METHODS FOR TREATING SOLVENT EFFECTS AND INTERMOLECULAR FORCES. Mark S. Gordon Iowa State University Ames Laboratory
|
|
|
- Lewis Buddy Rose
- 5 years ago
- Views:
Transcription

1 METHODS FOR TREATING SOLVENT EFFECTS AND INTERMOLECULAR FORCES Mark S. Gordon Iowa State University Ames Laboratory
2 OUTLINE Solvation Methods Explicit vs. implicit methods Explicit Methods TIP3P, TIP4P SPC, SPC/E EFP Method for Solvation Summary of EFP1 method for water Sample input files Monte Carlo method Example applications Generalized EFP Method (EFP2)
3 SOLVENT MODELS: CONTINUUM Mostly based on Onsager reaction field model Computationally efficient Sensitive to cavity size and shape Do not account for explicit solute-solvent interactions
4 SOLVATION MODELS: EXPLICIT Solvent molecules described using potentials Empirical potentials: lots of fitted parameters Ab initio potentials: computationally expensive Include explicit solute-solvent interactions Configurational sampling necessary: computationally demanding

5 Multi-Layered Approach to Solvation H H O H O H H O MM H H O H H O H Ab Initio Solute Continuum
6 DISCRETE/EXPLICIT MODELS TIP3P, TIP4P, TIP5P Jorgensen etal, JCP, 79, 926 (1983) Basically simple Lennard-Jones model m n ε mn = [q i q j / r ij ] + A / R 12 C / R 6 i j ε mn =interaction energy between waters m,n q O <0 = -2q H, A, C fitted to bulk properties Rigid molecules TIP4P adds a 4th center inside O toward Hs Very popular model; doesn t get OO radial distribution function right: misses 2nd peak
7 DISCRETE/EXPLICIT MODELS SPC (Simple Point Charge), SPC/E Berendsen etal, JPC, 91, 6269 (1987) SPC: m n ε mn = [q i q j / r ij ] + A / R 12 C / R 6 i j SPC same as TIP, but fitted to MD simulations of density and vaporization energy Rigid molecules Gets OO radial distribution function right Accounts for polarizability/induction
8 SPC/E RDF goo(r) Curve: SPC/E Points: exptl R, Angstroms
9 General Effective Fragment Potential Discrete solvation method Fragment potential is one electron contribution to the QM Hamiltonian if QM part is present Potentials are obtained by separate QM calculations depend on properties of isolated molecules can be systematically improved Internally frozen geometries
10 GOALS Develop general approach to accurately describe intermolecular interactions Solvent effects on Ions and molecules Chemical reaction mechanisms Spectral shifts Liquid behavior Liquid-gas, liquid-surface interface Van der Waals interactions Polymer interactions Enzyme interactions Incorporate all important physics No parameter fits Efficient

11
12 Effective Fragment Potential System is divided into an ab initio region for the solute and a fragment region for the solvent molecules. E = Eab initio + Einteraction
13 General EFP Method In the most general implementation EFP should include all relevant energy contributions: Einteraction = Ecoulomb + Epolarization + Eexch. rep. + Edispersion + Echarge transfer + Ehigher order terms
14 Interaction energy consists of : electrostatic, polarization and exchange repulsion/charge transfer term Einteraction= Ecoulomb + Epolarization + Eexchange repulsion/charge transfer K E interaction = V Elec k (µ,s) + V Pol Re l (µ,s) + V p m (µ, s) k =1 Hartree Fock based EFP L l =1 M m =1 Distributed Multipolar expansion Damping at small R LMO polarizability expansion Iterate to self-consistency Fit to Functional Form
15 Analytic gradients (first derivatives) have been derived and coded for all terms. So, one can perform geometry optimizations, transition state searches, dynamics
 Same general approach Separate fits for exchange repulsion+ct, dispersion More effective correlation, especially at long range EFP-EFP done, EFP-QM in")
16 HIGHER LEVELS OF EFP1 DFT-based EFP (Ivana Adamovic) Same general approach Based on B3LYP Adds some level of correlation MP2-based EFP (Lyuda Slipchenko,Jie Song) Same general approach Separate fits for exchange repulsion+ct, dispersion More effective correlation, especially at long range EFP-EFP done, EFP-QM in progress
17 EFP1/DFT: APPLICATION TO S N 2 Microsolvation of Cl - + CH 3 Br (Ronnie Bierbaum) Solute=DFT/B3LYP, MP2, CCSD(T) Water=DFT, MP2, EFP/DFT Preliminary analysis CCSD(T) has minor effect relative to MP2 (< 1 kcal/ mol) Basis set effects are small (<1 kcal/mol) Basis set used: aug-cc-pvdz

18 GAS PHASE 0.00 separate reactants Cl Br IRC -6.2 DFT (4.4) -1.5 MP2 (10.2) transition state IRC -7.0 DFT -6.1 MP2 products DFT MP2 ion-molecule reactant DFT MP2 ion-molecule product

19 DFT
20 Forward central barrier and relative differences as a function of n (kcal/mol) Forward E a ΔE a n=0 DFT n=1 EFP/DFT DFT n=2 EFP/DFT DFT n=3 (isomer 2) EFP/DFT DFT n=0 MP n=1 MP2/EFP MP n=2 MP2/EFP MP n=3 (isomer 2) MP2/EFP MP n=4 (isomer 5) MP2/EFP
21 Forward central barrier and relative differences as a function of n (kcal/mol) Forward E a ΔE a n=0 DFT n=1 EFP/DFT DFT n=2 EFP/DFT DFT n=3 (isomer 2) EFP/DFT DFT n=0 MP n=1 MP2/EFP MP n=2 MP2/EFP MP n=3 (isomer 2) MP2/EFP MP n=4 (isomer 5) MP2/EFP
22 Forward central barrier and relative differences as a function of n (kcal/mol) Forward E a ΔE a n=0 DFT n=1 EFP/DFT DFT n=2 EFP/DFT DFT n=3 (isomer 2) EFP/DFT DFT n=0 MP n=1 MP2/EFP MP n=2 MP2/EFP MP n=3 (isomer 2) MP2/EFP MP n=4 (isomer 5) MP2/EFP
23 CONCLUSIONS EFP reproduces QM at small fraction of cost DFT greatly underestimates central barrier DFT reproduces solvent effect on central barrier MP2 for solute with EFP for solvent good compromise Reproduces both absolute and solvent-shifted MP2 central barriers Suggests EFP1/MP2 will be just as good
24 Water hexamer isomers Binding DFT MP2 CCSD HF Energy B3LYP (T) prism cage book cyclic boat basis set: DH(d,p) -units: kcal/mol
25 Water hexamer isomers Binding Energy DFT B3LYP EFP1/ DFT MP2 CCSD (T) HF EFP1/ HF prism cage book cyclic boat basis set: DH(d,p) -units: kcal/mol
26 Water hexamer isomers Binding Energy DFT B3LYP EFP1/ DFT MP2 EFP1/ MP2 CCSD (T) HF EFP1/ HF prism cage book cyclic boat basis set: DH(d,p) -units: kcal/mol
27 Initial structure: 62 waters, at least 26 ps equilibration Timestep size = 1 fs 2.5 goo(r) EFP1/HF EFP1/DFT EFP1/MP2 SPC/E Exp (THG) r (Angstroms) Exp (THG): X-ray; Sorenson et. al. J. Chem. Phys. 113, 9149 (2000).
28 Initial structure: 512 waters, at least 5 ps equilibration Timestep size = 1 fs goo(r) EFP1/HF EFP1/DFT EFP1/MP2 SPC/E Exp (THG) r (Angstroms) Exp (THG): X-ray; Sorenson et. al. J. Chem. Phys. 113, 9149 (2000).


 JCP, 108,")
 Mol. Phys.")
29 GENERAL EFP METHOD: EFP2 Jan Jensen Mark Freitag Ivana Adamovic Hui Li Lyuda Slipchenko Dan Kemp Tony Smith Peng Xu Mol. Phys., 89, 1313 (1996) JCP, 108, 4772 (1999) JCP, 112, 7300 (2000) Mol. Phys., 103, 379 (2005) Mol. Phys., 107, 999 (2009)
30 Generalized EFP2 Method Interaction energy consists of : electrostatic, polarization, exchange repulsion, dispersion, and charge transfer terms Einteraction= ECoulomb + EPolarization + Eexrep + Edispersion + ECharge Transfer No Parameter Fits Distributed Multipolar expansion Screening: near field LMO polarizability expansion Iterate to self-consistency From first principles using LMO overlaps Distributed LMO dispersion from first principles
31 S ij 2 E exch 2 2 2ln S ij A B 2 S ij F ik S kj + F jl S li 2T ij π i A j B Summary If all approximations presented above are combined, E exch can be approximated as (provided LMOs are used) R ij i A j B S ij Z J R ij + 2 R il + Z I R Ij + 2 R kj R ij J B l B I B k A i A j B k A l B This equation requires only the computation of intermolecular overlap and electronic kinetic energy integrals, i.e. no two-electron integrals other than those calculated once for the isolated molecules. It contains no adjustable parameters, only fixed parameters computed for the isolated molecules, such as the LMOs in some AO basis, Fock matrices in the LMO bases, and the LMO centroids of charge Numerical Tests E exch E exch (kcal/mol) C s C 2v E exch (Morokuma) RHF/6-31++(d,p) Oxygen-Oxygen Distance (Å)
32 EFP2 Dispersion: Ivana Adamovic E disp = C 6 R 6 + C 7 R 7 + C 8 R Simplest approximation: terminate at C 6 term Calculate E disp using distributed LMO expansion, E disp = x,y,z k A j B αβγδ T αβ kj T γδ kj 0 dνα k αγ (iν) j α βδ (iν) = k A j B C 6 kj / R 6 kj In terms of dynamic polarizability over imaginary frequency range and field gradient components, T Accomplished using dynamic TDHF equations Amos et al., JPC, 89, 2186 (1985); CPL, 278, 278 (1997)
 ( (i, j ) = 1 S f dmp 2 ij m n=0 n/ 2 ij n! Mol. Phys., 107, 999 (2009); J. Phys. Chem.")
33 Overlap Damping Based on intermolecular overlap Already available for exchange repulsion No fitting required, no additional cost m 2ln S ) ( (i, j ) = 1 S f dmp 2 ij m n=0 n/ 2 ij n! Mol. Phys., 107, 999 (2009); J. Phys. Chem. 119, 2161 (2015)
34 Charge transfer (CT) HOMO E= LUMO E=0.011 Charge Transfer Virtual Virtual Occupied A B Occupied Ammonium nitrate RHF/6-31++G(d,p) - Excitation of electrons in A to the unoccupied orbitals of B - Significant in ionic systems



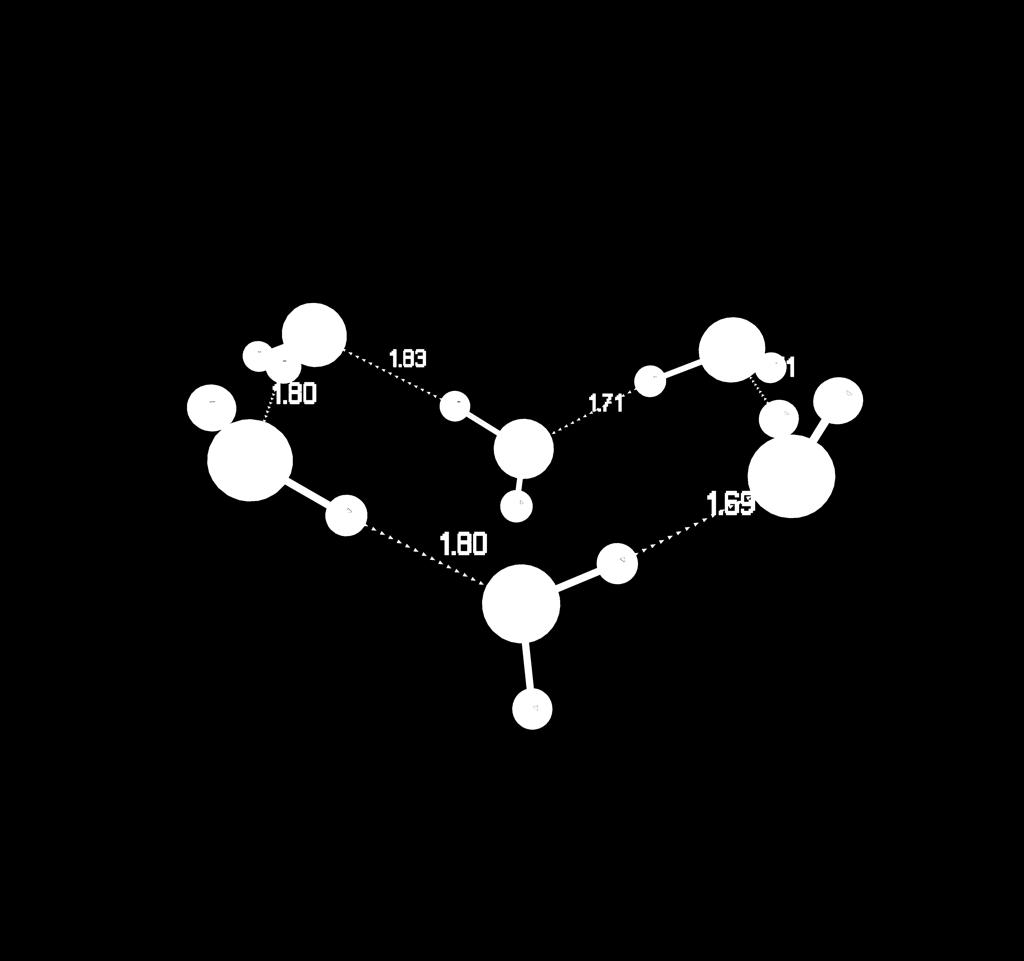
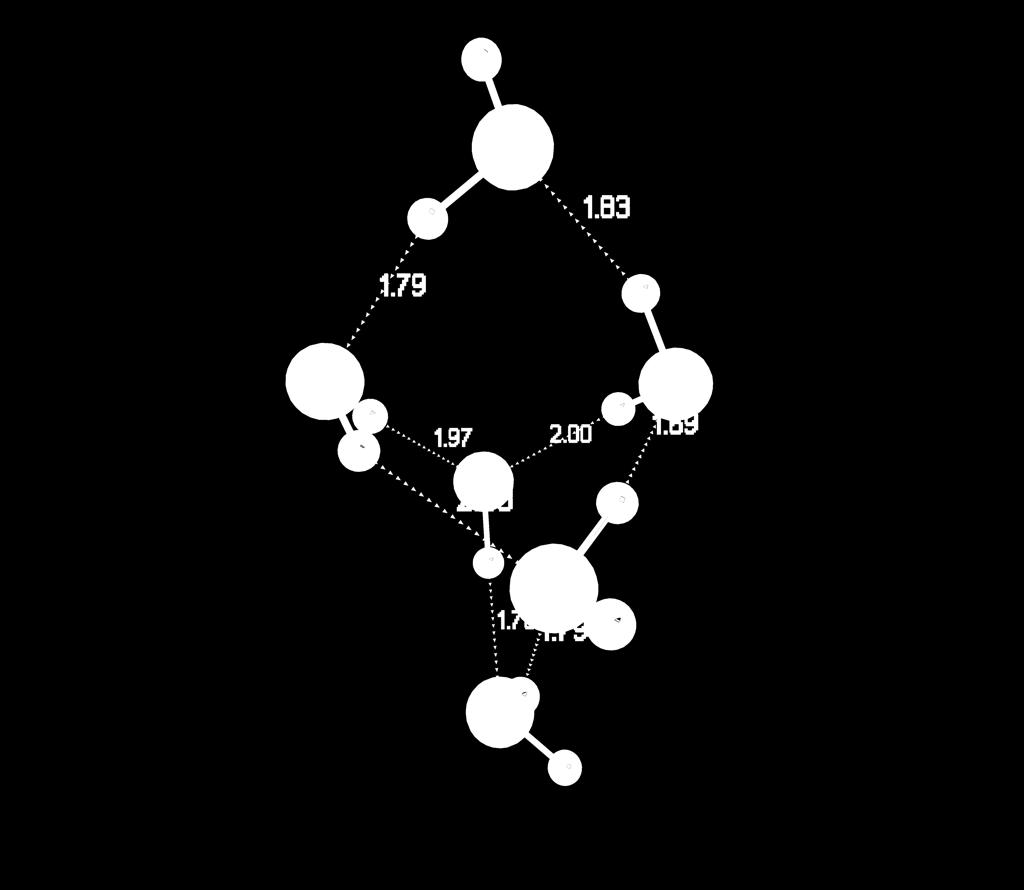
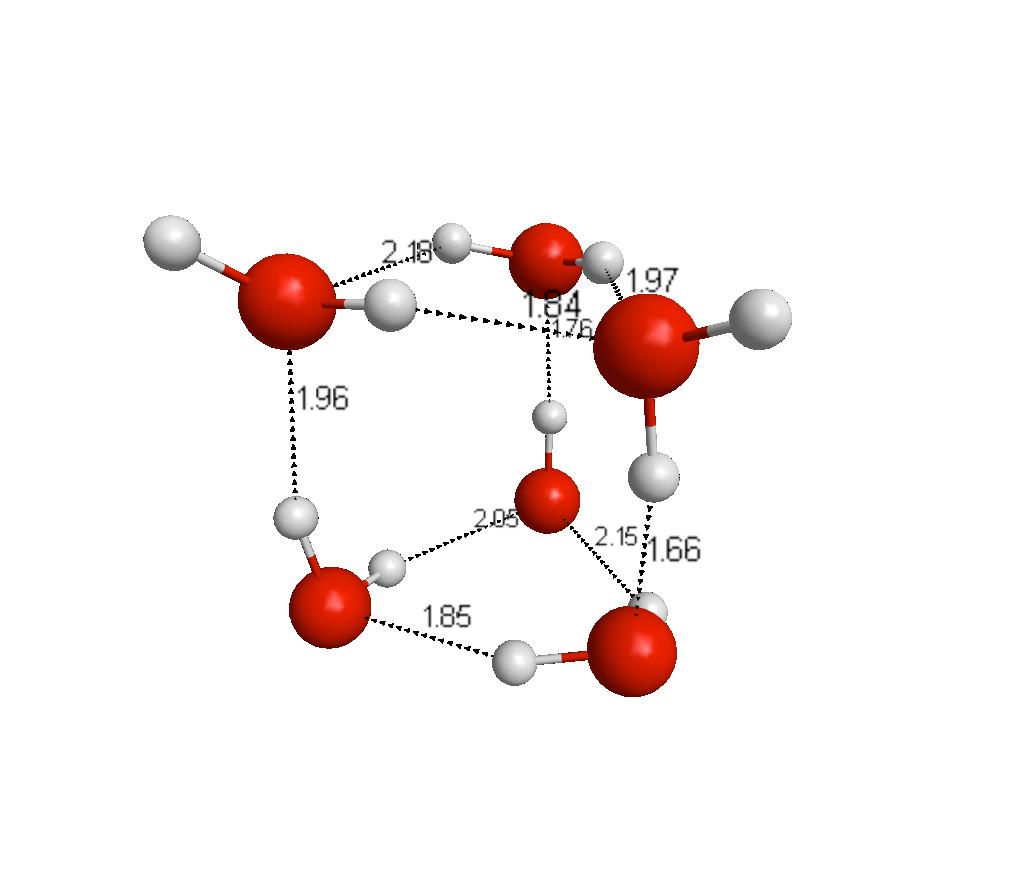
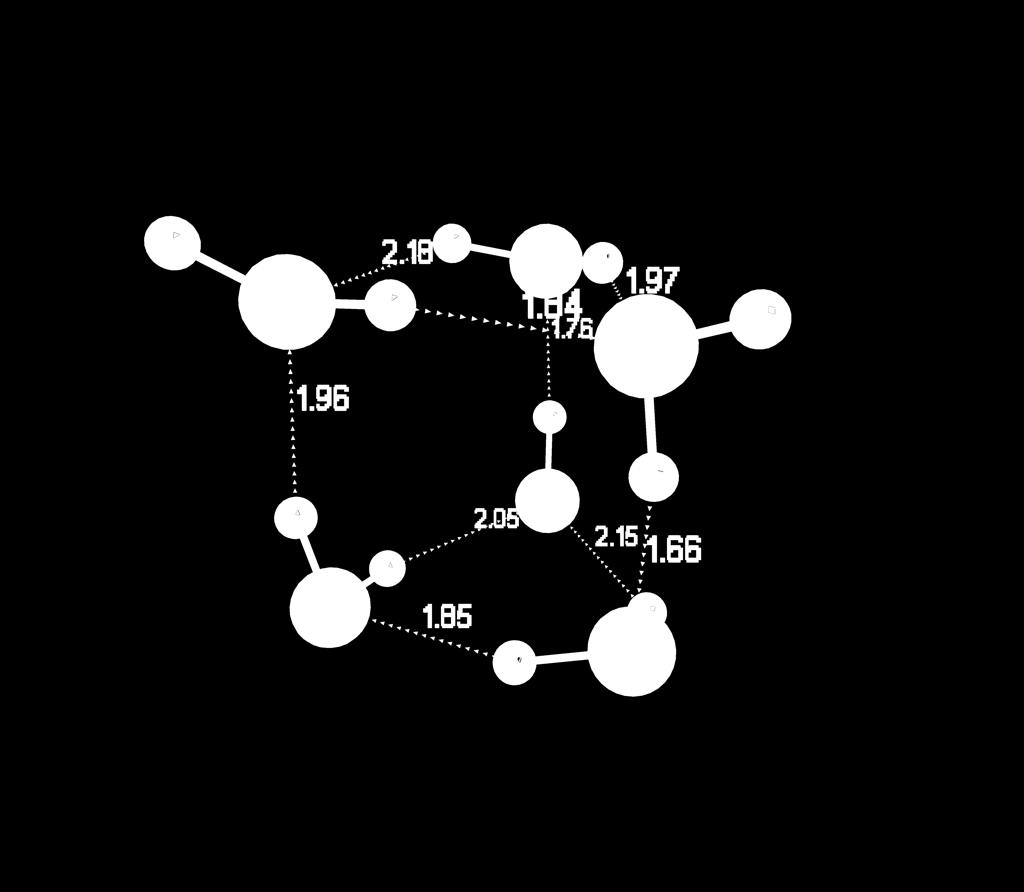

35 Water Hexamers bag boat book cage cyclic prism
36 Water Hexamer Errors in Total Interaction Energy (relative to CCSD(T)//MP2/aug-cc-pVTZ)

37 Water Hexamer Errors in Total Interaction Energy (relative to CCSD(T)//MP2/aug-cc-pVTZ)

38 Water Hexamer Errors in Total Interaction Energy (relative to CCSD(T)//MP2/aug-cc-pVTZ)

39 Water Hexamer (Prism) Contributions to the Total Interaction Energy
40 Water Hexamer (Prism) Contributions to the Total Interaction Energy

41 Water Hexamer (Prism) Contributions to the Total Interaction Energy

42 Relative CCSD(T) Interaction Energies of Water Hexamers
/aug-cc-pVTZ = -4.")


/aug-cc-pVQZ = -3.")
43 Ar Hexamer c2v Total Interaction Energy CCSD(T)/aug-cc-pVTZ = FMO2-CCSD(T)/aug-cc-pVQZ = d4h Total Interaction Energy CCSD(T)/aug-cc-pVTZ = FMO2-CCSD(T)/aug-cc-pVQZ = d3h Total Interaction Energy CCSD(T)/aug-cc-pVTZ = FMO2-CCSD(T)/aug-cc-pVQZ = -3.07
44 Total Interaction Energies Argon Hexamer Prism Bag Trigonal Planar Method D4h C2v D3h EFP MP2/ACCD MP2/ACCT MP2/ACCQ CC/ACCD CC/ACCT
45 2-Body Interaction Energy Argon Hexamer Prism Bag Trigonal Planar Method D4h C2v D3h EFP MP2/ACCD MP2/ACCT MP2/ACCQ CC/ACCD CC/ACCT
46 3-Body Interaction Energy Argon Hexamer Prism Bag Trigonal Planar Method D4h C2v D3h EFP MP2/ACCD MP2/ACCT MP2/ACCQ CC/ACCD CC/ACCT
 Relative to")
47 Ar Hexamer: Error (Total Interaction Energy) Relative to CC/ACCT

48 Error (Total Interaction Energy) Relative to CC/ ACCT - Ar Hexamer

49 Error (Total Interaction Energy) Relative to CC/ ACCT - Ar Hexamer
50 Benzene Dimer: Lyuda Slipchenko Some structures dominated by dispersion Especially π π interactions HF cannot get this right DFT only with special effort Requires correlated methods: MP2, CCSD(T) Good test of EFP
; M.O. Sinnokrot and C.D. Sherrill, JPC, 108, 10200 (2004)")
51 Low-energy configurations of benzene dimer R 2 R R R 1 sandwich T-shaped parallel-displaced High-level ab initio calculations: M.O. Sinnokrot, E.F. Valeev, and C.D. Sherrill, JACS, 124, (2002); M.O. Sinnokrot and C.D. Sherrill, JPC, 108, (2004)
52 Compare EFP2 with Symmetry Adapted Perturbation Theory Sandwich & T-shape: SAPT: aug-cc-pvdz M.O. Sinnokrot and C.D. Sherrill, JPC, 108, (2004) EFP2: G(3df,2p) electrostatic with charge-charge damping term exchange repulsion polarization dispersion
53 Electrostatic Term
54 Exchange Repulsion
55 Polarization
56 Dispersion
57 Total Binding Energy
58 Parallel-Displaced Configuration
59 Benzene Dimer: Summary method basis sandwich T-shape parallel-displaced R energy R energy R1 R2 energy MP2 aug-cc-pvdz MP2 aug-cc-pvtz MP2 aug-cc-pvqz CCSD(T ) CCSD(T ) EFP2 aug-cc-pvdz aug-cc-pvqz G (3df,2p) distances: angstroms, energies: kcal/mol EFP: ~.4 sec CCSD(T): ~10 days
60 Benzene Dimer: Summary method basis sandwich T-shape parallel-displaced R energy R energy R1 R2 energy MP2 aug-cc-pvdz MP2 aug-cc-pvtz MP2 aug-cc-pvqz CCSD(T ) CCSD(T ) EFP2 aug-cc-pvdz aug-cc-pvqz G (3df,2p) distances: angstroms, energies: kcal/mol EFP: ~.4 sec MP2/QZ: ~10 hours
61 Contributions to the Interaction Energy dimer Exchange Induction Dispersion Total sandwich T-shaped Electrostatic paralleldisplaced
62 DNA BASES: Q.T. SMITH Compare with Jurecka, P.; Hobza, P. J. Am. Chem. Soc. 2003, 125, 15608) RI-MP2 / ext. cc-pvtz or TZVPP geometries RI-MP2/aug-cc-pVXZ (X = D, T, Q) to CBS limit energies CCSD(T) correction added Counterpoise corrected for BSSE EFP generated with G(3df,2p) basis set
/CBS energy: -16.9 kcal/mol EFP COUL -43.3 REP 36.4 POL -14.7 DISP -10.7 TOTAL -32.3 EFP COUL -27.1 REP 26.7 POL -7.2 DISP -8.")
63 Guanine-Cytosine H-Bonded RI-MP2/TVZPP optimized Adenine-Thymine H-Bonded 1.76 Å 1.93 Å 1.91 Å 1.82 Å 1.92 Å CCSD(T)/CBS energy: kcal/mol CCSD(T)/CBS energy: kcal/mol EFP COUL REP 36.4 POL DISP TOTAL EFP COUL REP 26.7 POL -7.2 DISP -8.2 TOTAL -15.8
/CBS energy: -19.")

64 Guanine-Cytosine stacked RI-MP2/TVZPP optimized Adenine-Thymine Stacked RI-MP2/TVZPP optimized 3.40 Å 3.20 Å CCSD(T)/CBS energy: kcal/mol CCSD(T)/CBS energy: kcal/mol EFP COUL REP 20.6 POL -3.1 DISP TOTAL EFP COUL -7.8 REP 16.1 POL -0.7 DISP TOTAL -10.0
65 CURRENT LIMITATIONS EFP1 is for water only QM part can be any level of theory Solvent must be water EFP2 can be generated for any species QM-EFP energies are now done QM-EFP gradients are in progress
66 CONTINUUM SOLVATION MODELS Simplest is Onsager self-consistent reaction field (SCRF) model ($SCRF) Solute (QM) dipole moment µ polarizes medium (solvent) through dielectric ε Newly polarized solvent alters solute dipole moment Iterated to self-consistency Accomplished by adding new term to QM Hamiltonian
67 ONSAGER SCRF MODEL V σ = -r.r r = position vector R is proportional to molecular dipole moment R = gµ µ = dipole vector g = 2 (ε 1)/[(2ε+1)(a 3 )] a = radius of cavity Usually assume spherical cavity Radius can be pretty arbitrary Adds simple <χ i x χ j > integrals - very cheap
68 PCM MODEL Polarizable Continuum Model (PCM) Tomasi etal., Chem. Phys. Lett., 255, 327 (1996) Van der Waals type surface cavity Uses detailed knowledge of electrostatic potential Cavity dispersion potential determined from surface area Can include dispersion effects Interfaced with EFP in GAMESS: $PCM See both input and reference sections in manual
69 OTHER METHODS COSMO Initially developed by Klamt Further developed by Baldridge (U. Zurich) No gradients, not interfaced with EFP SVP Chipman (Notre Dame Radiation Laboratory) Accounts for charge leakage outside cavity SM5 Truhlar, Cramer (Minnesota) Highly parametrized, interfaced with GAMESS
CLUSTER-BASED APPROACHES TO SOLVATION. Mark S. Gordon Iowa State University Ames Laboratory
 CLUSTER-BASED APPROACHES TO SOLVATION Mark S. Gordon Iowa State University Ames Laboratory 1 OUTLINE Solvation Methods Explicit vs. implicit methods Explicit Methods TIP3P, TIP4P SPC, SPC/E EFP Method
CLUSTER-BASED APPROACHES TO SOLVATION Mark S. Gordon Iowa State University Ames Laboratory 1 OUTLINE Solvation Methods Explicit vs. implicit methods Explicit Methods TIP3P, TIP4P SPC, SPC/E EFP Method
Fragmentation methods
 Fragmentation methods Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method
Fragmentation methods Scaling of QM Methods HF, DFT scale as N 4 MP2 scales as N 5 CC methods scale as N 7 What if we could freeze the value of N regardless of the size of the system? Then each method
Alanine: Then There Was Water
 Chemistry Publications Chemistry 6-2009 Alanine: Then There Was Water Jonathan M. Mullin Iowa State University Mark S. Gordon Iowa State University, mgordon@iastate.edu Follow this and additional works
Chemistry Publications Chemistry 6-2009 Alanine: Then There Was Water Jonathan M. Mullin Iowa State University Mark S. Gordon Iowa State University, mgordon@iastate.edu Follow this and additional works
Non-covalent force fields computed ab initio
 Non-covalent force fields computed ab initio Supermolecule calculations Symmetry-adapted perturbation theory (SAPT) Supermolecule calculations Requirements: E = E AB E A E B. Include electron correlation,
Non-covalent force fields computed ab initio Supermolecule calculations Symmetry-adapted perturbation theory (SAPT) Supermolecule calculations Requirements: E = E AB E A E B. Include electron correlation,
Towards gas-phase accuracy for condensed phase problems
 Towards gas-phase accuracy for condensed phase problems Fred Manby Centre for Computational Chemistry, School of Chemistry University of Bristol STC 2006: Quantum Chemistry Methods and Applications Erkner,
Towards gas-phase accuracy for condensed phase problems Fred Manby Centre for Computational Chemistry, School of Chemistry University of Bristol STC 2006: Quantum Chemistry Methods and Applications Erkner,
Modeling in Solution. Important Observations. Energy Concepts
 Modeling in Solution 1 Important Observations Various electrostatic effects for a solvated molecule are often less important than for an isolated gaseous molecule when the molecule is dissolved in a solvent
Modeling in Solution 1 Important Observations Various electrostatic effects for a solvated molecule are often less important than for an isolated gaseous molecule when the molecule is dissolved in a solvent
Dispersion Interactions from the Exchange-Hole Dipole Moment
 Dispersion Interactions from the Exchange-Hole Dipole Moment Erin R. Johnson and Alberto Otero-de-la-Roza Chemistry and Chemical Biology, University of California, Merced E. R. Johnson (UC Merced) Dispersion
Dispersion Interactions from the Exchange-Hole Dipole Moment Erin R. Johnson and Alberto Otero-de-la-Roza Chemistry and Chemical Biology, University of California, Merced E. R. Johnson (UC Merced) Dispersion
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Electronic structure theory: Fundamentals to frontiers. VI. Analysis and more.
 Electronic structure theory: Fundamentals to frontiers. VI. Analysis and more. MARTIN HEAD-GORDON Department of Chemistry, University of California, Berkeley, and, Chemical Sciences Division, Lawrence
Electronic structure theory: Fundamentals to frontiers. VI. Analysis and more. MARTIN HEAD-GORDON Department of Chemistry, University of California, Berkeley, and, Chemical Sciences Division, Lawrence
Dispersion Correction Derived from First Principles for Density Functional Theory and Hartree Fock Theory
 Chemistry Publications Chemistry 2-2015 Dispersion Correction Derived from First Principles for Density Functional Theory and Hartree Fock Theory Emilie B. Guidez Iowa State University, eguidez@ameslab.gov
Chemistry Publications Chemistry 2-2015 Dispersion Correction Derived from First Principles for Density Functional Theory and Hartree Fock Theory Emilie B. Guidez Iowa State University, eguidez@ameslab.gov
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory
 Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Electronic structure theory: Fundamentals to frontiers. 2. Density functional theory MARTIN HEAD-GORDON, Department of Chemistry, University of California, and Chemical Sciences Division, Lawrence Berkeley
Water models in classical simulations
 Water models in classical simulations Maria Fyta Institut für Computerphysik, Universität Stuttgart Stuttgart, Germany Water transparent, odorless, tasteless and ubiquitous really simple: two H atoms attached
Water models in classical simulations Maria Fyta Institut für Computerphysik, Universität Stuttgart Stuttgart, Germany Water transparent, odorless, tasteless and ubiquitous really simple: two H atoms attached
Towards a force field based on density fitting
 THE JOURNAL OF CHEMICAL PHYSICS 124, 104101 2006 Towards a force field based on density fitting Jean-Philip Piquemal a and G. Andrés Cisneros Laboratory of Structural Biology, National Institute of Environmental
THE JOURNAL OF CHEMICAL PHYSICS 124, 104101 2006 Towards a force field based on density fitting Jean-Philip Piquemal a and G. Andrés Cisneros Laboratory of Structural Biology, National Institute of Environmental
Water Benzene Interactions: An Effective Fragment Potential and Correlated Quantum Chemistry Study
 Chemistry Publications Chemistry 2009 Water Benzene Interactions: An Effective Fragment Potential and Correlated Quantum Chemistry Study Lyudmila V. Slipchenko Iowa State University Mark S. Gordon Iowa
Chemistry Publications Chemistry 2009 Water Benzene Interactions: An Effective Fragment Potential and Correlated Quantum Chemistry Study Lyudmila V. Slipchenko Iowa State University Mark S. Gordon Iowa
PLEASE SCROLL DOWN FOR ARTICLE
 This article was downloaded by: [Purdue University] On: 30 March 009 Access details: Access Details: [subscription number 907055043] Publisher Taylor & Francis Informa Ltd Registered in England and Wales
This article was downloaded by: [Purdue University] On: 30 March 009 Access details: Access Details: [subscription number 907055043] Publisher Taylor & Francis Informa Ltd Registered in England and Wales
Investigating Popular Water Models
 Investigating Popular Water odels Albert DeFusco, Ken Jordan Center for olecular and aterials Simulations What are we modeling? OOOOOLLLLLDDDDDEDEEEENENNNNNN OOLL D E OOOOOLLLLLDDDDDEDEEEENENNNNNN OOLL
Investigating Popular Water odels Albert DeFusco, Ken Jordan Center for olecular and aterials Simulations What are we modeling? OOOOOLLLLLDDDDDEDEEEENENNNNNN OOLL D E OOOOOLLLLLDDDDDEDEEEENENNNNNN OOLL
Spectroscopy of the Cyano Radical in an Aqueous Environment
 4854 J. Phys. Chem. A 2006, 110, 4854-4865 Spectroscopy of the Cyano Radical in an Aqueous Environment Piotr A. Pieniazek, Stephen E. Bradforth,* and Anna I. Krylov* Department of Chemistry, UniVersity
4854 J. Phys. Chem. A 2006, 110, 4854-4865 Spectroscopy of the Cyano Radical in an Aqueous Environment Piotr A. Pieniazek, Stephen E. Bradforth,* and Anna I. Krylov* Department of Chemistry, UniVersity
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
Uptake of OH radical to aqueous aerosol: a computational study
 Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Uptake of OH radical to aqueous aerosol: a computational study Grigory Andreev Karpov Institute of Physical Chemistry 10 Vorontsovo pole, Moscow, 105064, Russia Institute of Physical Chemistry and Electrochemistry
Specific Ion Solvtion in Ethylene Carbonate and Propylene Carbonate
 Specific Ion Solvtion in Ethylene Carbonate and Propylene Carbonate A. Arslanargin, A. Powers, S. Rick, T. Pollard, T. Beck Univ Cincinnati Chemistry Support: NSF, OSC TSRC 2016 November 2, 2016 A. Arslanargin,
Specific Ion Solvtion in Ethylene Carbonate and Propylene Carbonate A. Arslanargin, A. Powers, S. Rick, T. Pollard, T. Beck Univ Cincinnati Chemistry Support: NSF, OSC TSRC 2016 November 2, 2016 A. Arslanargin,
Advanced in silico drug design
 Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced Electronic Structure Theory Density functional theory. Dr Fred Manby
 Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
List of Figures Page Figure No. Figure Caption No. Figure 1.1.
 List of Figures Figure No. Figure Caption Page No. Figure 1.1. Cation- interactions and their modulations. 4 Figure 1.2. Three conformations of benzene dimer, S is not a minimum on the potential energy
List of Figures Figure No. Figure Caption Page No. Figure 1.1. Cation- interactions and their modulations. 4 Figure 1.2. Three conformations of benzene dimer, S is not a minimum on the potential energy
The Atmospheric Significance of Water. Clusters and Ozone-water Complexes.
 The Atmospheric Significance of Water Clusters and Ozone-water Complexes. Josep M. Anglada, a,* Gerald J. Hoffman, b Lyudmila V. Slipchenko, c Marilia M.Costa, d Manuel F. Ruiz-López, d and Joseph S. Francisco.
The Atmospheric Significance of Water Clusters and Ozone-water Complexes. Josep M. Anglada, a,* Gerald J. Hoffman, b Lyudmila V. Slipchenko, c Marilia M.Costa, d Manuel F. Ruiz-López, d and Joseph S. Francisco.
Subject of the Lecture:
 Subject of the Lecture: Conceptual basis for the development of force fields. Implementation/validation Water - a worked example Extensions - combining molecular mechanics and quantum mechanics (QM/MM)
Subject of the Lecture: Conceptual basis for the development of force fields. Implementation/validation Water - a worked example Extensions - combining molecular mechanics and quantum mechanics (QM/MM)
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs
 Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs Founded in 1993, Q-Chem strives to bring its customers state-ofthe-art methods and algorithms for performing quantum chemistry calculations. Cutting-edge
Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs Founded in 1993, Q-Chem strives to bring its customers state-ofthe-art methods and algorithms for performing quantum chemistry calculations. Cutting-edge
Why Is Molecular Interaction Important in Our Life
 Why Is Molecular Interaction Important in ur Life QuLiS and Graduate School of Science iroshima University http://www.nabit.hiroshima-u.ac.jp/iwatasue/indexe.htm Suehiro Iwata Sept. 29, 2007 Department
Why Is Molecular Interaction Important in ur Life QuLiS and Graduate School of Science iroshima University http://www.nabit.hiroshima-u.ac.jp/iwatasue/indexe.htm Suehiro Iwata Sept. 29, 2007 Department
Chemistry 4560/5560 Molecular Modeling Fall 2014
 Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Computational Chemistry I
 Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Q-Chem Workshop. Doubletree Hotel 2085 S. Harbor Boulevard Anaheim, CA March 26, Schedule
 Q-Chem Workshop Doubletree Hotel 2085 S. Harbor Boulevard Anaheim, CA 92802 March 26, 2011 1 8:30 Schedule Welcome remarks, Prof. Peter Gill, Australian National Univ and President of Q-Chem 8:45-9:15
Q-Chem Workshop Doubletree Hotel 2085 S. Harbor Boulevard Anaheim, CA 92802 March 26, 2011 1 8:30 Schedule Welcome remarks, Prof. Peter Gill, Australian National Univ and President of Q-Chem 8:45-9:15
Feet on the potential energy surface, head in the π clouds
 Graduate Theses and Dissertations Iowa State University Capstones, Theses and Dissertations 2011 Feet on the potential energy surface, head in the π clouds Quentin Anthony Smith Iowa State University Follow
Graduate Theses and Dissertations Iowa State University Capstones, Theses and Dissertations 2011 Feet on the potential energy surface, head in the π clouds Quentin Anthony Smith Iowa State University Follow
Charge-Transfer and Dispersion Energies in Water Clusters
 II.26 Charge-Transfer and Dispersion Energies in Water Clusters Suehiro Iwata 1,2, Pradipta Bandyopadhyay 3, Sotiris S. Xantheas 4 1)Toyota Physical and Chemical Research Institute (2008-2012, fellow)
II.26 Charge-Transfer and Dispersion Energies in Water Clusters Suehiro Iwata 1,2, Pradipta Bandyopadhyay 3, Sotiris S. Xantheas 4 1)Toyota Physical and Chemical Research Institute (2008-2012, fellow)
Intermolecular Forces in Density Functional Theory
 Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
Intermolecular Forces in Density Functional Theory Problems of DFT Peter Pulay at WATOC2005: There are 3 problems with DFT 1. Accuracy does not converge 2. Spin states of open shell systems often incorrect
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Evaluation of Charge Penetration between Distributed Multipolar Expansions
 Chemistry Publications Chemistry 5-2000 Evaluation of Charge Penetration between Distributed Multipolar Expansions Mark Alan Freitag Iowa State University Mark S. Gordon Iowa State University, mgordon@iastate.edu
Chemistry Publications Chemistry 5-2000 Evaluation of Charge Penetration between Distributed Multipolar Expansions Mark Alan Freitag Iowa State University Mark S. Gordon Iowa State University, mgordon@iastate.edu
DFT calculations of NMR indirect spin spin coupling constants
 DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
Phase Equilibria and Molecular Solutions Jan G. Korvink and Evgenii Rudnyi IMTEK Albert Ludwig University Freiburg, Germany
 Phase Equilibria and Molecular Solutions Jan G. Korvink and Evgenii Rudnyi IMTEK Albert Ludwig University Freiburg, Germany Preliminaries Learning Goals Phase Equilibria Phase diagrams and classical thermodynamics
Phase Equilibria and Molecular Solutions Jan G. Korvink and Evgenii Rudnyi IMTEK Albert Ludwig University Freiburg, Germany Preliminaries Learning Goals Phase Equilibria Phase diagrams and classical thermodynamics
A CSOV Study of the Difference between HF and DFT Intermolecular Interaction Energy Values: The Importance of the Charge Transfer Contribution
 A CSOV Study of the Difference between HF and DFT Intermolecular Interaction Energy Values: The Importance of the Charge Transfer Contribution JEAN-PHILIP PIQUEMAL, 1 * ANTONIO MARQUEZ, 2 OLIVIER PARISEL,
A CSOV Study of the Difference between HF and DFT Intermolecular Interaction Energy Values: The Importance of the Charge Transfer Contribution JEAN-PHILIP PIQUEMAL, 1 * ANTONIO MARQUEZ, 2 OLIVIER PARISEL,
k θ (θ θ 0 ) 2 angles r i j r i j
 2 angles r i j r i j") 1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
Density Func,onal Theory (Chapter 6, Jensen)
") Chem 580: DFT Density Func,onal Theory (Chapter 6, Jensen) Hohenberg- Kohn Theorem (Phys. Rev., 136,B864 (1964)): For molecules with a non degenerate ground state, the ground state molecular energy and
Chem 580: DFT Density Func,onal Theory (Chapter 6, Jensen) Hohenberg- Kohn Theorem (Phys. Rev., 136,B864 (1964)): For molecules with a non degenerate ground state, the ground state molecular energy and
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
On the Unusual Properties of Halogen Bonds: A Detailed ab Initio Study of X 2 -(H 2 O) 1-5 clusters (X ) Cl and Br)
 1-5 clusters (X ) Cl and Br)") 5496 J. Phys. Chem. A 2009, 113, 5496 5505 On the Unusual Properties of Halogen Bonds: A Detailed ab Initio Study of X 2 -(H 2 O) 1-5 clusters (X ) Cl and Br) Margarita I. Bernal-Uruchurtu* and Ramón Hernández-Lamoneda
5496 J. Phys. Chem. A 2009, 113, 5496 5505 On the Unusual Properties of Halogen Bonds: A Detailed ab Initio Study of X 2 -(H 2 O) 1-5 clusters (X ) Cl and Br) Margarita I. Bernal-Uruchurtu* and Ramón Hernández-Lamoneda
Quantum Monte Carlo backflow calculations of benzene dimers
 Quantum Monte Carlo backflow calculations of benzene dimers Kathleen Schwarz*, Cyrus Umrigar**, Richard Hennig*** *Cornell University Department of Chemistry, **Cornell University Department of Physics,
Quantum Monte Carlo backflow calculations of benzene dimers Kathleen Schwarz*, Cyrus Umrigar**, Richard Hennig*** *Cornell University Department of Chemistry, **Cornell University Department of Physics,
Predictive Computing for Solids and Liquids
 Predictive Computing for Solids and Liquids So Hirata Department of Chemistry May 214 Blue Waters Symposium 1 Schrödinger equation for a water molecule 1-particle, 3-dimensional partial differential equation
Predictive Computing for Solids and Liquids So Hirata Department of Chemistry May 214 Blue Waters Symposium 1 Schrödinger equation for a water molecule 1-particle, 3-dimensional partial differential equation
Michael W. Mahoney Department of Physics, Yale University, New Haven, Connecticut 06520
 JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 23 15 DECEMBER 2001 Quantum, intramolecular flexibility, and polarizability effects on the reproduction of the density anomaly of liquid water by simple potential
JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 23 15 DECEMBER 2001 Quantum, intramolecular flexibility, and polarizability effects on the reproduction of the density anomaly of liquid water by simple potential
ELECTRONIC STRUCTURE AND SPECTROSCOPY IN THE GAS AND CONDENSED PHASE: METHODOLOGY AND APPLICATIONS. Vitalii Vanovschi
 ELECTRONIC STRUCTURE AND SPECTROSCOPY IN THE GAS AND CONDENSED PHASE: METHODOLOGY AND APPLICATIONS. by Vitalii Vanovschi A Dissertation Presented to the FACULTY OF THE GRADUATE SCHOOL UNIVERSITY OF SOUTHERN
ELECTRONIC STRUCTURE AND SPECTROSCOPY IN THE GAS AND CONDENSED PHASE: METHODOLOGY AND APPLICATIONS. by Vitalii Vanovschi A Dissertation Presented to the FACULTY OF THE GRADUATE SCHOOL UNIVERSITY OF SOUTHERN
Dielectric polarization of 2-pyrrolidinone molecules in benzene solution - a quantum-chemical study
 Dielectric polarization of 2-pyrrolidinone molecules in benzene solution - a quantum-chemical study L. Gorb* ), J. Jadżyn $) and K. W. Wojciechowski #) Institute of Molecular Physics, Polish Academy of
Dielectric polarization of 2-pyrrolidinone molecules in benzene solution - a quantum-chemical study L. Gorb* ), J. Jadżyn $) and K. W. Wojciechowski #) Institute of Molecular Physics, Polish Academy of
Electron Correlation Methods
 Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Reactive Empirical Force Fields
 Reactive Empirical Force Fields Jason Quenneville jasonq@lanl.gov X-1: Solid Mechanics, EOS and Materials Properties Applied Physics Division Los Alamos National Laboratory Timothy C. Germann, Los Alamos
Reactive Empirical Force Fields Jason Quenneville jasonq@lanl.gov X-1: Solid Mechanics, EOS and Materials Properties Applied Physics Division Los Alamos National Laboratory Timothy C. Germann, Los Alamos
General Physical Chemistry II
 General Physical Chemistry II Lecture 13 Aleksey Kocherzhenko October 16, 2014" Last time " The Hückel method" Ø Used to study π systems of conjugated molecules" Ø π orbitals are treated separately from
General Physical Chemistry II Lecture 13 Aleksey Kocherzhenko October 16, 2014" Last time " The Hückel method" Ø Used to study π systems of conjugated molecules" Ø π orbitals are treated separately from
Accurate van der Waals interactions from ground state electron density
 Accurate van der Waals interactions from ground state electron density Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute EXCITCM09,
Accurate van der Waals interactions from ground state electron density Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute EXCITCM09,
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Role of Charge Transfer in the Structure and Dynamics of the Hydrated Proton
 Article Role of Charge Transfer in the Structure and Dynamics of the Hydrated Proton Jessica M. J. Swanson, and Jack Simons Subscriber access provided by UNIV OF UTAH J. Phys. Chem. B, 2009, 113 (15),
Article Role of Charge Transfer in the Structure and Dynamics of the Hydrated Proton Jessica M. J. Swanson, and Jack Simons Subscriber access provided by UNIV OF UTAH J. Phys. Chem. B, 2009, 113 (15),
QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C
 Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
Chemistry 460 Fall 2017 Dr. Jean M. Standard November 6, 2017 QUANTUM CHEMISTRY PROJECT 3: PARTS B AND C PART B: POTENTIAL CURVE, SPECTROSCOPIC CONSTANTS, AND DISSOCIATION ENERGY OF DIATOMIC HYDROGEN (20
TDDFT in Chemistry and Biochemistry III
 TDDFT in Chemistry and Biochemistry III Dmitrij Rappoport Department of Chemistry and Chemical Biology Harvard University TDDFT Winter School Benasque, January 2010 Dmitrij Rappoport (Harvard U.) TDDFT
TDDFT in Chemistry and Biochemistry III Dmitrij Rappoport Department of Chemistry and Chemical Biology Harvard University TDDFT Winter School Benasque, January 2010 Dmitrij Rappoport (Harvard U.) TDDFT
Tailoring the Properties of Quadruplex Nucleobases for Biological and Nanomaterial Applications
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting Information for: Tailoring the Properties of Quadruplex Nucleobases
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2014 Supporting Information for: Tailoring the Properties of Quadruplex Nucleobases
MP2 Basis Set Limit Binding Energy Estimates of Hydrogen-bonded Complexes from Extrapolation-oriented Basis Sets
 386 Bull. Korean Chem. Soc. 2007, Vol. 28, No. 3 Young Choon Park and Jae Shin Lee MP2 Basis Set Limit Binding Energy Estimates of Hydrogen-bonded Complexes from Extrapolation-oriented Basis Sets Young
386 Bull. Korean Chem. Soc. 2007, Vol. 28, No. 3 Young Choon Park and Jae Shin Lee MP2 Basis Set Limit Binding Energy Estimates of Hydrogen-bonded Complexes from Extrapolation-oriented Basis Sets Young
Supporting Information
 Supporting Information Computational Evidence of Inversion of 1 L a and 1 L b -Derived Excited States in Naphthalene Excimer Formation from ab Initio Multireference Theory with Large Active Space: DMRG-CASPT2
Supporting Information Computational Evidence of Inversion of 1 L a and 1 L b -Derived Excited States in Naphthalene Excimer Formation from ab Initio Multireference Theory with Large Active Space: DMRG-CASPT2
计算物理作业二. Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit.
 计算物理作业二 Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit. In this exercise, basis indicates one of the following basis sets: STO-3G, cc-pvdz, cc-pvtz, cc-pvqz
计算物理作业二 Excercise 1: Illustration of the convergence of the dissociation energy for H 2 toward HF limit. In this exercise, basis indicates one of the following basis sets: STO-3G, cc-pvdz, cc-pvtz, cc-pvqz
Coupled-cluster and perturbation methods for macromolecules
 Coupled-cluster and perturbation methods for macromolecules So Hirata Quantum Theory Project and MacroCenter Departments of Chemistry & Physics, University of Florida Contents Accurate electronic structure
Coupled-cluster and perturbation methods for macromolecules So Hirata Quantum Theory Project and MacroCenter Departments of Chemistry & Physics, University of Florida Contents Accurate electronic structure
Computational Chemistry. An Introduction to Molecular Dynamic Simulations
 Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Characteristics of the interaction in azulene (H 2 X) n=1,2 (X=O,S) clusters.
 n=1,2 (X=O,S) clusters.") Characteristics of the interaction in azulene (H 2 X) n=1,2 (X=O,S) clusters. Enrique M. Cabaleiro-Lago (a), Ángeles Peña-Gallego (b), Jesús Rodríguez-Otero (b), M. Merced Montero-Campillo (b) (a) Departamento
Characteristics of the interaction in azulene (H 2 X) n=1,2 (X=O,S) clusters. Enrique M. Cabaleiro-Lago (a), Ángeles Peña-Gallego (b), Jesús Rodríguez-Otero (b), M. Merced Montero-Campillo (b) (a) Departamento
High-level Quantum Chemistry Methods and Benchmark Datasets for Molecules
 High-level Quantum Chemistry Methods and Benchmark Datasets for Molecules Markus Schneider Fritz Haber Institute of the MPS, Berlin, Germany École Polytechnique Fédérale de Lausanne, Switzerland دانشگاه
High-level Quantum Chemistry Methods and Benchmark Datasets for Molecules Markus Schneider Fritz Haber Institute of the MPS, Berlin, Germany École Polytechnique Fédérale de Lausanne, Switzerland دانشگاه
Atomic and molecular interaction forces in biology
 Atomic and molecular interaction forces in biology 1 Outline Types of interactions relevant to biology Van der Waals interactions H-bond interactions Some properties of water Hydrophobic effect 2 Types
Atomic and molecular interaction forces in biology 1 Outline Types of interactions relevant to biology Van der Waals interactions H-bond interactions Some properties of water Hydrophobic effect 2 Types
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D.
 3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
Noncovalent interactions in biochemistry
 Noncovalent interactions in biochemistry Kevin E. Riley 1 and Pavel Hobza 2,3 Noncovalent interactions are known to play a key role in biochemistry. The knowledge of stabilization (relative) energies and
Noncovalent interactions in biochemistry Kevin E. Riley 1 and Pavel Hobza 2,3 Noncovalent interactions are known to play a key role in biochemistry. The knowledge of stabilization (relative) energies and
Electric properties of molecules
 Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
AN INTRODUCTION TO QUANTUM CHEMISTRY. Mark S. Gordon Iowa State University
 AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Modeling Chemical Reactions in Aqueous Solutions
 University of Arkansas, Fayetteville ScholarWorks@UARK Theses and Dissertations 8-2013 Modeling Chemical Reactions in Aqueous Solutions Osman Uner University of Arkansas, Fayetteville Follow this and additional
University of Arkansas, Fayetteville ScholarWorks@UARK Theses and Dissertations 8-2013 Modeling Chemical Reactions in Aqueous Solutions Osman Uner University of Arkansas, Fayetteville Follow this and additional
Introduction. Claudio A. Morgado,, Petr Jurečka,, Daniel Svozil, Pavel Hobza,, and
 1524 J. Chem. Theory Comput. 2009, 5, 1524 1544 Balance of Attraction and Repulsion in Nucleic-Acid Base Stacking: CCSD(T)/Complete-Basis-Set-Limit Calculations on Uracil Dimer and a Comparison with the
1524 J. Chem. Theory Comput. 2009, 5, 1524 1544 Balance of Attraction and Repulsion in Nucleic-Acid Base Stacking: CCSD(T)/Complete-Basis-Set-Limit Calculations on Uracil Dimer and a Comparison with the
Introduction to Computational Chemistry for Experimental Chemists... (Part 2/2)
") 12 th PhD seminar, Garching, October 31 st 2008 Introduction to Computational Chemistry for Experimental Chemists... (Part 2/2) Dr. Markus Drees, TU München Universität Regensburg Universität Augsburg
12 th PhD seminar, Garching, October 31 st 2008 Introduction to Computational Chemistry for Experimental Chemists... (Part 2/2) Dr. Markus Drees, TU München Universität Regensburg Universität Augsburg
An Interpretation of the Enhancement of the Water Dipole Moment Due to the Presence of Other Water Molecules
 J. Phys. Chem. A 2008, 112, 4885 4894 4885 An Interpretation of the Enhancement of the Water Dipole Moment Due to the Presence of Other Water Molecules Daniel D. Kemp and Mark S. Gordon* Department of
J. Phys. Chem. A 2008, 112, 4885 4894 4885 An Interpretation of the Enhancement of the Water Dipole Moment Due to the Presence of Other Water Molecules Daniel D. Kemp and Mark S. Gordon* Department of
Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia
 University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
Practical Advice for Quantum Chemistry Computations. C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology
 Practical Advice for Quantum Chemistry Computations C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Choice of Basis Set STO-3G is too small 6-31G* or 6-31G** 6 probably
Practical Advice for Quantum Chemistry Computations C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Choice of Basis Set STO-3G is too small 6-31G* or 6-31G** 6 probably
A theoretical investigation of the relative stability of hydrated glycine and methylcarbamic acid from water clusters to interstellar icesw
 PCCP Dynamic Article Links Cite this: Phys. Chem. Chem. Phys., 2012, 14, 4942 4958 www.rsc.org/pccp PAPER A theoretical investigation of the relative stability of hydrated glycine and methylcarbamic acid
PCCP Dynamic Article Links Cite this: Phys. Chem. Chem. Phys., 2012, 14, 4942 4958 www.rsc.org/pccp PAPER A theoretical investigation of the relative stability of hydrated glycine and methylcarbamic acid
Chemistry Publications
 Chemistry Publications Chemistry 3-1998 An Approximate Formula for the Intermolecular Pauli Repulsion Between Closed Shell Molecules. II. Application to the Effective Fragment Potential Method Jan H. Jensen
Chemistry Publications Chemistry 3-1998 An Approximate Formula for the Intermolecular Pauli Repulsion Between Closed Shell Molecules. II. Application to the Effective Fragment Potential Method Jan H. Jensen
Why the Sulfinyl Group is special in DMSO? Chao Lv June 4, 2014
 Why the Sulfinyl Group is special in DMSO? Chao Lv June 4, 2014 The Parameterization of Dimethyl Sulfoxide (DMSO) Nucleic Acids are known to be difficult to be parameterized because: 1. The interac
Why the Sulfinyl Group is special in DMSO? Chao Lv June 4, 2014 The Parameterization of Dimethyl Sulfoxide (DMSO) Nucleic Acids are known to be difficult to be parameterized because: 1. The interac
Chemistry 334 Part 2: Computational Quantum Chemistry
 Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Computational and spectroscopic investigation of 7-azaindole: Solvation and intermolecular interactions
 Computational and spectroscopic investigation of 7-azaindole: Solvation and intermolecular interactions Michael Kamrath, Krista Cruse, Nathan Erickson, Molly Beernink Abstract We report results of an experimental
Computational and spectroscopic investigation of 7-azaindole: Solvation and intermolecular interactions Michael Kamrath, Krista Cruse, Nathan Erickson, Molly Beernink Abstract We report results of an experimental
Coupled-Cluster Perturbative Triples for Bond Breaking
 Coupled-Cluster Perturbative Triples for Bond Breaking Andrew G. Taube and Rodney J. Bartlett Quantum Theory Project University of Florida INT CC Meeting Seattle July 8, 2008 Why does chemistry need triples?
Coupled-Cluster Perturbative Triples for Bond Breaking Andrew G. Taube and Rodney J. Bartlett Quantum Theory Project University of Florida INT CC Meeting Seattle July 8, 2008 Why does chemistry need triples?
PH 548 Atomistic Simulation Techniques
 PH 548 Atomistic Simulation Techniques Lectures: Lab: 2-0-2-6 Tuesday 12-1 PM Thursday 12-1 PM Monday 2-5 PM P. K. Padmanabhan PH548: Two Excellent Books How to do it? 1 2 F 12 F 23 3 F i = F ij j i F
PH 548 Atomistic Simulation Techniques Lectures: Lab: 2-0-2-6 Tuesday 12-1 PM Thursday 12-1 PM Monday 2-5 PM P. K. Padmanabhan PH548: Two Excellent Books How to do it? 1 2 F 12 F 23 3 F i = F ij j i F
Oslo node. Highly accurate calculations benchmarking and extrapolations
 Oslo node Highly accurate calculations benchmarking and extrapolations Torgeir Ruden, with A. Halkier, P. Jørgensen, J. Olsen, W. Klopper, J. Gauss, P. Taylor Explicitly correlated methods Pål Dahle, collaboration
Oslo node Highly accurate calculations benchmarking and extrapolations Torgeir Ruden, with A. Halkier, P. Jørgensen, J. Olsen, W. Klopper, J. Gauss, P. Taylor Explicitly correlated methods Pål Dahle, collaboration
Alek Marenich Cramer -Truhlar Group. University of Minnesota
 and Computational Electrochemistry Alek Marenich Cramer -Truhlar Group University of Minnesota Outline Solvation and charge models Energetics and dynamics in solution Computational electrochemistry Future
and Computational Electrochemistry Alek Marenich Cramer -Truhlar Group University of Minnesota Outline Solvation and charge models Energetics and dynamics in solution Computational electrochemistry Future
Development and Application of Combined Quantum Mechanical and Molecular Mechanical Methods
 University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Student Research Projects, Dissertations, and Theses - Chemistry Department Chemistry, Department of Fall 12-2014 Development
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Student Research Projects, Dissertations, and Theses - Chemistry Department Chemistry, Department of Fall 12-2014 Development
New Perspective on structure and bonding in water using XAS and XRS
 New Perspective on structure and bonding in water using XAS and XRS Anders Nilsson Stanford Synchrotron Radiation Laboratory (SSRL) and Stockholm University, Sweden R. Ludwig Angew. Chem. 40, 1808 (2001)
New Perspective on structure and bonding in water using XAS and XRS Anders Nilsson Stanford Synchrotron Radiation Laboratory (SSRL) and Stockholm University, Sweden R. Ludwig Angew. Chem. 40, 1808 (2001)
Computational and Spectroscopic Investigation of Solution Phase Excited State Dynamics in 7 azaindole
 Computational and Spectroscopic Investigation of Solution Phase Excited State Dynamics in 7 azaindole Nathan Erickson, Molly Beernink, and Nathaniel Swenson Midwest Undergraduate Computational Chemistry
Computational and Spectroscopic Investigation of Solution Phase Excited State Dynamics in 7 azaindole Nathan Erickson, Molly Beernink, and Nathaniel Swenson Midwest Undergraduate Computational Chemistry
Fundamental Interactions: 6 Forces
 Fundamental Interactions: 6 Forces In nuclear and high-energy physics 6 fundamental forces are recognized, which describe the structure of matter. - the strong interaction - the weak interaction act inside
Fundamental Interactions: 6 Forces In nuclear and high-energy physics 6 fundamental forces are recognized, which describe the structure of matter. - the strong interaction - the weak interaction act inside
( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r)
Ψ el ( r;r) = E el ( R)Ψ el ( r;r)") Born Oppenheimer Approximation: Ĥ el ( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r) For a molecule with N electrons and M nuclei: Ĥ el What is E el (R)? s* potential surface Reaction Barrier Unstable intermediate
Born Oppenheimer Approximation: Ĥ el ( R)Ψ el ( r;r) = E el ( R)Ψ el ( r;r) For a molecule with N electrons and M nuclei: Ĥ el What is E el (R)? s* potential surface Reaction Barrier Unstable intermediate
Methods for van der Waals Interactions
 Methods for van der Waals Interactions Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute FHI DFT and Beyond Workshop, Jul.
Methods for van der Waals Interactions Alexandre Tkatchenko Theory Department, Fritz Haber Institut der MPG Berlin, Germany tkatchen@fhi berlin.mpg.de Haber Institute FHI DFT and Beyond Workshop, Jul.
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
Intermolecular Forces & Condensed Phases
 Intermolecular Forces & Condensed Phases CHEM 107 T. Hughbanks READING We will discuss some of Chapter 5 that we skipped earlier (Van der Waals equation, pp. 145-8), but this is just a segue into intermolecular
Intermolecular Forces & Condensed Phases CHEM 107 T. Hughbanks READING We will discuss some of Chapter 5 that we skipped earlier (Van der Waals equation, pp. 145-8), but this is just a segue into intermolecular
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
Valence electronic structure of isopropyl iodide investigated by electron momentum spectroscopy. --- Influence of intramolecular interactions
 Valence electronic structure of isopropyl iodide investigated by electron momentum spectroscopy --- Influence of intramolecular interactions Minfu Zhao, Xu Shan, Shanshan Niu, Yaguo Tang, Zhaohui Liu,
Valence electronic structure of isopropyl iodide investigated by electron momentum spectroscopy --- Influence of intramolecular interactions Minfu Zhao, Xu Shan, Shanshan Niu, Yaguo Tang, Zhaohui Liu,
Accurate Description of Intermolecular Interactions Involving Ions Using Symmetry-Adapted Perturbation Theory
 pubs.acs.org/jctc Accurate Description of Intermolecular Interactions Involving Ions Using Symmetry-Adapted Perturbation Theory Ka Un Lao, Rainer Scha ffer, Georg Jansen,*, and John M. Herbert*, Department
pubs.acs.org/jctc Accurate Description of Intermolecular Interactions Involving Ions Using Symmetry-Adapted Perturbation Theory Ka Un Lao, Rainer Scha ffer, Georg Jansen,*, and John M. Herbert*, Department
The Effective Fragment Potential: A General Method for Predicting Intermolecular Interactions
 CHAPTER 10 The Effective Fragment Potential: A General Method for Predicting Intermolecular Interactions Mark S. Gordon *, Lyudmilla Slipchenko *,HuiLi ** and Jan H. Jensen *** Contents 1. Introduction
CHAPTER 10 The Effective Fragment Potential: A General Method for Predicting Intermolecular Interactions Mark S. Gordon *, Lyudmilla Slipchenko *,HuiLi ** and Jan H. Jensen *** Contents 1. Introduction
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,