Q-Chem Workshop. Doubletree Hotel 2085 S. Harbor Boulevard Anaheim, CA March 26, Schedule
|
|
|
- Victor Bryan
- 6 years ago
- Views:
Transcription
1 Q-Chem Workshop Doubletree Hotel 2085 S. Harbor Boulevard Anaheim, CA March 26, :30 Schedule Welcome remarks, Prof. Peter Gill, Australian National Univ and President of Q-Chem 8:45-9:15 Prof. Peter Gill: New ways of thinking about old problems 9:15-9:45 Prof. Anna Krylov, University of Southern California: "Unleashing Q-Chem'sarsenal of correlated and excited-state methods to understand Green Fluorescent Protein photocycle" 9:45-10:15 Prof. Paul Ha-Yeon Cheong, Oregon State University: "Elucidation of the Mechanisms and Stereomechanics in Proline Sulfonamide Organocatalysis" 10:15-10:30 Coffee Break 10:30 11:00 Avogadro and QUI, Dr. Zhengting Gan 11:00-12:00 Tutorial I. DFT optimization, vibration analysis, TS search, Dr. Yihan Shao 12:00 1:00 Lunch 1:00 1:30 Prof. Martin Head-Gordon, University of California at Berkeley: "Electronic structure of molecules with strongly correlated electrons: New methods and example applications" 1:30 2:00 Prof. John Herbert, Ohio State University: "New developments in 'polarizable continuum' implicit solvent models for QM, MM, and QM/MM applications" 2:00 2:30 Dr. Jing Kong, CEO and Chief Scientist of Q-Chem: "Unlimited possibilities: Overview of Q-Chem 4.0 Features 2:30 2:45 Coffee Break 2:45 3:30 Tutorial II, MP2, CC, and excited states, Dr. Yihan Shao 3:30 4:15 Tutorial III, Intermolecular interactions, solvation effect, and QM/MM, Dr. Yihan Shao 2 4:15 4:30 Free discussion and concluding remarks, Dr. Jing Kong 1
2 History Q-Chem was founded in the early 1990s Q-Chem 2.1, 2000 Kong et al, J. Comput. Chem. 2000, 21, 1532 Q-Chem 3.0, 2006 Shao et al, Phys. Chem. Chem. Phys. 2006, 8, 3172 Q-Chem 4.0, this month 3 Features From over 100+ developers High efficiency on desktops, multicores, GPUs SCF (Hartree-Fock, DFT), MP2, CC Ground-state and excited States Single molecule and molecular complexes Gas-phase or condensed phase (solvent or biological environment) A complete list of our features can be found at 4 2
3 Outline Part 0, Software installation Part 1, Hartree-Fockand DFT Part 2, MP2, Coupled-cluster and Excited States Part 3, Intermolecular interaction and QM/MM 5 Software setup In this workshop, we will use three programs Q-Chem QUI, Q-Chem user interface Avogadro, molecular builder Please go to to download these programs 6 3


4 QUI Geometry can be inserted by hand or by Paste XYZ from Avogadro Calculation setup Preview of Q-Chem input file 7 QUI preferences 8 4

5 Builder Manipulate Select Avogadro Measurement Navigate (Left: rotate; Middle: zoom; Right: translate) 9 Part 1 Hartree-Fock and DFT Single point energy, geometry optimization Vibrational spectroscopy, NMR Transition state search, internal reaction path 10 5

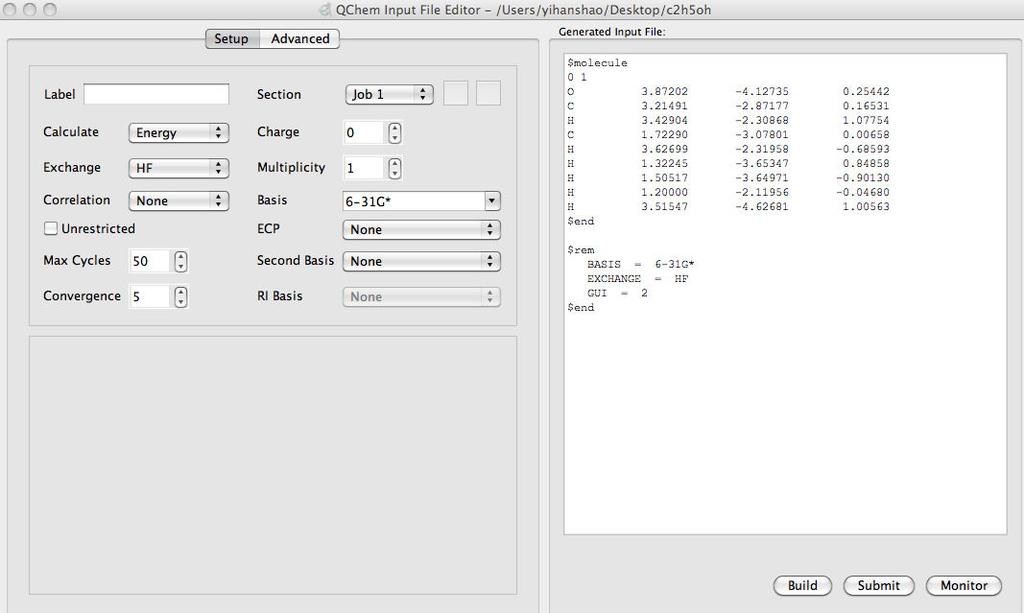
6 Build an ethanol molecule 11 Hartree-Fock//6-31G* calculation 12 6
7 SCF energy Final SCF energy 13 Mulliken charges 14 7


8 Natural orbital analysis 15 Natural orbital analysis 16 8




9 MO and density plots Make cube files that can be read by Avogardo HOMO and LUMO Nx, xmin, xmax Ny, ymin, ymax Nz, zmin, zmax Coordinates in Angstroms Ground-state density Number of MO, density, Transition density, Attachment/density plots 17 MO and density plots Run Q-Chem with qchem save input output scr $QCSCRATCH/scr/plots contains mo.13.cube, mo.14.cube and dens.0.cube 18 9


10 Geometry optimization 19 Vibrational analysis 20 10
11 NMR calculation 21 NMR shielding 22 11




12 Transition state search Make that there is one imaginary frequency Follow reaction path down to reactant and product 23 Reaction path 24 12
13 Exercises Exercise 1.1 Optimize the geometry of ethanol (C 2 H 5 OH) and its protonatedspecies (C 2 H 5 OH 2+ ) with B3LYP/6-31+G*. Compute Mullikencharges and natural atomic charges to see how the extra proton affects the charge distribution. Exercise 1.2 Optimize the geometry of formaldehyde (H 2 CO) run a frequency calculation at the optimized geometry use Avogadro to visualize its vibrational modes. 25 Part 2 MP2 methods Coupled-cluster methods Excited State Methods 26 13

14 MP2 27 MP

15 RIMP2 29 RIMP2 Compare this to MP2 energy:

16 Dual-basis RIMP2 31 Dual-basis RIMP2 Method SCF Energy Correlation Energy Total Energy SCF Time MP RIMP db-rimp Total Time 32 16

17 CCSD 33 CCSD 34 17

18 TDDFT 35 TDDFT calculation 36 18


19 Molecular Orbitals 37 Molecular Orbitals 38 19
20 Exercises Exercise 2.1 Optimize the geometry of cis-butadiene and transbutadiene with ωb97x-d (exchange = omegab97x- D)//6-31G* Compute the energy difference for the two optimized structures with B3LYP, B3LYP-D (add empirical_dispersion= true ) and ωb97x-d MP2//cc-pVTZ, RIMP2//cc-pVTZ, db-rimp2//cc-pvtz 39 Exercise 2.2 Exercises Optimize the CIS//6-31+G* geometry for the first excited state of formaldehyde (H 2 CO) cis_state_derivative = 1 Compare it to the ground-state geometry 40 20

21 Part 3 Intermoleculear Interaction Solvation QM/MM 41 Formamide dimer Benchmark Energy and Geometry DataBase


22 BSSE calculation 43 Energy Decomposition Analysis 44 22
23 EDA 45 BSSE results CCSD(T)//CBS: kcal/mol = kJ/mol B3LYP B3LYP-D 46 23


24 SM8 47 SM8 results 48 24

25 QM/MM 49 Exercises Exercise 3.1 Pick a molecular complex from the S22 dataset ( compute its binding energy with DFT and RIMP2 Exercise 3.2 Optimize the geometry of the complex above with SM8 solvation model Compare the optimized solvated geometry to the original gas-phase geometry 50 25
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations 1 / 30
 Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
Tutorial I: IQ MOL and Basic DFT and MP2 Calculations Q-Chem User Workshop, Denver March 21, 2015 1 / 30 2 / 30 Introduction to IQMOL DFT and MP2 Calculations 3 / 30 IQMOL and Q-CHEM IQMOL is an open-source
Jack Smith. Center for Environmental, Geotechnical and Applied Science. Marshall University
 Jack Smith Center for Environmental, Geotechnical and Applied Science Marshall University -- Division of Science and Research WV Higher Education Policy Commission WVU HPC Summer Institute June 20, 2014
Jack Smith Center for Environmental, Geotechnical and Applied Science Marshall University -- Division of Science and Research WV Higher Education Policy Commission WVU HPC Summer Institute June 20, 2014
Q-Chem Workshop Examples Part 2
 Marek Freindorf Q-Chem Workshop Examples Part 2 Louisville, KY March 2010 CO, MP2 Calculations Open Avogadro using the "Build" button of QUI Page 2 CO, MP2 Calculations Using Avogadro to create coordinates
Marek Freindorf Q-Chem Workshop Examples Part 2 Louisville, KY March 2010 CO, MP2 Calculations Open Avogadro using the "Build" button of QUI Page 2 CO, MP2 Calculations Using Avogadro to create coordinates
Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs
 Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs Founded in 1993, Q-Chem strives to bring its customers state-ofthe-art methods and algorithms for performing quantum chemistry calculations. Cutting-edge
Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs Founded in 1993, Q-Chem strives to bring its customers state-ofthe-art methods and algorithms for performing quantum chemistry calculations. Cutting-edge
计算物理作业二. Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit.
 计算物理作业二 Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit. In this exercise, basis indicates one of the following basis sets: STO-3G, cc-pvdz, cc-pvtz, cc-pvqz
计算物理作业二 Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit. In this exercise, basis indicates one of the following basis sets: STO-3G, cc-pvdz, cc-pvtz, cc-pvqz
Q-Chem 4.0: Expanding the Frontiers. Jing Kong Q-Chem Inc. Pittsburgh, PA
 Q-Chem 4.0: Expanding the Frontiers Jing Kong Q-Chem Inc. Pittsburgh, PA Q-Chem: Profile Q-Chem is a high performance quantum chemistry program; Contributed by best quantum chemists from 40 universities
Q-Chem 4.0: Expanding the Frontiers Jing Kong Q-Chem Inc. Pittsburgh, PA Q-Chem: Profile Q-Chem is a high performance quantum chemistry program; Contributed by best quantum chemists from 40 universities
IFM Chemistry Computational Chemistry 2010, 7.5 hp LAB2. Computer laboratory exercise 1 (LAB2): Quantum chemical calculations
: Quantum chemical calculations") Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Computer laboratory exercise 1 (LAB2): Quantum chemical calculations Introduction: The objective of the second computer laboratory exercise is to get acquainted with a program for performing quantum chemical
Electronic structure theory: Fundamentals to frontiers. VI. Analysis and more.
 Electronic structure theory: Fundamentals to frontiers. VI. Analysis and more. MARTIN HEAD-GORDON Department of Chemistry, University of California, Berkeley, and, Chemical Sciences Division, Lawrence
Electronic structure theory: Fundamentals to frontiers. VI. Analysis and more. MARTIN HEAD-GORDON Department of Chemistry, University of California, Berkeley, and, Chemical Sciences Division, Lawrence
SUPPLEMENTARY INFORMATION
 Calculations predict a stable molecular crystal of N 8 : Barak Hirshberg a, R. Benny Gerber a,b, and Anna I. Krylov c a Institute of Chemistry and The Fritz Haber Center for Molecular Dynamics, The Hebrew
Calculations predict a stable molecular crystal of N 8 : Barak Hirshberg a, R. Benny Gerber a,b, and Anna I. Krylov c a Institute of Chemistry and The Fritz Haber Center for Molecular Dynamics, The Hebrew
Q-Chem Workshop Tasks
 Marek Freindorf Q-Chem Workshop Tasks Washington DC August 2009 Basic Calculations Carbon Dioxide, Example 1A 1. Calculate an optimal geometry of carbon dioxide using the B3LYP/6-31+G* level of theory
Marek Freindorf Q-Chem Workshop Tasks Washington DC August 2009 Basic Calculations Carbon Dioxide, Example 1A 1. Calculate an optimal geometry of carbon dioxide using the B3LYP/6-31+G* level of theory
Density Functional Theory
 Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Supporting Information
 Supporting Information Computational Evidence of Inversion of 1 L a and 1 L b -Derived Excited States in Naphthalene Excimer Formation from ab Initio Multireference Theory with Large Active Space: DMRG-CASPT2
Supporting Information Computational Evidence of Inversion of 1 L a and 1 L b -Derived Excited States in Naphthalene Excimer Formation from ab Initio Multireference Theory with Large Active Space: DMRG-CASPT2
6. Arthur Adamson Postdoctoral Recognition Award, University of Southern California
 Kaushik D. Nanda Address: SSC-401C, University of Southern California, Los Angeles, CA 90089-0482 Email: kaushikdnanda@gmail.com; Phone: 408-406-0690 (Cell) EDUCATION AND DEGREES Ph.D. in Chemistry (2013)
Kaushik D. Nanda Address: SSC-401C, University of Southern California, Los Angeles, CA 90089-0482 Email: kaushikdnanda@gmail.com; Phone: 408-406-0690 (Cell) EDUCATION AND DEGREES Ph.D. in Chemistry (2013)
Chemistry 4560/5560 Molecular Modeling Fall 2014
 Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
List of Figures Page Figure No. Figure Caption No. Figure 1.1.
 List of Figures Figure No. Figure Caption Page No. Figure 1.1. Cation- interactions and their modulations. 4 Figure 1.2. Three conformations of benzene dimer, S is not a minimum on the potential energy
List of Figures Figure No. Figure Caption Page No. Figure 1.1. Cation- interactions and their modulations. 4 Figure 1.2. Three conformations of benzene dimer, S is not a minimum on the potential energy
QUANTUM CHEMISTRY WITH GAUSSIAN : A VERY BRIEF INTRODUCTION (PART 2)
") QUANTUM CHEMISTRY WITH GAUSSIAN : A VERY BRIEF INTRODUCTION (PART 2) TARAS V. POGORELOV AND MIKE HALLOCK SCHOOL OF CHEMICAL SCIENCES, UIUC This tutorial continues introduction to Gaussian [2]. Here we
QUANTUM CHEMISTRY WITH GAUSSIAN : A VERY BRIEF INTRODUCTION (PART 2) TARAS V. POGORELOV AND MIKE HALLOCK SCHOOL OF CHEMICAL SCIENCES, UIUC This tutorial continues introduction to Gaussian [2]. Here we
Computational and spectroscopic investigation of 7-azaindole: Solvation and intermolecular interactions
 Computational and spectroscopic investigation of 7-azaindole: Solvation and intermolecular interactions Michael Kamrath, Krista Cruse, Nathan Erickson, Molly Beernink Abstract We report results of an experimental
Computational and spectroscopic investigation of 7-azaindole: Solvation and intermolecular interactions Michael Kamrath, Krista Cruse, Nathan Erickson, Molly Beernink Abstract We report results of an experimental
Fragmentation methods
 Fragmentation methods Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method
Fragmentation methods Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method
Introduc)on to IQmol: Part I.!!! Shirin Faraji, Ilya Kaliman, and Anna Krylov
on to IQmol: Part I.!!! Shirin Faraji, Ilya Kaliman, and Anna Krylov") Introduc)on to IQmol: Part I!!! Shirin Faraji, Ilya Kaliman, and Anna Krylov! 1 Resources! Written by Dr. Andrew Gilbert Keep yourself up to date with IQmol website: http://iqmol.org! IQmol Youtube channel:
Introduc)on to IQmol: Part I!!! Shirin Faraji, Ilya Kaliman, and Anna Krylov! 1 Resources! Written by Dr. Andrew Gilbert Keep yourself up to date with IQmol website: http://iqmol.org! IQmol Youtube channel:
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Advanced Electronic Structure Theory Density functional theory. Dr Fred Manby
 Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Project 1 Report File: Chem4PB3_project_1_2017-solutions last changed: 02-Feb-2017
 Project 1 Report File: Chem4PB3_project_1_2017-solutions last changed: 02-Feb-2017 1. Formaldehyde comparison of results from different methods Yellow shaded boxes are closest to experimental Method #e-
Project 1 Report File: Chem4PB3_project_1_2017-solutions last changed: 02-Feb-2017 1. Formaldehyde comparison of results from different methods Yellow shaded boxes are closest to experimental Method #e-
QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C
 Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
Using Web-Based Computations in Organic Chemistry
 10/30/2017 1 Using Web-Based Computations in Organic Chemistry John Keller UAF Department of Chemistry & Biochemistry The UAF WebMO site Practical aspects of computational chemistry theory and nomenclature
10/30/2017 1 Using Web-Based Computations in Organic Chemistry John Keller UAF Department of Chemistry & Biochemistry The UAF WebMO site Practical aspects of computational chemistry theory and nomenclature
Literature values: ΔH f, gas = % error Source: ΔH f, solid = % error. For comparison, your experimental value was ΔH f = phase:
 1 Molecular Calculations Lab: Some guideline given at the bottom of page 3. 1. Use the semi-empirical AM1 method to calculate ΔH f for the compound you used in the heat of combustion experiment. Be sure
1 Molecular Calculations Lab: Some guideline given at the bottom of page 3. 1. Use the semi-empirical AM1 method to calculate ΔH f for the compound you used in the heat of combustion experiment. Be sure
one ν im: transition state saddle point
 Hypothetical Potential Energy Surface Ethane conformations Hartree-Fock theory, basis set stationary points all ν s >0: minimum eclipsed one ν im: transition state saddle point multiple ν im: hilltop 1
Hypothetical Potential Energy Surface Ethane conformations Hartree-Fock theory, basis set stationary points all ν s >0: minimum eclipsed one ν im: transition state saddle point multiple ν im: hilltop 1
Investigating excited state dynamics in 7 azaindole. Nathan Erickson, Molly Beernink, and Nathaniel Swenson
 Investigating excited state dynamics in 7 azaindole Nathan Erickson, Molly Beernink, and Nathaniel Swenson 1 Background I 7AI Dimer Previous studies have shown that 7 azaindole (7AI) readily forms H bonded
Investigating excited state dynamics in 7 azaindole Nathan Erickson, Molly Beernink, and Nathaniel Swenson 1 Background I 7AI Dimer Previous studies have shown that 7 azaindole (7AI) readily forms H bonded
Basic introduction of NWChem software
 Basic introduction of NWChem software Background NWChem is part of the Molecular Science Software Suite Designed and developed to be a highly efficient and portable Massively Parallel computational chemistry
Basic introduction of NWChem software Background NWChem is part of the Molecular Science Software Suite Designed and developed to be a highly efficient and portable Massively Parallel computational chemistry
Department of Chemistry. The Intersection of Computational Chemistry and Experiment
 Department of Chemistry The Intersection of Computational Chemistry and Experiment Structure, Vibrational and Electronic Spectra of Organic Molecules Angelo R. Rossi Department of Chemistry The University
Department of Chemistry The Intersection of Computational Chemistry and Experiment Structure, Vibrational and Electronic Spectra of Organic Molecules Angelo R. Rossi Department of Chemistry The University
Towards gas-phase accuracy for condensed phase problems
 Towards gas-phase accuracy for condensed phase problems Fred Manby Centre for Computational Chemistry, School of Chemistry University of Bristol STC 2006: Quantum Chemistry Methods and Applications Erkner,
Towards gas-phase accuracy for condensed phase problems Fred Manby Centre for Computational Chemistry, School of Chemistry University of Bristol STC 2006: Quantum Chemistry Methods and Applications Erkner,
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
METHODS FOR TREATING SOLVENT EFFECTS AND INTERMOLECULAR FORCES. Mark S. Gordon Iowa State University Ames Laboratory
 METHODS FOR TREATING SOLVENT EFFECTS AND INTERMOLECULAR FORCES Mark S. Gordon Iowa State University Ames Laboratory OUTLINE Solvation Methods Explicit vs. implicit methods Explicit Methods TIP3P, TIP4P
METHODS FOR TREATING SOLVENT EFFECTS AND INTERMOLECULAR FORCES Mark S. Gordon Iowa State University Ames Laboratory OUTLINE Solvation Methods Explicit vs. implicit methods Explicit Methods TIP3P, TIP4P
Figure 1: Transition State, Saddle Point, Reaction Pathway
 Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
Computational Chemistry Workshops West Ridge Research Building-UAF Campus 9:00am-4:00pm, Room 009 Electronic Structure - July 19-21, 2016 Molecular Dynamics - July 26-28, 2016 Potential Energy Surfaces
Tools for QM studies of large systems
 Tools for QM studies of large systems Automated, hessian-free saddle point search & characterization QM/MM implementation for zeolites Shaama Mallikarjun Sharada Advisors: Prof. Alexis T Bell, Prof. Martin
Tools for QM studies of large systems Automated, hessian-free saddle point search & characterization QM/MM implementation for zeolites Shaama Mallikarjun Sharada Advisors: Prof. Alexis T Bell, Prof. Martin
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Lec20 Fri 3mar17
 564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
Graphical User Interface Simplified Graphics Requests. The most commonly requested graphics (HOMO,
 Spartan 10 ( Spartan ) has been designed to address the ever increasing role that calculations play in chemistry and related fields. It represents a continued collaboration between Wavefunction, Inc.,
Spartan 10 ( Spartan ) has been designed to address the ever increasing role that calculations play in chemistry and related fields. It represents a continued collaboration between Wavefunction, Inc.,
Supporting Information for Transition State Charge-Transfer Reveals Electrophilic, Ambiphilic, and Nucleophilic Carbon-Hydrogen Bond Activation
 Supporting Information for Transition State Charge-Transfer Reveals Electrophilic, Ambiphilic, and Nucleophilic Carbon-Hydrogen Bond Activation by Daniel H. Ess, Robert J. Nielsen, William A. Goddard,
Supporting Information for Transition State Charge-Transfer Reveals Electrophilic, Ambiphilic, and Nucleophilic Carbon-Hydrogen Bond Activation by Daniel H. Ess, Robert J. Nielsen, William A. Goddard,
Introduction to Computational Chemistry for Experimental Chemists... (Part 2/2)
") 12 th PhD seminar, Garching, October 31 st 2008 Introduction to Computational Chemistry for Experimental Chemists... (Part 2/2) Dr. Markus Drees, TU München Universität Regensburg Universität Augsburg
12 th PhD seminar, Garching, October 31 st 2008 Introduction to Computational Chemistry for Experimental Chemists... (Part 2/2) Dr. Markus Drees, TU München Universität Regensburg Universität Augsburg
High-level Quantum Chemistry Methods and Benchmark Datasets for Molecules
 High-level Quantum Chemistry Methods and Benchmark Datasets for Molecules Markus Schneider Fritz Haber Institute of the MPS, Berlin, Germany École Polytechnique Fédérale de Lausanne, Switzerland دانشگاه
High-level Quantum Chemistry Methods and Benchmark Datasets for Molecules Markus Schneider Fritz Haber Institute of the MPS, Berlin, Germany École Polytechnique Fédérale de Lausanne, Switzerland دانشگاه
Practical Advice for Quantum Chemistry Computations. C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology
 Practical Advice for Quantum Chemistry Computations C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Choice of Basis Set STO-3G is too small 6-31G* or 6-31G** 6 probably
Practical Advice for Quantum Chemistry Computations C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Choice of Basis Set STO-3G is too small 6-31G* or 6-31G** 6 probably
Valence electronic structure of isopropyl iodide investigated by electron momentum spectroscopy. --- Influence of intramolecular interactions
 Valence electronic structure of isopropyl iodide investigated by electron momentum spectroscopy --- Influence of intramolecular interactions Minfu Zhao, Xu Shan, Shanshan Niu, Yaguo Tang, Zhaohui Liu,
Valence electronic structure of isopropyl iodide investigated by electron momentum spectroscopy --- Influence of intramolecular interactions Minfu Zhao, Xu Shan, Shanshan Niu, Yaguo Tang, Zhaohui Liu,
Study of Ozone in Tribhuvan University, Kathmandu, Nepal. Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal
 Study of Ozone in Tribhuvan University, Kathmandu, Nepal Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal 1 Country of the Mt Everest 2 View of the Mt Everest 3 4 5
Study of Ozone in Tribhuvan University, Kathmandu, Nepal Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal 1 Country of the Mt Everest 2 View of the Mt Everest 3 4 5
Computational and Spectroscopic Investigation of Solution Phase Excited State Dynamics in 7 azaindole
 Computational and Spectroscopic Investigation of Solution Phase Excited State Dynamics in 7 azaindole Nathan Erickson, Molly Beernink, and Nathaniel Swenson Midwest Undergraduate Computational Chemistry
Computational and Spectroscopic Investigation of Solution Phase Excited State Dynamics in 7 azaindole Nathan Erickson, Molly Beernink, and Nathaniel Swenson Midwest Undergraduate Computational Chemistry
17. Computational Chemistry Research Unit
 17. Computational Chemistry Research Unit 17.1. Unit members Kimihiko Hirao (Unit Leader) Jong-Won Song (Research Scientist) Rahul Kar (Postdoctoral Researcher) Takao Tsuneda (Senior Visiting Scientist)
17. Computational Chemistry Research Unit 17.1. Unit members Kimihiko Hirao (Unit Leader) Jong-Won Song (Research Scientist) Rahul Kar (Postdoctoral Researcher) Takao Tsuneda (Senior Visiting Scientist)
Role of Charge Transfer in the Structure and Dynamics of the Hydrated Proton
 Article Role of Charge Transfer in the Structure and Dynamics of the Hydrated Proton Jessica M. J. Swanson, and Jack Simons Subscriber access provided by UNIV OF UTAH J. Phys. Chem. B, 2009, 113 (15),
Article Role of Charge Transfer in the Structure and Dynamics of the Hydrated Proton Jessica M. J. Swanson, and Jack Simons Subscriber access provided by UNIV OF UTAH J. Phys. Chem. B, 2009, 113 (15),
Benzene Dimer: dispersion forces and electronic correlation
 Benzene Dimer: dispersion forces and electronic correlation Introduction The Benzene dimer is an ideal example of a system bound by π-π interaction, which is in several cases present in many biologically
Benzene Dimer: dispersion forces and electronic correlation Introduction The Benzene dimer is an ideal example of a system bound by π-π interaction, which is in several cases present in many biologically
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
Introduction to Computational Chemistry Exercise 2
 Introduction to Computational Chemistry Exercise 2 Intermolecular interactions and vibrational motion Lecturer: Antti Lignell Name Introduction In this computer exercise, we model intermolecular interactions
Introduction to Computational Chemistry Exercise 2 Intermolecular interactions and vibrational motion Lecturer: Antti Lignell Name Introduction In this computer exercise, we model intermolecular interactions
Non-covalent force fields computed ab initio
 Non-covalent force fields computed ab initio Supermolecule calculations Symmetry-adapted perturbation theory (SAPT) Supermolecule calculations Requirements: E = E AB E A E B. Include electron correlation,
Non-covalent force fields computed ab initio Supermolecule calculations Symmetry-adapted perturbation theory (SAPT) Supermolecule calculations Requirements: E = E AB E A E B. Include electron correlation,
Molecular Modelling for Medicinal Chemistry (F13MMM) Room A36
 Room A36") Molecular Modelling for Medicinal Chemistry (F13MMM) jonathan.hirst@nottingham.ac.uk Room A36 http://comp.chem.nottingham.ac.uk Assisted reading Molecular Modelling: Principles and Applications. Andrew
Molecular Modelling for Medicinal Chemistry (F13MMM) jonathan.hirst@nottingham.ac.uk Room A36 http://comp.chem.nottingham.ac.uk Assisted reading Molecular Modelling: Principles and Applications. Andrew
Computational Chemistry Using the University of Alaska WebMO Site
 2/7/2017 1 Computational Chemistry Using the University of Alaska WebMO Site John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks Intro and operation of WebMO and MOPAC Basic
2/7/2017 1 Computational Chemistry Using the University of Alaska WebMO Site John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks Intro and operation of WebMO and MOPAC Basic
Spectroscopy of the Cyano Radical in an Aqueous Environment
 4854 J. Phys. Chem. A 2006, 110, 4854-4865 Spectroscopy of the Cyano Radical in an Aqueous Environment Piotr A. Pieniazek, Stephen E. Bradforth,* and Anna I. Krylov* Department of Chemistry, UniVersity
4854 J. Phys. Chem. A 2006, 110, 4854-4865 Spectroscopy of the Cyano Radical in an Aqueous Environment Piotr A. Pieniazek, Stephen E. Bradforth,* and Anna I. Krylov* Department of Chemistry, UniVersity
NMR and IR spectra & vibrational analysis
 Lab 5: NMR and IR spectra & vibrational analysis A brief theoretical background 1 Some of the available chemical quantum methods for calculating NMR chemical shifts are based on the Hartree-Fock self-consistent
Lab 5: NMR and IR spectra & vibrational analysis A brief theoretical background 1 Some of the available chemical quantum methods for calculating NMR chemical shifts are based on the Hartree-Fock self-consistent
Assignment 1: Molecular Mechanics (PART 2 25 points)
") Chemistry 380.37 Fall 2015 Dr. Jean M. Standard September 2, 2015 Assignment 1: Molecular Mechanics (PART 2 25 points) In this assignment, you will perform some additional molecular mechanics calculations
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard September 2, 2015 Assignment 1: Molecular Mechanics (PART 2 25 points) In this assignment, you will perform some additional molecular mechanics calculations
Supplementary Information for: Hydrogen abstraction by photoexcited benzophenone: consequences for DNA photosensitization
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2016 Supplementary Information for: Hydrogen abstraction by photoexcited benzophenone:
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2016 Supplementary Information for: Hydrogen abstraction by photoexcited benzophenone:
John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks
 10/15/2016 1 WebMO & Gaussian John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks Corrections and updates 9-5-2017 SCHEDULE 9-10 Intro and basic operation of WebMO and MOPAC
10/15/2016 1 WebMO & Gaussian John Keller Department of Chemistry & Biochemistry University of Alaska Fairbanks Corrections and updates 9-5-2017 SCHEDULE 9-10 Intro and basic operation of WebMO and MOPAC
CHEM 344 Molecular Modeling
 CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Part 1: Molecular Orbital Theory, Hybridization, & Formal Charge * all calculation data obtained
CHEM 344 Molecular Modeling The Use of Computational Chemistry to Support Experimental Organic Chemistry Part 1: Molecular Orbital Theory, Hybridization, & Formal Charge * all calculation data obtained
Molecular tailoring: a possible synthetic route to hexasilabenzene
 SUPPORTING INFORMATION Molecular tailoring: a possible synthetic route to hexasilabenzene Zsolt Benedek, Tibor Szilvási and Tamás Veszprémi* Department of Inorganic and Analytical Chemistry, Budapest University
SUPPORTING INFORMATION Molecular tailoring: a possible synthetic route to hexasilabenzene Zsolt Benedek, Tibor Szilvási and Tamás Veszprémi* Department of Inorganic and Analytical Chemistry, Budapest University
2. A(nother) BRIEF TOUR OF COMPUTATIONAL CHEMISTRY METHODS
 BRIEF TOUR OF COMPUTATIONAL CHEMISTRY METHODS") 1. PURPOSE OF THE EXPERIMET This project will further your exposure to PC-Spartan for modeling molecular properties. In particular, you will investigate the electronic structure of polyenes and porphyrins
1. PURPOSE OF THE EXPERIMET This project will further your exposure to PC-Spartan for modeling molecular properties. In particular, you will investigate the electronic structure of polyenes and porphyrins
Fast and accurate Coulomb calculation with Gaussian functions
 Fast and accurate Coulomb calculation with Gaussian functions László Füsti-Molnár and Jing Kong Q-CHEM Inc., Pittsburgh, Pennysylvania 15213 THE JOURNAL OF CHEMICAL PHYSICS 122, 074108 2005 Received 8
Fast and accurate Coulomb calculation with Gaussian functions László Füsti-Molnár and Jing Kong Q-CHEM Inc., Pittsburgh, Pennysylvania 15213 THE JOURNAL OF CHEMICAL PHYSICS 122, 074108 2005 Received 8
Molecular Orbitals for Ozone
 Molecular Orbitals for Ozone Purpose: In this exercise you will do semi-empirical molecular orbital calculations on ozone with the goal of understanding the molecular orbital print out provided by Spartan
Molecular Orbitals for Ozone Purpose: In this exercise you will do semi-empirical molecular orbital calculations on ozone with the goal of understanding the molecular orbital print out provided by Spartan
SERS and NMR Studies of Typical Aggregation-induced. Emission Molecules
 Supplemental information SERS and NMR Studies of Typical Aggregation-induced Emission Molecules Cheng Fang, Yujun Xie, Martin R. Johnston, Yinlan Ruan, Ben Zhong Tang 5, *, Qian Peng, *, Youhong Tang 6,
Supplemental information SERS and NMR Studies of Typical Aggregation-induced Emission Molecules Cheng Fang, Yujun Xie, Martin R. Johnston, Yinlan Ruan, Ben Zhong Tang 5, *, Qian Peng, *, Youhong Tang 6,
(Some of the) Quantum Chemical Advances in the Q- Chem 4.0 Program Package
 Quantum Chemical Advances in the Q- Chem 4.0 Program Package") (Some of the) Quantum Chemical Advances in the Q- Chem 4.0 Program Package Yihan Shao yihan.shao@gmail.com Texas A&M Workshop October 26, 2012 Outline About Q- Chem A brief review of quantum chemistry
(Some of the) Quantum Chemical Advances in the Q- Chem 4.0 Program Package Yihan Shao yihan.shao@gmail.com Texas A&M Workshop October 26, 2012 Outline About Q- Chem A brief review of quantum chemistry
AUTOMATED CONFORMATIONAL ANALYSIS
 Spartan 18 Spartan 18 Parallel Suite is the latest version of Wavefunction s Spartan line of molecular modeling software for research and education, representing a significant improvement in access to
Spartan 18 Spartan 18 Parallel Suite is the latest version of Wavefunction s Spartan line of molecular modeling software for research and education, representing a significant improvement in access to
Interfacing Q-Chem and CHARMM to Perform QM/MM Reaction Path Calculations*
 Interfacing Q-Chem and CHARMM to Perform QM/MM Reaction Path Calculations* H. LEE WOODCOCK III, 1 MILAN HODO S CEK, 2 ANDREW T. B. GILBERT, 3 PETER M. W. GILL, 3 HENRY F. SCHAEFER III, 4 BERNARD R. BROOKS
Interfacing Q-Chem and CHARMM to Perform QM/MM Reaction Path Calculations* H. LEE WOODCOCK III, 1 MILAN HODO S CEK, 2 ANDREW T. B. GILBERT, 3 PETER M. W. GILL, 3 HENRY F. SCHAEFER III, 4 BERNARD R. BROOKS
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Table of Contents. Table of Contents Spin-orbit splitting of semiconductor band structures
 Table of Contents Table of Contents Spin-orbit splitting of semiconductor band structures Relavistic effects in Kohn-Sham DFT Silicon band splitting with ATK-DFT LSDA initial guess for the ground state
Table of Contents Table of Contents Spin-orbit splitting of semiconductor band structures Relavistic effects in Kohn-Sham DFT Silicon band splitting with ATK-DFT LSDA initial guess for the ground state
Introduction to Hartree-Fock calculations in Spartan
 EE5 in 2008 Hannes Jónsson Introduction to Hartree-Fock calculations in Spartan In this exercise, you will get to use state of the art software for carrying out calculations of wavefunctions for molecues,
EE5 in 2008 Hannes Jónsson Introduction to Hartree-Fock calculations in Spartan In this exercise, you will get to use state of the art software for carrying out calculations of wavefunctions for molecues,
This is a simple input file for the calculation of NMR chemical shieldings for a given molecule using the B3LYP functional and def2-tzvpp basis set:
 Computing NMR parameters using ORCA This practical comes with a short lecture on the basics of the computation of NMR parameters using standard electronic structure theory methods. By now you should have
Computing NMR parameters using ORCA This practical comes with a short lecture on the basics of the computation of NMR parameters using standard electronic structure theory methods. By now you should have
A Theoretical Analysis of the Catalytic Cycle of. a Ni Cross-Coupling Process: Application of the. Energetic Span Model
 Supporting Information for: A Theoretical Analysis of the Catalytic Cycle of a Ni Cross-Coupling Process: Application of the Energetic Span Model Sebastian Kozuch*, Sophia E. Lee, Sason Shaik* [*] Institute
Supporting Information for: A Theoretical Analysis of the Catalytic Cycle of a Ni Cross-Coupling Process: Application of the Energetic Span Model Sebastian Kozuch*, Sophia E. Lee, Sason Shaik* [*] Institute
Advanced in silico drug design
 Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
Department of Chemistry, University of Rhode Island, Kingston, RI USA
 Supporting Information for Controlled Organocatalytic Ring-Opening Polymerization of - Thionocaprolactone Partha P. Datta and Matthew K. Kiesewetter Department of Chemistry, University of Rhode Island,
Supporting Information for Controlled Organocatalytic Ring-Opening Polymerization of - Thionocaprolactone Partha P. Datta and Matthew K. Kiesewetter Department of Chemistry, University of Rhode Island,
Linking electronic and molecular structure: Insight into aqueous chloride solvation. Supplementary Information
 Linking electronic and molecular structure: Insight into aqueous chloride solvation Ling Ge, Leonardo Bernasconi, and Patricia Hunt Department of Chemistry, Imperial College London, London SW7 2AZ, United
Linking electronic and molecular structure: Insight into aqueous chloride solvation Ling Ge, Leonardo Bernasconi, and Patricia Hunt Department of Chemistry, Imperial College London, London SW7 2AZ, United
Performance of B3PW91, PBE1PBE and OPBE Functionals in Comparison to B3LYP for 13C NMR Chemical Shift Calculations
 Journal of Science and Technology Ubon Ratchathani University : Special Issue November 17 Performance of B3PW91, PBE1PBE and OPBE Functionals in Comparison to B3LYP for 13C NMR Chemical Shift Calculations
Journal of Science and Technology Ubon Ratchathani University : Special Issue November 17 Performance of B3PW91, PBE1PBE and OPBE Functionals in Comparison to B3LYP for 13C NMR Chemical Shift Calculations
Advanced Electronic Structure Theory Density functional theory. Dr Fred Manby
 Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ Course overview This is a course about density functional theory (DFT)
Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ Course overview This is a course about density functional theory (DFT)
Integrating CML, FoX, Avogadro, NWChem, and EMSLHub to develop a computational chemistry knowledge and discovery base
 Integrating CML, FoX, Avogadro, NWChem, and EMSLHub to develop a computational chemistry knowledge and discovery base Wibe A. de Jong, David M. Brown, Andrew Walker, Marcus D. Hanwell Data is key to scientific
Integrating CML, FoX, Avogadro, NWChem, and EMSLHub to develop a computational chemistry knowledge and discovery base Wibe A. de Jong, David M. Brown, Andrew Walker, Marcus D. Hanwell Data is key to scientific
7 Infrared, Thermochemistry, UV-Vis, and NMR
 7 Infrared, Thermochemistry, UV-Vis, and NMR Exercise 1 Method Dependence and Scaling for the Infrared Spectrum of Formaldehyde. Build a molecule of formaldehyde using sp 2 C and atoms. Clean up the structure
7 Infrared, Thermochemistry, UV-Vis, and NMR Exercise 1 Method Dependence and Scaling for the Infrared Spectrum of Formaldehyde. Build a molecule of formaldehyde using sp 2 C and atoms. Clean up the structure
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
New Algorithms for Conventional and. Jing Kong ( 孔静 ) Q-Chem Inc. Pittsburgh, PA
 Q-Chem Inc. Pittsburgh, PA") New Algorithms for Conventional and Nondynamic DFT Jing Kong ( 孔静 ) Q-Chem Inc. Pittsburgh, PA XC Numerical Integration mrxc: multiresolution exchage-correlation Chunming Chang Nick Russ Phys. Rev. A.,
New Algorithms for Conventional and Nondynamic DFT Jing Kong ( 孔静 ) Q-Chem Inc. Pittsburgh, PA XC Numerical Integration mrxc: multiresolution exchage-correlation Chunming Chang Nick Russ Phys. Rev. A.,
CHEM 2010 Symmetry, Electronic Structure and Bonding Winter Numbering of Chapters and Assigned Problems
 CHEM 2010 Symmetry, Electronic Structure and Bonding Winter 2011 Numbering of Chapters and Assigned Problems The following table shows the correspondence between the chapter numbers in the full book (Physical
CHEM 2010 Symmetry, Electronic Structure and Bonding Winter 2011 Numbering of Chapters and Assigned Problems The following table shows the correspondence between the chapter numbers in the full book (Physical
Uptake of OH radical to aqueous aerosol: a computational study
 Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Modeling Ultrafast Deactivation in Oligothiophenes via Nonadiabatic Dynamics
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Supplementary Data for Modeling Ultrafast Deactivation in Oligothiophenes via Nonadiabatic
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Supplementary Data for Modeling Ultrafast Deactivation in Oligothiophenes via Nonadiabatic
Application Note. U. Heat of Formation of Ethyl Alcohol and Dimethyl Ether. Introduction
 Application Note U. Introduction The molecular builder (Molecular Builder) is part of the MEDEA standard suite of building tools. This tutorial provides an overview of the Molecular Builder s basic functionality.
Application Note U. Introduction The molecular builder (Molecular Builder) is part of the MEDEA standard suite of building tools. This tutorial provides an overview of the Molecular Builder s basic functionality.
Advanced usage of MOLCAS. Ex. 1. Usage of Symmetry in Molcas
 Advanced usage of MOLCAS Ex 1. Symmetry in Molcas (20 min). Ex 2. Transition state optimization (30 min). Ex 3. Ways to run RASSCF program (30 min). Ex 4. Optional. Using EXPBAS module (20 min) Ex 5. Optional.
Advanced usage of MOLCAS Ex 1. Symmetry in Molcas (20 min). Ex 2. Transition state optimization (30 min). Ex 3. Ways to run RASSCF program (30 min). Ex 4. Optional. Using EXPBAS module (20 min) Ex 5. Optional.
XYZ file format Protein Data Bank (pdb) file format Z - matrix
 file format Z - matrix") Chemistry block (exercise 1) In this exercise, students will be introduced how to preform simple quantum chemical calculations. Input files for Gaussian09. Output file structure. Geometry optimization,
Chemistry block (exercise 1) In this exercise, students will be introduced how to preform simple quantum chemical calculations. Input files for Gaussian09. Output file structure. Geometry optimization,
Electric properties of molecules
 Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
INVESTIGATION OF THE ABSORPTION OF CO 2 IN IONIC LIQUID. Kalyan Dhar 1 * and Syed Fahim 1
 Bangladesh J. Sci. Res. 29(1): 41-46, 2016 (June) INVESTIGATION OF THE ABSORPTION OF CO 2 IN IONIC LIQUID Kalyan Dhar 1 * and Syed Fahim 1 Dept. di Chimica Materiali e Ingegneria chimica G. Natta, Politecnico
Bangladesh J. Sci. Res. 29(1): 41-46, 2016 (June) INVESTIGATION OF THE ABSORPTION OF CO 2 IN IONIC LIQUID Kalyan Dhar 1 * and Syed Fahim 1 Dept. di Chimica Materiali e Ingegneria chimica G. Natta, Politecnico
The use of solvation models and the ONIOM layered approach in Gaussian.
 The use of solvation models and the NIM layered approach in Gaussian. In this lab we will consider two techniques that are very useful to model larger systems: the use of solvation models to mimic systems
The use of solvation models and the NIM layered approach in Gaussian. In this lab we will consider two techniques that are very useful to model larger systems: the use of solvation models to mimic systems
Supporting Information. Engineering the Composition and Crystallinity of Molybdenum Sulfide for High-performance Electrocatalytic Hydrogen Evolution
 Supporting Information Engineering the Composition and Crystallinity of Molybdenum Sulfide for High-performance Electrocatalytic Hydrogen Evolution Yanpeng Li 1,2 *, Yifei Yu 2, Robert A. Nielsen 3, William
Supporting Information Engineering the Composition and Crystallinity of Molybdenum Sulfide for High-performance Electrocatalytic Hydrogen Evolution Yanpeng Li 1,2 *, Yifei Yu 2, Robert A. Nielsen 3, William
Jet-FTIR-spectroscopy of pyrrole clusters
 , Corey A. Rice and Martin A. Suhm Institute of Physical Chemistry, University Göttingen Tammannstr. 6, 37077 Göttingen, Germany 61 st Ohio State University International Symposium on Molecular Spectroscopy
, Corey A. Rice and Martin A. Suhm Institute of Physical Chemistry, University Göttingen Tammannstr. 6, 37077 Göttingen, Germany 61 st Ohio State University International Symposium on Molecular Spectroscopy
Prediction of absorption spectra: conjugated chains
 Prediction of absorption spectra: Exploration of TDDFT and PCM on conjugated chains John T. O Connor and Craig M. Teague* Cornell College Mount Vernon, IA MU3C Winter Conference, February 2014 Purpose
Prediction of absorption spectra: Exploration of TDDFT and PCM on conjugated chains John T. O Connor and Craig M. Teague* Cornell College Mount Vernon, IA MU3C Winter Conference, February 2014 Purpose
Jaguar DFT Optimizations and Transition State Searches
 Jaguar DFT Optimizations and Transition State Searches Density Functional Theory (DFT) is a quantum mechanical (QM) method that gives results superior to Hartree Fock (HF) in less computational time. A
Jaguar DFT Optimizations and Transition State Searches Density Functional Theory (DFT) is a quantum mechanical (QM) method that gives results superior to Hartree Fock (HF) in less computational time. A
Theoretical Prediction of Nuclear Magnetic Shielding Constants of Acetonitrile
 Theoretical Prediction of Nuclear Magnetic Shielding Constants of Acetonitrile Ahmad Y. Adam Thesis submitted to the Faculty of the Virginia Polytechnic Institute and State University in partial fulfillment
Theoretical Prediction of Nuclear Magnetic Shielding Constants of Acetonitrile Ahmad Y. Adam Thesis submitted to the Faculty of the Virginia Polytechnic Institute and State University in partial fulfillment
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
Adrian W. Lange and John M. Herbert Department of Chemistry, The Ohio State University, Columbus, OH February 3, 2009
 Supporting Information for: Both intra- and interstrand charge-transfer excited states in aqueous B-DNA are present at energies comparable to, or just above, the 1 ππ excitonic bright states Adrian W.
Supporting Information for: Both intra- and interstrand charge-transfer excited states in aqueous B-DNA are present at energies comparable to, or just above, the 1 ππ excitonic bright states Adrian W.
Quantum Chemistry. NC State University. Lecture 5. The electronic structure of molecules Absorption spectroscopy Fluorescence spectroscopy
 Quantum Chemistry Lecture 5 The electronic structure of molecules Absorption spectroscopy Fluorescence spectroscopy NC State University 3.5 Selective absorption and emission by atmospheric gases (source:
Quantum Chemistry Lecture 5 The electronic structure of molecules Absorption spectroscopy Fluorescence spectroscopy NC State University 3.5 Selective absorption and emission by atmospheric gases (source:
Charge transfer interaction in the acetic acid benzene cation complex
 JOURNAL OF CHEMICAL PHYSICS VOLUME 114, NUMBER 11 15 MARCH 2001 Charge transfer interaction in the acetic acid benzene cation complex Kentaroh Kosugi, Yoshiya Inokuchi, and Nobuyuki Nishi a) Institute
JOURNAL OF CHEMICAL PHYSICS VOLUME 114, NUMBER 11 15 MARCH 2001 Charge transfer interaction in the acetic acid benzene cation complex Kentaroh Kosugi, Yoshiya Inokuchi, and Nobuyuki Nishi a) Institute