Intro to ab initio methods
|
|
|
- Jane Briggs
- 6 years ago
- Views:
Transcription
1 Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1
2 ab initio ab initio - from the beginning The concise Oxford Dictionary, Oxford University Press, 2001 ab initio calculation - A method of calculating atomic and molecular structure directly from the first principles of quantum mechanics, without using quantities derived from experiment (such as ionization energies found by spectroscopy) as parameters. A Dictionary of Chemistry, Oxford University Press, 2000 The most chemically accurate, physically precise computation possible. The holy grail of computational chemists. 2
3 Fundamentals The postulates and theorems of quantum mechanics form the rigorous foundation for the prediction of observable chemical and physical properties of matter from first principles. Any model of a material and its behavior, regardless of its source, must ultimately find its basis in quantum mechanics. All models of materials include QM either explicitly or implicitly. 3
4 Fundamentals Matter is made of atoms, which are composed of electrons, protons, and neutrons. Over 75 years ago the laws of QM as formulated by Schrodinger, Dirac and others made it theoretically possible to understand and calculate how electrons and atomic nuclei interact to form matter. However, the solution of the governing equations of QM is too difficult to solve exactly for anything but the simplest of systems (like the hydrogen atom). 4
5 QM Methods Over the past 10 years, ground-breaking advances in the development of QM techniques now allow QM calculations on molecular systems of real, practical interest. Strength of carbon nanotubes Optical spectra of quantum dots Interaction of biological molecules (docking) with surfaces, other bio-molecules, etc. Energy of nanostructures on surfaces Molecular structure and reactivity of complex molecules (e.g. buckyballs and related carbonaceous molecules) Bond strengths, angles for macromolecules. Fracture of inorganic matter. 5
6 QM Methods Doing a QM simulation or calculation means including the electrons explicitly. With QM methods, we can calculate properties that depend upon the electronic distribution, and to study processes like chemical reactions in which bonds are formed and broken. The explicit consideration of electrons distinguishes QM models and methods from classical force field models and methods. 6
7 Different QM Methods Several approaches exist. The two main ones are: Molecular orbital theory Came from chemistry, since primarily developed for individual molecules, gases and now liquids. Two flavors Ab initio: all electrons included (considered exact) Semi-empirical: only valence electrons included Density functional theory Came from physics and materials science community, since originally conceived for solids. All electrons included via electronic density (considered exact). 7
8 Fundamentals The fundamental postulates of QM assert that microscopic systems are describable by wave functions that completely characterize all of the physical properties of the system. A wavefunction squared is a probability density. There are QM operators corresponding to every physical observable that, when applied to a wave function, allow the prediction of the probability of finding the system to exhibit a particular value or range of values for that observable. x Ψ = xψ Eigenvalue eqn for position. 8
9 Fundamentals The operator that returns the system energy is called the Hamiltonian operator H. Eigenvalue equation for system energy HΨ = EΨ Time-independent Schrodinger Equation H = " $ i h 2 # 2 i " 2m e Kinetic energy of electrons $ k h 2 # 2 k " 2m k Kinetic energy of nuclei $ i $ k Potential energy of electrons & nuclei e 2 Z k r ik + $ i< j e 2 r ij + Potential energy of electrons $ k<l e 2 Z k Z l r kl Potential energy of nuclei 9
10 Born-0ppenheimer Approximation Need to simplify! Neutrons & protons are >1800 times more massive than electrons, and therefore move much more slowly. Thus, electronic relaxation is for all practical purposes instantaneous with respect to nuclear motion. We can decouple the motion, and consider the electronelectron interactions independently of the nuclear interactions. This is the Born-Oppenheimer approximation. For nearly all situations relevant to soft matter, this assumption is entirely justified. (H el +V n )Ψ el (q i ;q k ) = E el Ψ el (q i ;q k ) The Electronic Schrodinger Equation 10
11 QM Methods Goal of all QM methods in use today: Solve the electronic Schrodinger equation for the ground state energy of a system and the wavefunction that describes the positions of all the electrons. The energy is calculated for a given trial wavefunction, and the best wavefunction is found as that wavefunction that minimizes the energy. 11
12 QM Methods Solving SE is not so easy! Anything containing more than two elementary particles (i.e. one e- and one nucleon) can t be solved exactly: the many-body problem. Even after invoking Born-Oppenheimer, still can t solve exactly for anything containing more than two electrons. So -- all QM methods used today are APPROXIMATE after all, even if considered exact! That is, they provide approximate solutions to the Schrodinger equation. Some are more approximate than others. 12
13 Molecular Orbital Theory MOT is expressed in terms of molecular wave functions called molecular orbitals. Most popular implementation: write molecular orbital as a linear combination of atomic orbitals φ (LCAO): Eq in Leach K $ µ=1 " i = a µi # µ K = # atomic orbitals Many different ways of writing basis set, which leads to many different methods and implementations of MOT. 13
14 Molecular Orbital Theory Dozens of approaches for writing basis sets (e.g. in terms of Gaussian wavefunctions, or as linear combos of Gaussians). Different implementations retain different numbers of terms. Semi-empirical MOT methods consider only valence electrons. Some methods include electron exchange. Some methods include electron correlation. 14
15 Density Functional Theory A different approach for solving Schrodinger s equation for the ground state energies of matter. Based on theory of Hohenberg and Kohn (1964) which states that it is not necessary to consider the motion of each individual electron in the system. Instead, it suffices to know the average number of electrons at any one point in space. The HK theorem enables us to write E el as a functional of the electron density ρ. To perform a DFT calculation, one optimizes the energy with respect to the electron probability density, rather than with respect to the electronic wave function. For a given density, the lowest energy is the best one. 15
16 Density Functional Theory In the commonly used Kohn-Sham implementation, the density is written in terms of one-electron molecular orbitals called Kohn-Sham orbitals. This allows the energy to be optimized by solving a set of one-electron Schrodinger equations (the KS equations), but with electron correlation included. This is a key advantage of the DFT method - it s easier to include electron correlation. 16
17 Density Functional Theory In DFT calculations, the MO s are written as linear combinations of atomic orbitals (LCAO) or basis functions which can be represented using Gaussian functions, plane waves, etc. N $ i=1 " = a i # i 17
18 Density Functional Theory Different choices of basis sets, how many terms to use, of what type, contribute to difficulty of calculation. More than a few hundred light atoms is still too time-consuming, even on big computers. For molecules or systems with large numbers of electrons, pseudopotentials are used to represent the wavefunctions of valence electrons, and the core is treated in a simplified way. This is a basic introductory summary of the DFT method. 18
19 Implementing MOT & DFT in Computer Code John Pople at Northwestern is a pioneer in computational quantum chemistry. In 1970 Pople developed Gaussian, a quantum chemistry code that solves approximations of the SE for molecules. In 1990, he included DFT in Gaussian. This brought state-of-the-art QM computational methods to the masses. Ab initio and semi-empirical methods (especially MOT methods) have revolutionized the Pharmaceuticals Industry, and are now playing a major role in materials R&D. 19


20 1998 Nobel Prize in Chemistry Walter Kohn "for his development of the density-functional theory" John Pople "for his development of computational methods in quantum chemistry" 20
21 QM Codes for Materials Research Commonly used codes: Gaussian: ab initio MOT and DFT Available in Cerius 2 from Accelrys NWChem: ab initio MOT and DFT 21
22 DFT Codes for Materials Research DFT codes used in materials research: VASP - Vienna ab initio simulation package Siesta - from Spain Abinit Gaussian Castep - Cambridge sequential total energy package DMol 3 22


23 Applications of ab initio computations using DMol 3 23


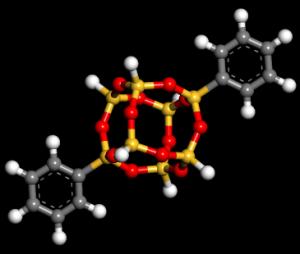
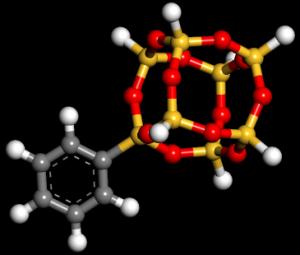
24 Electronic band structure of POSS cubes functionalized with n benzene molecules (n = 0-8) Energy (ev) 24



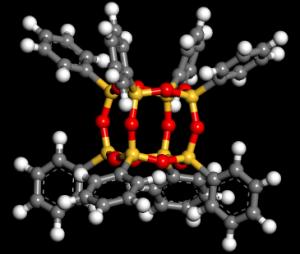
25 Electronic band structure of POSS cubes functionalized with acene molecules (benzene, naphtalene, anthracene, tetracene, and pentacene) Energy (ev) Pure poss 25

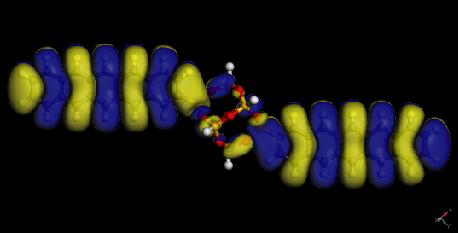
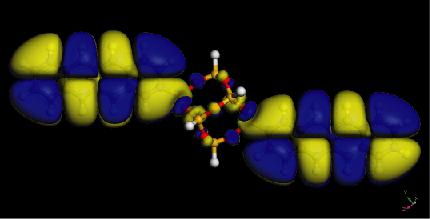
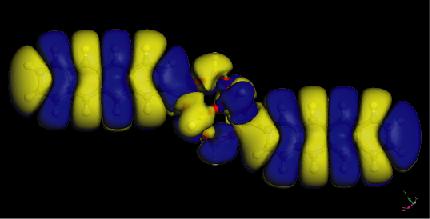
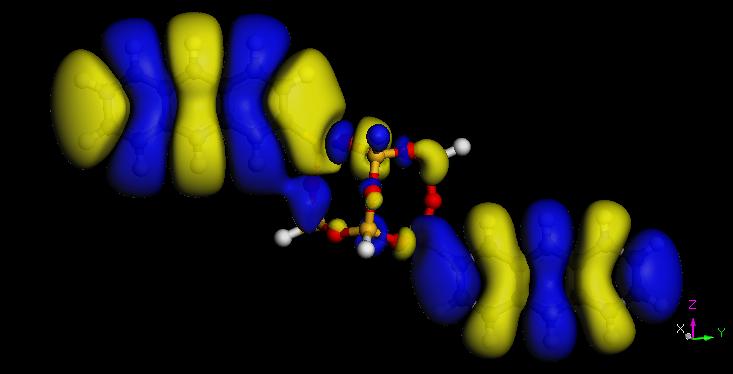

26 Electron densities of acene-functionalized POSS Molecule Band Gap (ev) Pure Acene (ev) P-POSS T-POSS A-POSS N-POSS B-POSS POSS HOMO LUMO 26


27 Electron densities of silica nanotubes HOMO LUMO 27
28 Future developments of ab initio methods Used for calculating quantities like reaction rates, bond strengths and angles, heats of formation, solubility, etc.. Any properties that depend critically on electron distribution. Lots of activity in MOT and DFT methods. Order N methods: The Holy Grail Ab initio - computational effort now scales with the number of electrons to a power n<4. Semi-empirical - with only valence electrons, can get order N scaling? 28
29 Too slow! Even with all of the advances of the past 10 years in ab initio methods ignoring some of the electrons (pseudopotentials) implementation of ab initio codes on parallel machines it is still not possible to use solely QM methods to simulate systems that contain more than a few thousand atoms (a chunk of matter containing less than 1 nm on a side), or for more than a picosecond for a very small number of atoms. 29
30 Still too slow! Ten years from now we may gain an order of magnitude in what can be simulated, but this is still not sufficient for many problems in soft matter. Assembly (especially if hierarchical) Mechanical properties of composites Rheology Development of structure on length scales of 10 to 100 s of nanometers. Enter classical force fields. 30
31 Classical vs. ab initio methods Classical No electronic properties ab initio Electronic details included Phenomenological potential energy surface (typically 2-body contributions) Difficult to describe bond breaking/formation Can do up to a billion particles Potential energy surface calculated directly from Schrodinger equation (many body terms included automatically) Describes bond breaking/formation Limited to several hundred atoms with significant dynamics 31
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Additional background material on the Nobel Prize in Chemistry 1998
 Additional background material on the Nobel Prize in Chemistry 1998 The Royal Swedish Academy of Sciences has decided to award the 1998 Nobel Prize in Chemistry with one half to Professor WALTER KOHN,
Additional background material on the Nobel Prize in Chemistry 1998 The Royal Swedish Academy of Sciences has decided to award the 1998 Nobel Prize in Chemistry with one half to Professor WALTER KOHN,
Chem 3502/4502 Physical Chemistry II (Quantum Mechanics) 3 Credits Spring Semester 2006 Christopher J. Cramer. Lecture 17, March 1, 2006
 3 Credits Spring Semester 2006 Christopher J. Cramer. Lecture 17, March 1, 2006") Chem 3502/4502 Physical Chemistry II (Quantum Mechanics) 3 Credits Spring Semester 2006 Christopher J. Cramer Lecture 17, March 1, 2006 (Some material in this lecture has been adapted from Cramer, C. J.
Chem 3502/4502 Physical Chemistry II (Quantum Mechanics) 3 Credits Spring Semester 2006 Christopher J. Cramer Lecture 17, March 1, 2006 (Some material in this lecture has been adapted from Cramer, C. J.
GEM4 Summer School OpenCourseWare
 GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
Introduction to Hartree-Fock Molecular Orbital Theory
 Introduction to Hartree-Fock Molecular Orbital Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Origins of Mathematical Modeling in Chemistry Plato (ca. 428-347
Introduction to Hartree-Fock Molecular Orbital Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Origins of Mathematical Modeling in Chemistry Plato (ca. 428-347
Density Functional Theory (DFT)
") Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
Density Functional Theory (DFT) An Introduction by A.I. Al-Sharif Irbid, Aug, 2 nd, 2009 Density Functional Theory Revolutionized our approach to the electronic structure of atoms, molecules and solid
The electronic structure of materials 2 - DFT
 Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
Quantum mechanics 2 - Lecture 9 December 19, 2012 1 Density functional theory (DFT) 2 Literature Contents 1 Density functional theory (DFT) 2 Literature Historical background The beginnings: L. de Broglie
Set the initial conditions r i. Update neighborlist. Get new forces F i
 v Set the initial conditions r i ( t 0 ), v i ( t 0 ) Update neighborlist Quantum mechanical models Get new forces F i ( r i ) Solve the equations of motion numerically over time step Δt : r i ( t n )
v Set the initial conditions r i ( t 0 ), v i ( t 0 ) Update neighborlist Quantum mechanical models Get new forces F i ( r i ) Solve the equations of motion numerically over time step Δt : r i ( t n )
Lecture 10. Born-Oppenheimer approximation LCAO-MO application to H + The potential energy surface MOs for diatomic molecules. NC State University
 Chemistry 431 Lecture 10 Diatomic molecules Born-Oppenheimer approximation LCAO-MO application to H + 2 The potential energy surface MOs for diatomic molecules NC State University Born-Oppenheimer approximation
Chemistry 431 Lecture 10 Diatomic molecules Born-Oppenheimer approximation LCAO-MO application to H + 2 The potential energy surface MOs for diatomic molecules NC State University Born-Oppenheimer approximation
Electron Correlation
 Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
Quantum Chemistry. NC State University. Lecture 5. The electronic structure of molecules Absorption spectroscopy Fluorescence spectroscopy
 Quantum Chemistry Lecture 5 The electronic structure of molecules Absorption spectroscopy Fluorescence spectroscopy NC State University 3.5 Selective absorption and emission by atmospheric gases (source:
Quantum Chemistry Lecture 5 The electronic structure of molecules Absorption spectroscopy Fluorescence spectroscopy NC State University 3.5 Selective absorption and emission by atmospheric gases (source:
Density Functional Theory
 Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard October 28, 2015 Density Functional Theory What is a Functional? A functional is a general mathematical quantity that represents a rule to convert a function
(Refer Slide Time: 5:21 min)
") Engineering Chemistry - 1 Prof. K. Mangala Sunder Department of Chemistry Indian Institute of Technology, Madras Module 1: Atoms and Molecules Lecture - 10 Model Problems in Quantum Chemistry Born-Oppenheimer
Engineering Chemistry - 1 Prof. K. Mangala Sunder Department of Chemistry Indian Institute of Technology, Madras Module 1: Atoms and Molecules Lecture - 10 Model Problems in Quantum Chemistry Born-Oppenheimer
Molecular Mechanics: The Ab Initio Foundation
 Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry room B430, Chemicum 4th floor vesa.hanninen@helsinki.fi September 3, 2013 Introduction and theoretical backround September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry room B430, Chemicum 4th floor vesa.hanninen@helsinki.fi September 3, 2013 Introduction and theoretical backround September
Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering
 Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering Outline PART 1: Fundamentals of Density functional theory (DFT)
Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering Outline PART 1: Fundamentals of Density functional theory (DFT)
Computational Chemistry. An Introduction to Molecular Dynamic Simulations
 Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
CHEM 545 Theory and Practice of Molecular Electronic Structure. Anna I. Krylov. DON T PANIC.
 CHEM 545 Theory and Practice of Molecular Electronic Structure Anna I. Krylov http://iopenshell.usc.edu/chem545/ DON T PANIC USC Fall 2014 Things to do: 1. Install IQmol (by this Thursday). http://iqmol.org/.
CHEM 545 Theory and Practice of Molecular Electronic Structure Anna I. Krylov http://iopenshell.usc.edu/chem545/ DON T PANIC USC Fall 2014 Things to do: 1. Install IQmol (by this Thursday). http://iqmol.org/.
Lecture 8: Introduction to Density Functional Theory
 Lecture 8: Introduction to Density Functional Theory Marie Curie Tutorial Series: Modeling Biomolecules December 6-11, 2004 Mark Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Science
Lecture 8: Introduction to Density Functional Theory Marie Curie Tutorial Series: Modeling Biomolecules December 6-11, 2004 Mark Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Science
Modeling & Simulation of Glass Structure
 Modeling & Simulation of Glass Structure VCG Lecture 21 John Kieffer Department of Materials Science and Engineering University of Michigan 1 Overview Historical perspective Simulation methodologies Theoretical
Modeling & Simulation of Glass Structure VCG Lecture 21 John Kieffer Department of Materials Science and Engineering University of Michigan 1 Overview Historical perspective Simulation methodologies Theoretical
Density Functional Theory
 Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Quantum Chemical Simulations and Descriptors. Dr. Antonio Chana, Dr. Mosè Casalegno
 Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Electronic structure, plane waves and pseudopotentials
 Electronic structure, plane waves and pseudopotentials P.J. Hasnip Spectroscopy Workshop 2009 We want to be able to predict what electrons and nuclei will do from first principles, without needing to know
Electronic structure, plane waves and pseudopotentials P.J. Hasnip Spectroscopy Workshop 2009 We want to be able to predict what electrons and nuclei will do from first principles, without needing to know
Chemistry 334 Part 2: Computational Quantum Chemistry
 Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Introduction to first-principles modelling and CASTEP
 to first-principles modelling and Phil Hasnip to + Atomistic Simulations If we know what the bonding in a material is beforehand, then we can often find good expressions for the forces between atoms, e.g.
to first-principles modelling and Phil Hasnip to + Atomistic Simulations If we know what the bonding in a material is beforehand, then we can often find good expressions for the forces between atoms, e.g.
CHEM3023: Spins, Atoms and Molecules
 CHEM3023: Spins, Atoms and Molecules CHEM3006P or similar background knowledge is required for this course. This course has two parts: Part 1: Quantum Chemistry techniques for simulations of molecular
CHEM3023: Spins, Atoms and Molecules CHEM3006P or similar background knowledge is required for this course. This course has two parts: Part 1: Quantum Chemistry techniques for simulations of molecular
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
Introduction to Density Functional Theory
 Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Born-Oppenheimer Approximation
 Born-Oppenheimer Approximation Adiabatic Assumption: Nuclei move so much more slowly than electron that the electrons that the electrons are assumed to be obtained if the nuclear kinetic energy is ignored,
Born-Oppenheimer Approximation Adiabatic Assumption: Nuclei move so much more slowly than electron that the electrons that the electrons are assumed to be obtained if the nuclear kinetic energy is ignored,
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory.
 Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Electronic structure calculations: fundamentals George C. Schatz Northwestern University
 Electronic structure calculations: fundamentals George C. Schatz Northwestern University Electronic Structure (often called Quantum Chemistry) calculations use quantum mechanics to determine the wavefunctions
Electronic structure calculations: fundamentals George C. Schatz Northwestern University Electronic Structure (often called Quantum Chemistry) calculations use quantum mechanics to determine the wavefunctions
Density Functional Theory
 Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Electrochemistry project, Chemistry Department, November Ab-initio Molecular Dynamics Simulation
 Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Key concepts in Density Functional Theory (I) Silvana Botti
 Silvana Botti") From the many body problem to the Kohn-Sham scheme European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address: Centre
From the many body problem to the Kohn-Sham scheme European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address: Centre
Introduction to Density Functional Theory (DFT)
") Introduction to Density Functional Theory (DFT) Brad Malone, Sadas Shankar The Problem: What's the big deal? Materials we are often interested in contain a macroscopically large number of particles (~1023
Introduction to Density Functional Theory (DFT) Brad Malone, Sadas Shankar The Problem: What's the big deal? Materials we are often interested in contain a macroscopically large number of particles (~1023
Algorithms and Computational Aspects of DFT Calculations
 Algorithms and Computational Aspects of DFT Calculations Part I Juan Meza and Chao Yang High Performance Computing Research Lawrence Berkeley National Laboratory IMA Tutorial Mathematical and Computational
Algorithms and Computational Aspects of DFT Calculations Part I Juan Meza and Chao Yang High Performance Computing Research Lawrence Berkeley National Laboratory IMA Tutorial Mathematical and Computational
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
CHEMISTRY Topic #1: Bonding What Holds Atoms Together? Spring 2012 Dr. Susan Lait
 CHEMISTRY 2000 Topic #1: Bonding What Holds Atoms Together? Spring 2012 Dr. Susan Lait Why Do Bonds Form? An energy diagram shows that a bond forms between two atoms if the overall energy of the system
CHEMISTRY 2000 Topic #1: Bonding What Holds Atoms Together? Spring 2012 Dr. Susan Lait Why Do Bonds Form? An energy diagram shows that a bond forms between two atoms if the overall energy of the system
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
Multi-Scale Modeling from First Principles
 m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
Introduction and theoretical background
 1 Introduction and theoretical background 1.1 The Schrödinger equation and models of chemistry The Schrödinger equation and its elements As early as 1929, the noted physicist P. A. M. Dirac wrote 1 The
1 Introduction and theoretical background 1.1 The Schrödinger equation and models of chemistry The Schrödinger equation and its elements As early as 1929, the noted physicist P. A. M. Dirac wrote 1 The
This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry.
 1 Computational Chemistry (Quantum Chemistry) Primer This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry. TABLE OF CONTENTS Methods...1 Basis
1 Computational Chemistry (Quantum Chemistry) Primer This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry. TABLE OF CONTENTS Methods...1 Basis
Key concepts in Density Functional Theory
 From the many body problem to the Kohn-Sham scheme ILM (LPMCN) CNRS, Université Lyon 1 - France European Theoretical Spectroscopy Facility (ETSF) December 12, 2012 Lyon Outline 1 The many-body problem
From the many body problem to the Kohn-Sham scheme ILM (LPMCN) CNRS, Université Lyon 1 - France European Theoretical Spectroscopy Facility (ETSF) December 12, 2012 Lyon Outline 1 The many-body problem
Computational Modeling of Protein-Ligand Interactions
 Computational Modeling of Protein-Ligand Interactions Steven R. Gwaltney Department of Chemistry Mississippi State University Mississippi State, MS 39762 Auguste Comte, 1830 Every attempt to refer chemical
Computational Modeling of Protein-Ligand Interactions Steven R. Gwaltney Department of Chemistry Mississippi State University Mississippi State, MS 39762 Auguste Comte, 1830 Every attempt to refer chemical
Lecture 19: Building Atoms and Molecules
 Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r y even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r y even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
Solid State Theory: Band Structure Methods
 Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:00-12:30 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Who am I? Assistant Professor, Institute for Theoretical and Computational Physics,
Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:00-12:30 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Who am I? Assistant Professor, Institute for Theoretical and Computational Physics,
Electron bands in crystals Pseudopotentials, Plane Waves, Local Orbitals
 Electron bands in crystals Pseudopotentials, Plane Waves, Local Orbitals Richard M. Martin UIUC Lecture at Summer School Hands-on introduction to Electronic Structure Materials Computation Center University
Electron bands in crystals Pseudopotentials, Plane Waves, Local Orbitals Richard M. Martin UIUC Lecture at Summer School Hands-on introduction to Electronic Structure Materials Computation Center University
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić
 Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Introduction to density-functional theory. Emmanuel Fromager
 Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Institut
Institut de Chimie, Strasbourg, France Page 1 Emmanuel Fromager Institut de Chimie de Strasbourg - Laboratoire de Chimie Quantique - Université de Strasbourg /CNRS M2 lecture, Strasbourg, France. Institut
Introduction to Computational Chemistry Computational (chemistry education) and/or. (Computational chemistry) education
 and/or. (Computational chemistry) education") Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools to help increase student understanding of material
Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools to help increase student understanding of material
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC
 Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC Sergey V. Churakov sergey.churakov@psi.ch Paul Scherrer Institute Switzerland Cement Phase Composition C-S-H H Solid Solution Model
Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC Sergey V. Churakov sergey.churakov@psi.ch Paul Scherrer Institute Switzerland Cement Phase Composition C-S-H H Solid Solution Model
Lecture 4: Band theory
 Lecture 4: Band theory Very short introduction to modern computational solid state chemistry Band theory of solids Molecules vs. solids Band structures Analysis of chemical bonding in Reciprocal space
Lecture 4: Band theory Very short introduction to modern computational solid state chemistry Band theory of solids Molecules vs. solids Band structures Analysis of chemical bonding in Reciprocal space
Hartree-Fock Theory Variational Principle (Rayleigh-Ritz method)
") Hartree-Fock Theory Variational Principle (Rayleigh-Ritz method) (note) (note) Schrodinger equation: Example: find an approximate solution for AHV Trial wave function: (note) b opt Mean-Field Approximation
Hartree-Fock Theory Variational Principle (Rayleigh-Ritz method) (note) (note) Schrodinger equation: Example: find an approximate solution for AHV Trial wave function: (note) b opt Mean-Field Approximation
MSE8210 Advanced Topics in Theoretical Surface and Interface Science
 MSE8210 Advanced Topics in Theoretical Surface and Interface Science Aloysius Soon 알로이시우스손 aloysius.soon@yonsei.ac.kr Course outline An introduction to fundamental concepts in theoretical surface science
MSE8210 Advanced Topics in Theoretical Surface and Interface Science Aloysius Soon 알로이시우스손 aloysius.soon@yonsei.ac.kr Course outline An introduction to fundamental concepts in theoretical surface science
Key concepts in Density Functional Theory (II)
") Kohn-Sham scheme and band structures European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Present Address: LPMCN Université
Kohn-Sham scheme and band structures European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Present Address: LPMCN Université
Simulation Methods II
 Simulation Methods II Maria Fyta Institute for Computational Physics Universität Stuttgart Summer Term 2018 SM II - contents First principles methods Hartree-Fock and beyond Density-funtional-theory Ab
Simulation Methods II Maria Fyta Institute for Computational Physics Universität Stuttgart Summer Term 2018 SM II - contents First principles methods Hartree-Fock and beyond Density-funtional-theory Ab
Computational Chemistry I
 Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Same idea for polyatomics, keep track of identical atom e.g. NH 3 consider only valence electrons F(2s,2p) H(1s)
 H(1s)") XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
XIII 63 Polyatomic bonding -09 -mod, Notes (13) Engel 16-17 Balance: nuclear repulsion, positive e-n attraction, neg. united atom AO ε i applies to all bonding, just more nuclei repulsion biggest at low
Time-dependent density functional theory (TDDFT)
") Advanced Workshop on High-Performance & High-Throughput Materials Simulations using Quantum ESPRESSO ICTP, Trieste, Italy, January 16 to 27, 2017 Time-dependent density functional theory (TDDFT) Ralph
Advanced Workshop on High-Performance & High-Throughput Materials Simulations using Quantum ESPRESSO ICTP, Trieste, Italy, January 16 to 27, 2017 Time-dependent density functional theory (TDDFT) Ralph
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Introduction to Electronic Structure Theory
 Introduction to Electronic Structure Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology June 2002 Last Revised: June 2003 1 Introduction The purpose of these
Introduction to Electronic Structure Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology June 2002 Last Revised: June 2003 1 Introduction The purpose of these
Quantum Mechanical Simulations
 Quantum Mechanical Simulations Prof. Yan Wang Woodruff School of Mechanical Engineering Georgia Institute of Technology Atlanta, GA 30332, U.S.A. yan.wang@me.gatech.edu Topics Quantum Monte Carlo Hartree-Fock
Quantum Mechanical Simulations Prof. Yan Wang Woodruff School of Mechanical Engineering Georgia Institute of Technology Atlanta, GA 30332, U.S.A. yan.wang@me.gatech.edu Topics Quantum Monte Carlo Hartree-Fock
Chemistry 483 Lecture Topics Fall 2009
 Chemistry 483 Lecture Topics Fall 2009 Text PHYSICAL CHEMISTRY A Molecular Approach McQuarrie and Simon A. Background (M&S,Chapter 1) Blackbody Radiation Photoelectric effect DeBroglie Wavelength Atomic
Chemistry 483 Lecture Topics Fall 2009 Text PHYSICAL CHEMISTRY A Molecular Approach McQuarrie and Simon A. Background (M&S,Chapter 1) Blackbody Radiation Photoelectric effect DeBroglie Wavelength Atomic
VALENCE Hilary Term 2018
 VALENCE Hilary Term 2018 8 Lectures Prof M. Brouard Valence is the theory of the chemical bond Outline plan 1. The Born-Oppenheimer approximation 2. Bonding in H + 2 the LCAO approximation 3. Many electron
VALENCE Hilary Term 2018 8 Lectures Prof M. Brouard Valence is the theory of the chemical bond Outline plan 1. The Born-Oppenheimer approximation 2. Bonding in H + 2 the LCAO approximation 3. Many electron
Introduction to DFT and Density Functionals. by Michel Côté Université de Montréal Département de physique
 Introduction to DFT and Density Functionals by Michel Côté Université de Montréal Département de physique Eamples Carbazole molecule Inside of diamant Réf: Jean-François Brière http://www.phys.umontreal.ca/~michel_
Introduction to DFT and Density Functionals by Michel Côté Université de Montréal Département de physique Eamples Carbazole molecule Inside of diamant Réf: Jean-François Brière http://www.phys.umontreal.ca/~michel_
Lecture 19: Building Atoms and Molecules
 Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r ψ even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
Lecture 19: Building Atoms and Molecules +e r n = 3 n = 2 n = 1 +e +e r ψ even Lecture 19, p 1 Today Nuclear Magnetic Resonance Using RF photons to drive transitions between nuclear spin orientations in
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 19122
 CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
CLIMBING THE LADDER OF DENSITY FUNCTIONAL APPROXIMATIONS JOHN P. PERDEW DEPARTMENT OF PHYSICS TEMPLE UNIVERSITY PHILADELPHIA, PA 191 THANKS TO MANY COLLABORATORS, INCLUDING SY VOSKO DAVID LANGRETH ALEX
The successful wavefunction can be written as a determinant: # 1 (2) # 2 (2) Electrons. This can be generalized to our 2N-electron wavefunction:
 # 2 (2) Electrons. This can be generalized to our 2N-electron wavefunction:") T2. CNDO to AM1: The Semiempirical Molecular Orbital Models The discussion in sections T2.1 T2.3 applies also to ab initio molecular orbital calculations. T2.1 Slater Determinants Consider the general
T2. CNDO to AM1: The Semiempirical Molecular Orbital Models The discussion in sections T2.1 T2.3 applies also to ab initio molecular orbital calculations. T2.1 Slater Determinants Consider the general
Chemistry 881 Lecture Topics Fall 2001
 Chemistry 881 Lecture Topics Fall 2001 Texts PHYSICAL CHEMISTRY A Molecular Approach McQuarrie and Simon MATHEMATICS for PHYSICAL CHEMISTRY, Mortimer i. Mathematics Review (M, Chapters 1,2,3 & 4; M&S,
Chemistry 881 Lecture Topics Fall 2001 Texts PHYSICAL CHEMISTRY A Molecular Approach McQuarrie and Simon MATHEMATICS for PHYSICAL CHEMISTRY, Mortimer i. Mathematics Review (M, Chapters 1,2,3 & 4; M&S,
5.61 Physical Chemistry Exam III 11/29/12. MASSACHUSETTS INSTITUTE OF TECHNOLOGY Department of Chemistry Chemistry Physical Chemistry.
 MASSACHUSETTS INSTITUTE OF TECHNOLOGY Department of Chemistry Chemistry - 5.61 Physical Chemistry Exam III (1) PRINT your name on the cover page. (2) It is suggested that you READ THE ENTIRE EXAM before
MASSACHUSETTS INSTITUTE OF TECHNOLOGY Department of Chemistry Chemistry - 5.61 Physical Chemistry Exam III (1) PRINT your name on the cover page. (2) It is suggested that you READ THE ENTIRE EXAM before
I. CSFs Are Used to Express the Full N-Electron Wavefunction
 Chapter 11 One Must be Able to Evaluate the Matrix Elements Among Properly Symmetry Adapted N- Electron Configuration Functions for Any Operator, the Electronic Hamiltonian in Particular. The Slater-Condon
Chapter 11 One Must be Able to Evaluate the Matrix Elements Among Properly Symmetry Adapted N- Electron Configuration Functions for Any Operator, the Electronic Hamiltonian in Particular. The Slater-Condon
Time-dependent density functional theory (TDDFT)
") 04/05/16 Hands-on workshop and Humboldt-Kolleg: Density-Functional Theory and Beyond - Basic Principles and Modern Insights Isfahan University of Technology, Isfahan, Iran, May 2 to 13, 2016 Time-dependent
04/05/16 Hands-on workshop and Humboldt-Kolleg: Density-Functional Theory and Beyond - Basic Principles and Modern Insights Isfahan University of Technology, Isfahan, Iran, May 2 to 13, 2016 Time-dependent
Introduction to Computational Quantum Chemistry: Theory
 Introduction to Computational Quantum Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC 3108 Course Lectures 2007 Introduction Hartree Fock Theory Configuration Interaction Lectures 1 Introduction
Introduction to Computational Quantum Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC 3108 Course Lectures 2007 Introduction Hartree Fock Theory Configuration Interaction Lectures 1 Introduction
Chemistry 2000 Lecture 1: Introduction to the molecular orbital theory
 Chemistry 2000 Lecture 1: Introduction to the molecular orbital theory Marc R. Roussel January 5, 2018 Marc R. Roussel Introduction to molecular orbitals January 5, 2018 1 / 24 Review: quantum mechanics
Chemistry 2000 Lecture 1: Introduction to the molecular orbital theory Marc R. Roussel January 5, 2018 Marc R. Roussel Introduction to molecular orbitals January 5, 2018 1 / 24 Review: quantum mechanics
7.1 Variational Principle
 7.1 Variational Principle Suppose that you want to determine the ground-state energy E g for a system described by H, but you are unable to solve the time-independent Schrödinger equation. It is possible
7.1 Variational Principle Suppose that you want to determine the ground-state energy E g for a system described by H, but you are unable to solve the time-independent Schrödinger equation. It is possible
QUANTUM MECHANICS AND MOLECULAR STRUCTURE
 6 QUANTUM MECHANICS AND MOLECULAR STRUCTURE 6.1 Quantum Picture of the Chemical Bond 6.2 Exact Molecular Orbital for the Simplest Molecule: H + 2 6.3 Molecular Orbital Theory and the Linear Combination
6 QUANTUM MECHANICS AND MOLECULAR STRUCTURE 6.1 Quantum Picture of the Chemical Bond 6.2 Exact Molecular Orbital for the Simplest Molecule: H + 2 6.3 Molecular Orbital Theory and the Linear Combination
Instructor background for the discussion points of Section 2
 Supplementary Information for: Orbitals Some fiction and some facts Jochen Autschbach Department of Chemistry State University of New York at Buffalo Buffalo, NY 14260 3000, USA Instructor background for
Supplementary Information for: Orbitals Some fiction and some facts Jochen Autschbach Department of Chemistry State University of New York at Buffalo Buffalo, NY 14260 3000, USA Instructor background for
Introduction to Computational Chemistry: Theory
 Introduction to Computational Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC andrew.gilbert@anu.edu.au 3023 Course Lectures Introduction Hartree Fock Theory Basis Sets Lecture 1 1 Introduction
Introduction to Computational Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC andrew.gilbert@anu.edu.au 3023 Course Lectures Introduction Hartree Fock Theory Basis Sets Lecture 1 1 Introduction
CHEM6085: Density Functional Theory
 Lecture 5 CHEM6085: Density Functional Theory Orbital-free (or pure ) DFT C.-K. Skylaris 1 Consists of three terms The electronic Hamiltonian operator Electronic kinetic energy operator Electron-Electron
Lecture 5 CHEM6085: Density Functional Theory Orbital-free (or pure ) DFT C.-K. Skylaris 1 Consists of three terms The electronic Hamiltonian operator Electronic kinetic energy operator Electron-Electron
Molecular Simulation I
 Molecular Simulation I Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation I Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Quantum mechanics can be used to calculate any property of a molecule. The energy E of a wavefunction Ψ evaluated for the Hamiltonian H is,
 Chapter : Molecules Quantum mechanics can be used to calculate any property of a molecule The energy E of a wavefunction Ψ evaluated for the Hamiltonian H is, E = Ψ H Ψ Ψ Ψ 1) At first this seems like
Chapter : Molecules Quantum mechanics can be used to calculate any property of a molecule The energy E of a wavefunction Ψ evaluated for the Hamiltonian H is, E = Ψ H Ψ Ψ Ψ 1) At first this seems like
Chemistry 3502 Physical Chemistry II (Quantum Mechanics) 3 Credits Fall Semester 2003 Christopher J. Cramer. Lecture 25, November 5, 2003
 3 Credits Fall Semester 2003 Christopher J. Cramer. Lecture 25, November 5, 2003") Chemistry 3502 Physical Chemistry II (Quantum Mechanics) 3 Credits Fall Semester 2003 Christopher J. Cramer Lecture 25, November 5, 2003 (Some material in this lecture has been adapted from Cramer, C.
Chemistry 3502 Physical Chemistry II (Quantum Mechanics) 3 Credits Fall Semester 2003 Christopher J. Cramer Lecture 25, November 5, 2003 (Some material in this lecture has been adapted from Cramer, C.
Solid State Theory: Band Structure Methods
 Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
Exp. 4. Quantum Chemical calculation: The potential energy curves and the orbitals of H2 +
 Exp. 4. Quantum Chemical calculation: The potential energy curves and the orbitals of H2 + 1. Objectives Quantum chemical solvers are used to obtain the energy and the orbitals of the simplest molecules
Exp. 4. Quantum Chemical calculation: The potential energy curves and the orbitals of H2 + 1. Objectives Quantum chemical solvers are used to obtain the energy and the orbitals of the simplest molecules
Module 6 1. Density functional theory
 Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
23 The Born-Oppenheimer approximation, the Many Electron Hamiltonian and the molecular Schrödinger Equation M I
 23 The Born-Oppenheimer approximation, the Many Electron Hamiltonian and the molecular Schrödinger Equation 1. Now we will write down the Hamiltonian for a molecular system comprising N nuclei and n electrons.
23 The Born-Oppenheimer approximation, the Many Electron Hamiltonian and the molecular Schrödinger Equation 1. Now we will write down the Hamiltonian for a molecular system comprising N nuclei and n electrons.
Computational Modeling Software and their applications
 Computational Modeling Software and their applications June 21, 2011 Damilola Daramola Center for Electrochemical Engineering Research ABC s of electrochemistry Introduction Computational Modeling the
Computational Modeling Software and their applications June 21, 2011 Damilola Daramola Center for Electrochemical Engineering Research ABC s of electrochemistry Introduction Computational Modeling the
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Density Functional Theory. Martin Lüders Daresbury Laboratory
 Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Electronic Structure Calculations and Density Functional Theory
 Electronic Structure Calculations and Density Functional Theory Rodolphe Vuilleumier Pôle de chimie théorique Département de chimie de l ENS CNRS Ecole normale supérieure UPMC Formation ModPhyChem Lyon,
Electronic Structure Calculations and Density Functional Theory Rodolphe Vuilleumier Pôle de chimie théorique Département de chimie de l ENS CNRS Ecole normale supérieure UPMC Formation ModPhyChem Lyon,
σ u * 1s g - gerade u - ungerade * - antibonding σ g 1s
 One of these two states is a repulsive (dissociative) state. Other excited states can be constructed using linear combinations of other orbitals. Some will be binding and others will be repulsive. Thus
One of these two states is a repulsive (dissociative) state. Other excited states can be constructed using linear combinations of other orbitals. Some will be binding and others will be repulsive. Thus
Lecture B6 Molecular Orbital Theory. Sometimes it's good to be alone.
 Lecture B6 Molecular Orbital Theory Sometimes it's good to be alone. Covalent Bond Theories 1. VSEPR (valence shell electron pair repulsion model). A set of empirical rules for predicting a molecular geometry
Lecture B6 Molecular Orbital Theory Sometimes it's good to be alone. Covalent Bond Theories 1. VSEPR (valence shell electron pair repulsion model). A set of empirical rules for predicting a molecular geometry
(1/2) M α 2 α, ˆTe = i. 1 r i r j, ˆV NN = α>β
 M α 2 α, ˆTe = i. 1 r i r j, ˆV NN = α>β") Chemistry 26 Spectroscopy Week # The Born-Oppenheimer Approximation, H + 2. Born-Oppenheimer approximation As for atoms, all information about a molecule is contained in the wave function Ψ, which is the
Chemistry 26 Spectroscopy Week # The Born-Oppenheimer Approximation, H + 2. Born-Oppenheimer approximation As for atoms, all information about a molecule is contained in the wave function Ψ, which is the
Density Functional Theory for Electrons in Materials
 Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
Lecture 08 Born Oppenheimer Approximation
 Chemistry II: Introduction to Molecular Spectroscopy Prof. Mangala Sunder Department of Chemistry and Biochemistry Indian Institute of Technology, Madras Lecture 08 Born Oppenheimer Approximation Welcome
Chemistry II: Introduction to Molecular Spectroscopy Prof. Mangala Sunder Department of Chemistry and Biochemistry Indian Institute of Technology, Madras Lecture 08 Born Oppenheimer Approximation Welcome
We can model covalent bonding in molecules in essentially two ways:
 CHEM 2060 Lecture 22: VB Theory L22-1 PART FIVE: The Covalent Bond We can model covalent bonding in molecules in essentially two ways: 1. Localized Bonds (retains electron pair concept of Lewis Structures)
CHEM 2060 Lecture 22: VB Theory L22-1 PART FIVE: The Covalent Bond We can model covalent bonding in molecules in essentially two ways: 1. Localized Bonds (retains electron pair concept of Lewis Structures)
Chemistry, Physics and the Born- Oppenheimer Approximation
 Chemistry, Physics and the Born- Oppenheimer Approximation Hendrik J. Monkhorst Quantum Theory Project University of Florida Gainesville, FL 32611-8435 Outline 1. Structure of Matter 2. Shell Models 3.
Chemistry, Physics and the Born- Oppenheimer Approximation Hendrik J. Monkhorst Quantum Theory Project University of Florida Gainesville, FL 32611-8435 Outline 1. Structure of Matter 2. Shell Models 3.
C/CS/Phys C191 Particle-in-a-box, Spin 10/02/08 Fall 2008 Lecture 11
 C/CS/Phys C191 Particle-in-a-box, Spin 10/0/08 Fall 008 Lecture 11 Last time we saw that the time dependent Schr. eqn. can be decomposed into two equations, one in time (t) and one in space (x): space
C/CS/Phys C191 Particle-in-a-box, Spin 10/0/08 Fall 008 Lecture 11 Last time we saw that the time dependent Schr. eqn. can be decomposed into two equations, one in time (t) and one in space (x): space