomistic Modeling of Bonding in Carbon-Based Nanostructures Timothy Stillings Department of Physics, Astronomy and Materials Science Missouri State University Advisor: Ridwan Sakidja Abstract