Modeling & Simulation of Glass Structure
|
|
|
- Edith Hollie Bishop
- 5 years ago
- Views:
Transcription
1 Modeling & Simulation of Glass Structure VCG Lecture 21 John Kieffer Department of Materials Science and Engineering University of Michigan 1
2 Overview Historical perspective Simulation methodologies Theoretical background Monte Carlo simulation Molecular dynamics simulation Reverse Monte Carlo Force fields Information retrieval Statistical mechanical formalisms Structural analyses Dynamics Application examples 2
 Induction interactions Between, for example, a permanent dipole and an induced dipole Dispersion interactions Long-range attraction that")

3 Force Fields Types of interactions between two molecules Electrostatic interactions Between charges (if ions) and between permanent dipoles, quadrupoles and higher multipole moments May be represented through partial charges Embedding terms (metals) Induction interactions Between, for example, a permanent dipole and an induced dipole Dispersion interactions Long-range attraction that has its origin in fluctuations of the charge distribution Valence (overlap) interactions Covalent bonds Repulsive interaction at short range which arises from exclusion principle Residual valence interactions Specific chemical forces giving rise to association and complex formation - e.g., hydrogen bonding Types of interactions within a molecule Bond stretch, bond-angle bending, torsional potential r q 3
 + - Johannes Diderik Van der")
4 Force Fields Non-bonded interactions are typically long-range and involve a large number of particles. Yet, contributions to the total energy and forces are considered as pairwise additive Non-bonded (electrostatic) + - Johannes Diderik Van der Waals Non-bonded (van der Waals) + 4
5 Metals Example: Morse force field Approximate physical behavior Computationally efficient 0.2 (r) r D 0 e 2 a r r 0 a r r 2 e 0 D 0 e a r r 0 2 e a r r Morse Harmonic r 5
 U ( r ) 4 r 12 r 6 Collision diameter typical cutoff at 2.5 minimum at r=1.")
6 Van der Waals interactions The most popular model of the van der Waals non-bonded interactions between atoms is the Lennard-Jones potential. U(r) U ( r ) 4 r 12 r 6 Collision diameter typical cutoff at 2.5 minimum at r=1.122 Sir John Edward Lennard-Jones r = r ij 6
7 Metals Embedded atom method E t F i 1 2 i i j i r ij i f r ij j j 7

8 Metals Embedded atom method f r f e e r r e 1 e e r r r e 1 F e 6 e e E c 1 ln e 8
9 Embedded atom method F e 6 e e E c 1 ln e B E c 1 2 E c = cohesive energy B = bulk modulus = atomic volume Other factors are model parameters that are determined based on lattice parameter, bulk modulus, shear modulus, cohesive energy, vacancy formation energy, and average electron density 9

10 Formation of dislocations in metals V. Bulatov, LLNL 10
11 Simulation of Laser Ablation on Metal Surface QuickTime and a TIFF (Un compressed) decompressor are needed to see this picture. G.H. Gilmer, LLNL atoms x 140 ps 11
12 Embedded atom method Simple closed form Computationally efficient Based on density functional theory concept Accurate for highly symmetric structures (FCC, BCC) Accurate for spherical electron orbitals Increasingly inaccurate for complex atomic electron configurations and low-symmetry structures 12
 force")
13 Range of interactions Short-range interactions: 1/2 N N C force calculations Long-range interactions: 1/2 N (N-1) force calculations 13
14 Ionic Materials Coulomb interactions are fundamental to atomistic systems Coulomb interactions have long-range effects CC 1 N N q i q j 4 0 i j i r ij 14

15 Ionic Materials Degree of charge localization can vary Charge polarization effects can be accounted for analytically CD N i N 1 q i 3 j r ij 4 0 r ij j i DC N i N j i q j r ij 3 i r ij DD N N i j i r ij 5 i r ij r j ij i j r ij 3 15
16 Ionic Materials Repulsion due to electron orbital overlap Repulsion due to interactions between nuclei handled by empirical function N NC BM A ij 1 z i n i j 1 i z j n j e i j r ij ij 16
17 The trouble with Coulomb interactions 5 CC (r ) ij 4 3 CC N i N j i q i q j r ij r ij 17
18 The trouble with Coulomb interactions Conditionally convergent I.e., depends on how one adds it up 18
19 Long-range interactions: Ewald summation method Total potential energy of one ion at the reference point. Summation decomposed into two parts, one to be evaluated in real space, the other in reciprocal space. b 1 2 a 1 a b r r l q l 3 2 e r r l
20 Reciprocal space 2 a 4 a a c G e igr G G G e igr G 2 c G e igr 4 G e igr G G c G 4 G G 2 20
21 Reciprocal space G G e igr r r l q l l l e r r l 21

22 Reciprocal space G e igr e igr d r G cell e igr d r q l cell cell l e r r l e igr d r 22

23 Reciprocal space G e igr e igr d r G cell e igr d r q t cell space t 3 2 e r r t 2 e igr d r 23
24 Reciprocal space G e igr e igr d r G q t cell space t 3 2 e r r t 2 e igr d r G q t 3 2 e igr t G t t q t e igr t space e 2 ig d r e G 2 4 S G e G
25 Reciprocal space a 4 S G G 2 e ig r G 2 4 At r = 0: Self-energy correction a 4 S G G 2 e G 2 4 b 4 r 2 rdr 2 q t b 1 4 S G G 2 e G q t
26 Real space q l 1 r l rdr r l r l l 0 r l r r dr 2 l q l r l erfc r l r l 26
27 Convergence in real and reciprocal space i 4 S G G 2 e G q i l q l r l erfc r l S(G) CC (r ) ij G r ij 27
28 Molecular Simulation Massively parallel molecular dynamics Spatial domain decomposition - large systems Replicated data - long simulation times 28
29 Spatial Decomposition and Ewald Sum 1 2 > 1 Requires larger G i 4 S G G 2 e G q i G 1 2 l q l r l erfc r l 29
30 Particle-Mesh Ewald Method S G q t e ig r t q t e i h x t k y t lz t t t r j r j f ij m+1 f ij r i m r i f [u] interpolation function S n n+1 G f u q e ig u q t e i h u k v lw t t 30
31 Example: fracture in brittle materials Quic ktime and a GIF decompressor are needed to see this picture. 31
32 Overview Historical perspective Simulation methodologies Theoretical background Monte Carlo simulation Molecular dynamics simulation Reverse Monte Carlo Force fields Information retrieval Statistical mechanical formalisms Structural analyses Dynamics Application examples 32
33 Ionic materials Fundamentally correct Accurate for strongly ionic compounds, including many ceramics and salts Long-range interactions cause computational expense Increasingly inaccurate when partially covalent interactions and charge transfer occurs 33
34 Questions? 34
35 Universal force fields Goal is to develop force field that can be to describe intra- and inter-molecular interactions for arbitrary molecules UFF (Universal force field) A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, "UFF, a Full Periodic-Table Force-Field for Molecular Mechanics and Molecular-Dynamics Simulations," J. Am. Chem. Soc, 114, (1992) Used for many nanoscale carbon structures (buckyballs, nanotubes) COMPASS (Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies) Sun, H; Rigby, D. "Polysiloxanes: Ab Initio Force Field And Structural, Conformational And Thermophysical Properties", Spectrochimica Acta (A),1997,53, 130, and subsequent references AMBER (Assisted Model Building with Energy Refinement) Both a force field and a simulation package for biomolecular simulations E total K r r r eq 2 K q bonds angles r r q 2 V n 1 cos n 2 dihedrals i j A ij R ij 12 B ij 6 R ij q i q j R ij H bonds C ij R ij 12 D ij R ij 10 35
36 Universal force fields CHARMM (Chemistry at HARvard Macromolecular Mechanics ) Both a force field and a simulation package for biomolecular simulations B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus, A Program for Macromolecular Energy, Minimization, and Dynamics Calculations, J. Comp. Chem. 4 (1983) (1983) Others GROMOS ( Jorgensen s Optimized Potential for Liquid Simulation (OPLS) ( DREIDING (Mayo, S. L., Olafson, B. D. & Goddard, W. A. Dreiding - a Generic Force- Field for Molecular Simulations. Journal of Physical Chemistry 94 (1990) ) Specific systems Alkanes and related systems SKS: B. Smit, S. Karaborni, and J.I. Siepmann, `Computer simulation of vaporliquid phase equilibria of n-alkanes', J. Chem. Phys. 102, (1995); `Erratum', 109, 352 (1998) TraPPE: Siepmann group ( 36
37 Latest trends: Charge transfer Charge localization depends on environment Chemical nature of surrounding atoms Local structure Electronegativity i 0 E q i i IP EA Atomic hardness J ii 0 2 E 2 q i i 0 IP EA IP = first ionization potential; EA = electron affinity 37
38 Charge transfer Energy of an isolated atom: E i q E i 0 i q i 1 2 J ii q i Energy of an N atoms: E E i 0 q 1,, q N N N N 0 q 1 2 J 0 q 2 i q i ii i i q j J ij i i j i N E i 0 i 0q i 1 2 i N i 1 N j 1 q i q j J ij 38
39 Charge transfer 5 J (r) ij 4 J ij r i r i 1 r i r j r j r j d r i d r j 3 2 Spherically symmetric Slater-type orbitals are assumed for (r) r ij 39
40 Charge transfer Chemical potential of electrons associated with atom i: E q i i i 0 N j 1 q j J ij Equilibrium requires 1 = 2 = 3 = = N, which yields N 1 equations. In addition, charge neutrality requires that N q j 0 j 1 40
41 Charge transfer Solving the system of equations yields the equilibrium charges 1 1 J 21 J 11 J 2 N J 1 N J N 1 J 11 J NN J 1 N q 1 q 2 q N N Equilibrium charges depend on the chemical nature of species and on the geometry of the configuration, by virtue of the fact that J ij r i r i 1 r i r j r j r j d r i d r j 41
42 (r ij ) (J ) C h a r g e tr a n s fe r Covalently bonded materials repulsive charge transfer function covalent attractive Coulomb total r ij (nm) N N C 3 B ij ik q ijk N C 1 i 1 j 1 k j torque Directional character of bonds q ijk Covalent bonding term (empirical), e.g., ij A ij e ( i j r ij ) ij ij e ij r ij ij r ij b ij 1 e 1 a ij 42
 2 (q Z q ijk ) 2 Z 0 e 0 N Z i ( e ( r ij j i a Z ) b Z 1) 1 L. P. Huang and J. Kieffer, J. Chem. Phys. 118, 1487 (2003) 43")
43 Reactive force fields Charge redistribution q i q i o N C e ij 1 e b ij r ij a ij j 1 1 Flexible coordination N 3 B ij ik q ijk N C 1 N C i 1 j 1 k j 1 q ijk C Z e A Z Z 0 3 ( Z 0 Z i ) 2 (q Z q ijk ) 2 Z 0 e 0 N Z i ( e ( r ij j i a Z ) b Z 1) 1 L. P. Huang and J. Kieffer, J. Chem. Phys. 118, 1487 (2003) 43
44 Force field parameterization Reproduce known structures (static properties) density at ambient pressure equation of state (density vs. pressure) mechanical properties (elastic moduli, strength, etc.) Reproduce dynamic properties melting temperatures vibrational spectra Compare with results from ab initio calculations bonding energies Interatomic forces 44


 High-P quartz at 25 GPa")


 a = 4.2 Å b = 4.")
 a")

45 Crystalline silica polymorphs one parameterization -quartz at 0 GPa 0 GPa (P ) a = 4.9 Å b = 4.9 Å c = 5.4 Å -cristobalite 14 GPa (P ) High-P quartz at 25 GPa 16 GPa (C222 1 ) a = 6.7 Å b = 5.7 Å c = 6.1 Å 20 GPa (P ) a = 4.2 Å b = 4.2 Å c = 6.0 Å X-I phase a c 25 GPa (P42/mnm) a = 4.2 Å b = 4.2 Å c = 2.8 Å stishovite 40 GPa (Pnnm) a = 3.7 Å b = 4.5 Å c = 2.8 Å post-stishovite Ideal high-p quartz with bcc anion sub-lattice Huang, Durandurdu and Kieffer, Nature Materials (in press) 45
 J. Chem.")

46 Parameter Optimization: Crystal Structures (Hexyl-POSS) J. Chem. Soc. Dalton Trans. 1994, K, NVT, 64 molecules 46
47 Parameter Optimization: IR Spectrum J. Chem. Soc. Dalton Trans. 1994,
48 Parameter Optimization: Fitting to Electronic Structure Calculations POSS-NaCl Potential Surface High Intermediate Low 48
49 Fitting Results Mulliken point shell EEM Si O H ~0.01 (E)

50 Charge Equilibration Method 50
51 Questions? 51
52 QM Methods Doing a QM simulation or calculation means including the electrons explicitly. With QM methods, we can calculate properties that depend upon the electronic distribution, and to study processes like chemical reactions in which bonds are formed and broken. The explicit consideration of electrons distinguishes QM models and methods from classical force field models and methods. 52
53 Different QM Methods Several approaches exist. The two main ones are: Molecular orbital theory Came from chemistry, since primarily developed for individual molecules, gases and now liquids. Two flavors Ab initio: all electrons included (considered exact) Semi-empirical: only valence electrons included Density functional theory Came from physics and materials science community, since originally conceived for solids. All electrons included via electronic density (considered exact). 53
54 Fundamentals The operator that returns the system energy is called the Hamiltonian operator H. Eigenvalue equation for system energy H = E Time-independent Schrodinger Equation H i 2 2 m e i 2 k m k k i k e 2 Z k r ik i j e 2 r ij k l e 2 Z k Z l r kl Kinetic energy of electrons Kinetic energy of nuclei Potential energy of electrons & nuclei Potential energy of electrons Potential energy of nuclei 54
55 Born-Oppenheimer Approximation Need to simplify! Neutrons & protons are >1800 times more massive than electrons, and therefore move much more slowly. Thus, electronic relaxation is for all practical purposes instantaneous with respect to nuclear motion. We can decouple the motion, and consider the electron-electron interactions independently of the nuclear interactions. This is the Born- Oppenheimer approximation. For nearly all situations relevant to soft matter, this assumption is entirely justified. (H el +V n ) el (q i ;q k ) = E el el (q i ;q k ) The Electronic Schrodinger Equation 55
56 QM Methods Goal of all QM methods in use today: Solve the electronic Schrodinger equation for the ground state energy of a system and the wavefunction that describes the positions of all the electrons. The energy is calculated for a given trial wavefunction, and the best wavefunction is found as that wavefunction that minimizes the energy. 56
57 QM Methods Solving SE is not so easy! Anything containing more than two elementary particles (i.e. one e- and one nucleon) can t be solved exactly: the many-body problem. Even after invoking Born-Oppenheimer, still can t solve exactly for anything containing more than two electrons. So -- all QM methods used today are APPROXIMATE after all, even if considered exact! That is, they provide approximate solutions to the Schrodinger equation. Some are more approximate than others. 57
58 Molecular Orbital Theory MOT is expressed in terms of molecular wave functions called molecular orbitals. Most popular implementation: write molecular orbital as a linear combination of atomic orbitals (LCAO): Eq in Leach K i a i 1 K = # atomic orbitals Many different ways of writing basis set, which leads to many different methods and implementations of MOT. 58
59 Molecular Orbital Theory Dozens of approaches for writing basis sets (e.g. in terms of Gaussian wavefunctions, or as linear combos of Gaussians). Different implementations retain different numbers of terms. Semi-empirical MOT methods consider only valence electrons. Some methods include electron exchange. Some methods include electron correlation. 59
60 Density Functional Theory A different approach for solving Schrodinger s equation for the ground state energies of matter. Based on theory of Hohenberg and Kohn (1964) which states that it is not necessary to consider the motion of each individual electron in the system. Instead, it suffices to know the average number of electrons at any one point in space. The HK theorem enables us to write E el as a functional of the electron density M A 1 Z A r 1 A r 2 d r 2 V X C r 1 r 1 2 i r i i i r i To perform a DFT calculation, one optimizes the energy with respect to the electron probability density, rather than with respect to the electronic wave function. For a given density, the lowest energy is the best one. 60
61 Density Functional Theory In the commonly used Kohn-Sham implementation, the density is written in terms of one-electron molecular orbitals called Kohn-Sham orbitals. This allows the energy to be optimized by solving a set of one-electron Schrodinger equations (the KS equations), but with electron correlation included. This is a key advantage of the DFT method - it s easier to include electron correlation. Different choices of basis sets, how many terms to use, of what type, contribute to difficulty of calculation. More than a few hundred light atoms is still too time-consuming, even on big computers. For molecules or systems with large numbers of electrons, pseudopotentials are used to represent the wavefunctions of valence electrons, and the core is treated in a simplified way. 61


62 Applications of ab initio computations using DMol 3 62





63 Electronic band structure of POSS cubes functionalized with n benzene molecules (n = 0-8) Energy (ev) 63




64 Electronic band structure of POSS cubes functionalized with acene molecules (benzene, naphtalene, anthracene, tetracene, and pentacene) Energy (ev) Pure poss 64




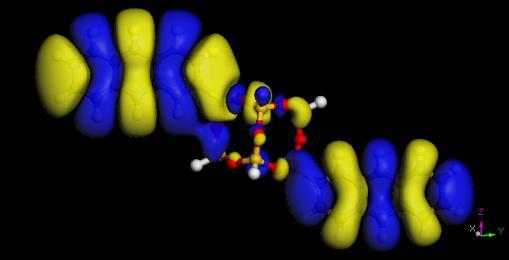
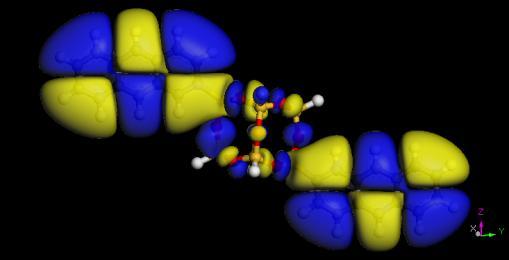
65 Electron densities of acene-functionalized POSS Molecule Band Gap (ev) Pure Acene (ev) P-POSS T-POSS A-POSS N-POSS B-POSS POSS HOMO LUMO 65


66 Electron densities of silica nanotubes HOMO LUMO 66
67 Classical vs. ab initio methods Classical No electronic properties ab initio Electronic details included Phenomenological potential energy surface (typically 2- body contributions) Difficult to describe bond breaking/formation Can do up to a billion particles Potential energy surface calculated directly from Schrodinger equation (many body terms included automatically) Describes bond breaking/formation Limited to several hundred atoms with significant dynamics 67
68 Editor s Note: Lecture 21 ended here 4/2/07 Questions? 68
69 Overview Historical perspective Simulation methodologies Theoretical background Monte Carlo simulation Molecular dynamics simulation Reverse Monte Carlo Force fields Information retrieval Statistical mechanical formalisms Structural analyses Dynamics Application examples 69
70 Simulation Observables: Thermodynamics In a classical, many body system, the temperature is defined from the equipartition theorem in terms of the average kinetic energy per degree of freedom: 1 2 m v x k B T In a simulation, we use this equation as an operational definition of T. Thus the lower the temperature, the lower the kinetic energy, and the slower the average velocity. 70
 k B ( 3 N N c ) N c = # of constraints (e.g. linear momentum) NVE 3N - N c = # of degrees of freedom time steps 71")
71 Simulation Observables: Thermodynamics Thermodynamic quantities T ( t ) N Calculate T,, p, E etc. depending on ensemble i 1 m i v i 2 ( t ) k B ( 3 N N c ) N c = # of constraints (e.g. linear momentum) NVE 3N - N c = # of degrees of freedom time steps 71

72 Simulation Observables: Thermodynamics Pressure NVE P 1 3V N i 1 m i v i 2 1 6V N i 1 N j 1 f ij r ij P is obtained from the virial thm of Clausius. Virial = exp. value of the sum of the products of the particle coords and the forces acting on them. W=x i f xi =-3Nk B T time steps 72
73 Measuring quantities in molecular simulations Suppose we wish to measure in an experiment the value of a property, A, of a system such as pressure or density or heat capacity. The property A will depend on the positions and momenta of the N atoms or molecules that comprise the system. The instantaneous value of A is: A(p N (t),r N (t)) = A(p 1x,p 1y,p 1z,, x 1,y 1,z 1, t). Over time, A fluctuates due to interactions between the atoms. 73
74 Measuring quantities in molecular simulations In any experiment, the value that we measure is an average of A over the time of the measurement (a time average). As the time, t, of the measurement increases to infinity, the true average value of A is attained: A lim 1 t 0 A p N ( t ), r N ( t ) dt Note that strictly speaking this is true only for ergodic systems in equilibrium. 74
75 Measuring quantities in molecular simulations To calculate properties in a molecular simulation, we average a property A over successive configurations in the trajectory generated by the equations of motion. A lim n steps n steps 1 A p N ( t ), r N ( t ) n steps t 0 The larger n steps is, the more accurate the estimate of the true time-averaged value of A. 75
76 Ensemble averages In statistical mechanics we learn that experiments and simulations may be performed in different ensembles. An ensemble is a collection of individual systems that can be studied en masse to yield macroscopic properties of the entire collection. Every equilibrium configuration we generate via MD represents a unique microstate of the ensemble. 76
77 Ensemble averages There are many different thermodynamic ensembles, each of which is characterized by thermodynamic variables that are fixed in the simulation. NVE - microcanonical NVT - canonical NPT - isobaric-isothermal VT - Gibbs N = number of particles, V = volume, E = total energy, T = temperature, P = pressure, = chemical potential The simplest MD simulation has constant N, V and E. The NVE ensemble is the microcanonical ensemble; all states are equally likely. In NVE MD, solve F=ma for an isolated system, one for which no exchange of energy or particles with the outside. 77
78 Molecular Dynamics Simulation: NVT Velocity Rescaling Recall KE = mv 2 /2 = 3Nk B T/2 in an unconstrained system. Simple way to alter T: rescale velocities every time step by a factor (t): If temperature at time t is T(t) and v is multiplied by, then the temperature change is T 1 2 N 2 m i ( v i ) 2 1 i 1 3 N k B 2 N i m i v i 2 N k B T T ta rg e t T (t ) ( 2 1)T (t ) T ta rg e t / T (t ) 78
79 Molecular Dynamics Simulation: NVT Velocity Rescaling This method does not generate configurations in the canonical ensemble. Why? Because system is not coupled to an external reservoir, or heat bath. Thus not a good method for controlling temperature, since it s unknown how rescaling affects properties of the system. Instead, use a proper thermostat. Using a weakly coupled thermostat can help eliminate drift in total energy due to algorithm inaccuracies and round-off error. Several types of thermostats; choose wisely depending on your problem! 79
80 Molecular Dynamics Simulation: NVT Stochastic collision method: Andersen thermostat Occasionally replace a randomly chosen particle s velocity with a velocity chosen randomly from a Maxwell-Boltzmann distribution at the desired T. p (v ix ) = m 2 k B T 1/2 exp (- 1 2 k B T m v ix 2 ) This replacement represents stochastic collisions with the heat bath. Equivalent to the system being in contact with a heat bath that randomly emits thermal particles that collide with the atoms in the system and change their velocity. 80
81 Molecular Dynamics Simulation: NVT Extended system method: Nose -Hoover thermostat Introduced by Nose and developed by Hoover in Considers the thermostat to be part of the system (hence extended system ). Based on extended Lagrangian approach in which an additional dynamical variable or degree of freedom s, representing the reservoir, imposes constraint of constant T. extended system reservoir physical system 81
82 Molecular Dynamics Simulation: NVT Extended system method: Nose -Hoover thermostat Reservoir has potential energy U res and kinetic energy K res. U res = (f+1)k B T ln s K res = (Q/2)(ds/dt) 2 reservoir physical system ( t ) sp s Q f = # of dof in physical system, and T is the desired temperature. Q has dimensions energy x (time) 2, and is considered the fictitious mass of the additional degree of freedom. The magnitude of Q controls the coupling between the physical system and the reservoir, and thus influences the temperature fluctuations. 82
83 Molecular Dynamics Simulation: NVT Extended system method: Nose -Hoover thermostat Q controls the energy flow between system and reservoir. If Q is large, the energy flow is slow; the limit of infinite Q corresponds to NVE MD since there is then no exchange of energy between system and reservoir. If Q is small, then the energy oscillates unphysically, causing equilibration problems. Nose suggested Q = Cfk B T, where C can be obtained by performing trial simulations for a series of test systems and observing how well the system maintains that desired T. Upshot: equations of motion are modified and t is multiplied by Q. Read in Frenkel and Smit for details if interested. 83
84 Molecular Dynamics Simulation: NVT Extended system method: Nose -Hoover thermostat Best and most used thermostat today for NVT simulations. Ensures that average total KE per particle is that of the equilibrium isothermal ensemble. Reproduces canonical ensemble in every respect (provided only the total energy of the extended system is conserved.) If momentum also conserved, NH still works only if COM fixed. Otherwise, must use the method of NH chains. More complicated than other methods, but can be used with velocity Verlet and predictor-corrector methods. Dynamics NOT independent of Q, but better than Andersen. 84
85 Molecular Dynamics Simulation: NPT Just as one might like to specify T in an MD simulation, one might also like to maintain the system at a fixed pressure P. Simulations in the isobaric-isothermal NPT ensemble are the most similar to experimental conditions. Certain phenomena may be more easily achieved under conditions of constant pressure, like certain phase transformations. 85
86 Molecular Dynamics Simulation: NPT Two types: Andersen Barostat Allows box size to fluctuate but not change shape. Appropriate for liquids, which do not support shear stress. Change volume by rescaling positions: r = r+r Rahman-Parinello Barostat Allows box size and shape to change. Use only for solids. Necessary for simulating phase transformations. Read in Frenkel and Smit for details if interested. 86
87 Revisit the equations of motion Recall Lagrangian L 1 2 N m r i Ý T r Ý r i i ij i 1 i 1 j i N N t L L r Ý i r i L r Ý i L m i Ý r i r ij t f i L m i r Ý i r Ý i r i j i r ij r ij m i r Ý i f i 87



88 Flexible simulation boxes Mechanical deformation Structural transitions 88

89 Raman-Parinello algorithm c Basis vectors are dynamical variables: h a b c b a 89
90 Raman-Parinello algorithm L 1 2 N m s i Ý T G s Ý 1 i i 2 i 1 W Tr N N h Ý T h Ý r p 1 ij 2 i 1 j i Tr G t L L s Ý i s i t L h Ý L h Raman-Parinello Nosé-Hoover algorithm L 1 2 N T m i &s i G &s i f 2 1 W T r h & T h & 2 i 1 Q 2 &f 2 N N r ij p 1 2 i 1 j i T r G g 1 k B T ln f 90
91 Simulation Observables: Thermodynamics Thermodynamic Response Functions: Heat Capacity At the end (less error?) C V E T V 1 k B T 2 E E 2 <E 2 > - <E> 2 On the fly NVE: Perform simulations at many different E for the same N & V, and measure T. Calculate C v from derivative of E(T) (numerically or fitting polynomial and then differentiating). NVT: (1) Fix N and V, perform simulations at different T and measure E. Then calculate C v from derivative of E(T). (2) Calculate C v from fluctuations in energy. 92
92 Simulation Observables: Thermodynamics Thermodynamic Response Functions: Adiabatic Compressibility S 1 V V P S P P 2 NVE: (1) Fix N and E, perform simulations at different V and measure P. Calculate S from derivative of V(P). (2) Calculate S from fluctuations in pressure. NVT: Calculate S from fluctuations in pressure. 93
93 Simulation Observables: Thermodynamics Thermodynamic Response Functions: Isothermal Compressibility T 1 V V P T 1 k B T V V 2 V 2 NVT: Fix N and T, perform simulations at different V and measure P. Calculate T from derivative of V(P). NPT: Fix N and T, perform simulations at different P and measure V. Calculate T from derivative of V(P). NPT: Calculate T from fluctuations in volume. VT: Calculate T from fluctuations in N. 94
94 Questions? 95
Intro to ab initio methods
 Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
MD Thermodynamics. Lecture 12 3/26/18. Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky
 MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
What is Classical Molecular Dynamics?
 What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Ab initio molecular dynamics. Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy. Bangalore, 04 September 2014
 Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Force fields in computer simulation of soft nanomaterials
 Lecture 2 Part B Force fields in computer simulation of soft nanomaterials Recommended reading: Leach Chapter 4 1 Force Field Methods Force field methods are simulation methods that use classical force
Lecture 2 Part B Force fields in computer simulation of soft nanomaterials Recommended reading: Leach Chapter 4 1 Force Field Methods Force field methods are simulation methods that use classical force
Molecular Dynamics Simulations. Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia
 Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Javier Junquera. Statistical mechanics
 Javier Junquera Statistical mechanics From the microscopic to the macroscopic level: the realm of statistical mechanics Computer simulations Thermodynamic state Generates information at the microscopic
Javier Junquera Statistical mechanics From the microscopic to the macroscopic level: the realm of statistical mechanics Computer simulations Thermodynamic state Generates information at the microscopic
Modeling & Simulation of Glass Structure
 Modeling & Simulation of Glass Structure VCG Lecture 19 John Kieffer Department of Materials Science and Engineering University of Michigan 1 Overview Historical perspective Simulation methodologies Theoretical
Modeling & Simulation of Glass Structure VCG Lecture 19 John Kieffer Department of Materials Science and Engineering University of Michigan 1 Overview Historical perspective Simulation methodologies Theoretical
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
 A Nobel Prize for Molecular Dynamics and QM/MM What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential
A Nobel Prize for Molecular Dynamics and QM/MM What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential
510 Subject Index. Hamiltonian 33, 86, 88, 89 Hamilton operator 34, 164, 166
 Subject Index Ab-initio calculation 24, 122, 161. 165 Acentric factor 279, 338 Activity absolute 258, 295 coefficient 7 definition 7 Atom 23 Atomic units 93 Avogadro number 5, 92 Axilrod-Teller-forces
Subject Index Ab-initio calculation 24, 122, 161. 165 Acentric factor 279, 338 Activity absolute 258, 295 coefficient 7 definition 7 Atom 23 Atomic units 93 Avogadro number 5, 92 Axilrod-Teller-forces
General Physical Chemistry II
 General Physical Chemistry II Lecture 13 Aleksey Kocherzhenko October 16, 2014" Last time " The Hückel method" Ø Used to study π systems of conjugated molecules" Ø π orbitals are treated separately from
General Physical Chemistry II Lecture 13 Aleksey Kocherzhenko October 16, 2014" Last time " The Hückel method" Ø Used to study π systems of conjugated molecules" Ø π orbitals are treated separately from
Interatomic Potentials. The electronic-structure problem
 Interatomic Potentials Before we can start a simulation, we need the model! Interaction between atoms and molecules is determined by quantum mechanics: Schrödinger Equation + Born-Oppenheimer approximation
Interatomic Potentials Before we can start a simulation, we need the model! Interaction between atoms and molecules is determined by quantum mechanics: Schrödinger Equation + Born-Oppenheimer approximation
Ab Ini'o Molecular Dynamics (MD) Simula?ons
 Simula?ons") Ab Ini'o Molecular Dynamics (MD) Simula?ons Rick Remsing ICMS, CCDM, Temple University, Philadelphia, PA What are Molecular Dynamics (MD) Simulations? Technique to compute statistical and transport properties
Ab Ini'o Molecular Dynamics (MD) Simula?ons Rick Remsing ICMS, CCDM, Temple University, Philadelphia, PA What are Molecular Dynamics (MD) Simulations? Technique to compute statistical and transport properties
The broad topic of physical metallurgy provides a basis that links the structure of materials with their properties, focusing primarily on metals.
 Physical Metallurgy The broad topic of physical metallurgy provides a basis that links the structure of materials with their properties, focusing primarily on metals. Crystal Binding In our discussions
Physical Metallurgy The broad topic of physical metallurgy provides a basis that links the structure of materials with their properties, focusing primarily on metals. Crystal Binding In our discussions
Potentials, periodicity
 Potentials, periodicity Lecture 2 1/23/18 1 Survey responses 2 Topic requests DFT (10), Molecular dynamics (7), Monte Carlo (5) Machine Learning (4), High-throughput, Databases (4) NEB, phonons, Non-equilibrium
Potentials, periodicity Lecture 2 1/23/18 1 Survey responses 2 Topic requests DFT (10), Molecular dynamics (7), Monte Carlo (5) Machine Learning (4), High-throughput, Databases (4) NEB, phonons, Non-equilibrium
Understanding Molecular Simulation 2009 Monte Carlo and Molecular Dynamics in different ensembles. Srikanth Sastry
 JNCASR August 20, 21 2009 Understanding Molecular Simulation 2009 Monte Carlo and Molecular Dynamics in different ensembles Srikanth Sastry Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore
JNCASR August 20, 21 2009 Understanding Molecular Simulation 2009 Monte Carlo and Molecular Dynamics in different ensembles Srikanth Sastry Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Atoms, electrons and Solids
 Atoms, electrons and Solids Shell model of an atom negative electron orbiting a positive nucleus QM tells that to minimize total energy the electrons fill up shells. Each orbit in a shell has a specific
Atoms, electrons and Solids Shell model of an atom negative electron orbiting a positive nucleus QM tells that to minimize total energy the electrons fill up shells. Each orbit in a shell has a specific
Introduction to DFTB. Marcus Elstner. July 28, 2006
 Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 1 David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Time/s Multi-Scale Modeling Based on SDSC Blue Horizon (SP3) 1.728 Tflops
1 CE 530 Molecular Simulation Lecture 1 David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Time/s Multi-Scale Modeling Based on SDSC Blue Horizon (SP3) 1.728 Tflops
Ab initio molecular dynamics and nuclear quantum effects
 Ab initio molecular dynamics and nuclear quantum effects Luca M. Ghiringhelli Fritz Haber Institute Hands on workshop density functional theory and beyond: First principles simulations of molecules and
Ab initio molecular dynamics and nuclear quantum effects Luca M. Ghiringhelli Fritz Haber Institute Hands on workshop density functional theory and beyond: First principles simulations of molecules and
Preface Introduction to the electron liquid
 Table of Preface page xvii 1 Introduction to the electron liquid 1 1.1 A tale of many electrons 1 1.2 Where the electrons roam: physical realizations of the electron liquid 5 1.2.1 Three dimensions 5 1.2.2
Table of Preface page xvii 1 Introduction to the electron liquid 1 1.1 A tale of many electrons 1 1.2 Where the electrons roam: physical realizations of the electron liquid 5 1.2.1 Three dimensions 5 1.2.2
Ab initio Molecular Dynamics Born Oppenheimer and beyond
 Ab initio Molecular Dynamics Born Oppenheimer and beyond Reminder, reliability of MD MD trajectories are chaotic (exponential divergence with respect to initial conditions), BUT... With a good integrator
Ab initio Molecular Dynamics Born Oppenheimer and beyond Reminder, reliability of MD MD trajectories are chaotic (exponential divergence with respect to initial conditions), BUT... With a good integrator
Introduction to atomic scale simulations
 Powder Technology course Autumn semester 2017 Introduction to atomic scale simulations Peter M Derlet Condensed Matter Theory Paul Scherrer Institut peter.derlet@psi.ch Outline of lecture The big picture
Powder Technology course Autumn semester 2017 Introduction to atomic scale simulations Peter M Derlet Condensed Matter Theory Paul Scherrer Institut peter.derlet@psi.ch Outline of lecture The big picture
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Computer Simulation of Shock Waves in Condensed Matter. Matthew R. Farrow 2 November 2007
 Computer Simulation of Shock Waves in Condensed Matter Matthew R. Farrow 2 November 2007 Outline of talk Shock wave theory Results Conclusion Computer simulation of shock waves Shock Wave Theory Shock
Computer Simulation of Shock Waves in Condensed Matter Matthew R. Farrow 2 November 2007 Outline of talk Shock wave theory Results Conclusion Computer simulation of shock waves Shock Wave Theory Shock
Computer simulation methods (1) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (1) Dr. Vania Calandrini Why computational methods To understand and predict the properties of complex systems (many degrees of freedom): liquids, solids, adsorption of molecules
Computer simulation methods (1) Dr. Vania Calandrini Why computational methods To understand and predict the properties of complex systems (many degrees of freedom): liquids, solids, adsorption of molecules
Molecular Aggregation
 Molecular Aggregation Structure Analysis and Molecular Simulation of Crystals and Liquids ANGELO GAVEZZOTTI University of Milano OXFORD UNIVERSITY PRESS Contents PART I FUNDAMENTALS 1 The molecule: structure,
Molecular Aggregation Structure Analysis and Molecular Simulation of Crystals and Liquids ANGELO GAVEZZOTTI University of Milano OXFORD UNIVERSITY PRESS Contents PART I FUNDAMENTALS 1 The molecule: structure,
Biomolecular modeling I
 2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
Lecture 6 - Bonding in Crystals
 Lecture 6 onding in Crystals inding in Crystals (Kittel Ch. 3) inding of atoms to form crystals A crystal is a repeated array of atoms Why do they form? What are characteristic bonding mechanisms? How
Lecture 6 onding in Crystals inding in Crystals (Kittel Ch. 3) inding of atoms to form crystals A crystal is a repeated array of atoms Why do they form? What are characteristic bonding mechanisms? How
Biomolecules are dynamic no single structure is a perfect model
 Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Introduction to Molecular Dynamics
 Introduction to Molecular Dynamics Dr. Kasra Momeni www.knanosys.com Overview of the MD Classical Dynamics Outline Basics and Terminology Pairwise interacting objects Interatomic potentials (short-range
Introduction to Molecular Dynamics Dr. Kasra Momeni www.knanosys.com Overview of the MD Classical Dynamics Outline Basics and Terminology Pairwise interacting objects Interatomic potentials (short-range
The electronic structure of materials 1
 Quantum mechanics 2 - Lecture 9 December 18, 2013 1 An overview 2 Literature Contents 1 An overview 2 Literature Electronic ground state Ground state cohesive energy equilibrium crystal structure phase
Quantum mechanics 2 - Lecture 9 December 18, 2013 1 An overview 2 Literature Contents 1 An overview 2 Literature Electronic ground state Ground state cohesive energy equilibrium crystal structure phase
Energy and Forces in DFT
 Energy and Forces in DFT Total Energy as a function of nuclear positions {R} E tot ({R}) = E DF T ({R}) + E II ({R}) (1) where E DF T ({R}) = DFT energy calculated for the ground-state density charge-density
Energy and Forces in DFT Total Energy as a function of nuclear positions {R} E tot ({R}) = E DF T ({R}) + E II ({R}) (1) where E DF T ({R}) = DFT energy calculated for the ground-state density charge-density
Basics of Statistical Mechanics
 Basics of Statistical Mechanics Review of ensembles Microcanonical, canonical, Maxwell-Boltzmann Constant pressure, temperature, volume, Thermodynamic limit Ergodicity (see online notes also) Reading assignment:
Basics of Statistical Mechanics Review of ensembles Microcanonical, canonical, Maxwell-Boltzmann Constant pressure, temperature, volume, Thermodynamic limit Ergodicity (see online notes also) Reading assignment:
Introduction to Simulation - Lectures 17, 18. Molecular Dynamics. Nicolas Hadjiconstantinou
 Introduction to Simulation - Lectures 17, 18 Molecular Dynamics Nicolas Hadjiconstantinou Molecular Dynamics Molecular dynamics is a technique for computing the equilibrium and non-equilibrium properties
Introduction to Simulation - Lectures 17, 18 Molecular Dynamics Nicolas Hadjiconstantinou Molecular Dynamics Molecular dynamics is a technique for computing the equilibrium and non-equilibrium properties
Atomic structure & interatomic bonding. Chapter two
 Atomic structure & interatomic bonding Chapter two 1 Atomic Structure Mass Charge Proton 1.67 х 10-27 kg + 1.60 х 10-19 C Neutron 1.67 х 10-27 kg Neutral Electron 9.11 х 10-31 kg - 1.60 х 10-19 C Electron
Atomic structure & interatomic bonding Chapter two 1 Atomic Structure Mass Charge Proton 1.67 х 10-27 kg + 1.60 х 10-19 C Neutron 1.67 х 10-27 kg Neutral Electron 9.11 х 10-31 kg - 1.60 х 10-19 C Electron
Gear methods I + 1/18
 Gear methods I + 1/18 Predictor-corrector type: knowledge of history is used to predict an approximate solution, which is made more accurate in the following step we do not want (otherwise good) methods
Gear methods I + 1/18 Predictor-corrector type: knowledge of history is used to predict an approximate solution, which is made more accurate in the following step we do not want (otherwise good) methods
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Chapter 3. Crystal Binding
 Chapter 3. Crystal Binding Energy of a crystal and crystal binding Cohesive energy of Molecular crystals Ionic crystals Metallic crystals Elasticity What causes matter to exist in three different forms?
Chapter 3. Crystal Binding Energy of a crystal and crystal binding Cohesive energy of Molecular crystals Ionic crystals Metallic crystals Elasticity What causes matter to exist in three different forms?
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
Modeling Materials. Continuum, Atomistic and Multiscale Techniques. gg CAMBRIDGE ^0 TADMOR ELLAD B. HHHHM. University of Minnesota, USA
 HHHHM Modeling Materials Continuum, Atomistic and Multiscale Techniques ELLAD B. TADMOR University of Minnesota, USA RONALD E. MILLER Carleton University, Canada gg CAMBRIDGE ^0 UNIVERSITY PRESS Preface
HHHHM Modeling Materials Continuum, Atomistic and Multiscale Techniques ELLAD B. TADMOR University of Minnesota, USA RONALD E. MILLER Carleton University, Canada gg CAMBRIDGE ^0 UNIVERSITY PRESS Preface
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
74 these states cannot be reliably obtained from experiments. In addition, the barriers between the local minima can also not be obtained reliably fro
 73 Chapter 5 Development of Adiabatic Force Field for Polyvinyl Chloride (PVC) and Chlorinated PVC (CPVC) 5.1 Introduction Chlorinated polyvinyl chloride has become an important specialty polymer due to
73 Chapter 5 Development of Adiabatic Force Field for Polyvinyl Chloride (PVC) and Chlorinated PVC (CPVC) 5.1 Introduction Chlorinated polyvinyl chloride has become an important specialty polymer due to
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Length Scales (c) ICAMS: http://www.icams.de/cms/upload/01_home/01_research_at_icams/length_scales_1024x780.png Goals for today: Background
André Schleife Department of Materials Science and Engineering Length Scales (c) ICAMS: http://www.icams.de/cms/upload/01_home/01_research_at_icams/length_scales_1024x780.png Goals for today: Background
Temperature and Pressure Controls
 Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Hyeyoung Shin a, Tod A. Pascal ab, William A. Goddard III abc*, and Hyungjun Kim a* Korea
 The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
Scientific Computing II
 Scientific Computing II Molecular Dynamics Simulation Michael Bader SCCS Summer Term 2015 Molecular Dynamics Simulation, Summer Term 2015 1 Continuum Mechanics for Fluid Mechanics? Molecular Dynamics the
Scientific Computing II Molecular Dynamics Simulation Michael Bader SCCS Summer Term 2015 Molecular Dynamics Simulation, Summer Term 2015 1 Continuum Mechanics for Fluid Mechanics? Molecular Dynamics the
From Atoms to Materials: Predictive Theory and Simulations
 From Atoms to Materials: Predictive Theory and Simulations Week 3 Lecture 4 Potentials for metals and semiconductors Ale Strachan strachan@purdue.edu School of Materials Engineering & Birck anotechnology
From Atoms to Materials: Predictive Theory and Simulations Week 3 Lecture 4 Potentials for metals and semiconductors Ale Strachan strachan@purdue.edu School of Materials Engineering & Birck anotechnology
Set the initial conditions r i. Update neighborlist. r i. Get new forces F i
 Set the initial conditions r i t 0, v i t 0 Update neighborlist Get new forces F i r i Solve the equations of motion numerically over time step t : r i t n r i t n + v i t n v i t n + Perform T, P scaling
Set the initial conditions r i t 0, v i t 0 Update neighborlist Get new forces F i r i Solve the equations of motion numerically over time step t : r i t n r i t n + v i t n v i t n + Perform T, P scaling
The micro-properties of [hmpy+] [Tf 2 N-] Ionic liquid: a simulation. study. 1. Introduction
![The micro-properties of [hmpy+] [Tf 2 N-] Ionic liquid: a simulation. study. 1. Introduction](/thumbs/72/66941683.jpg "The micro-properties of [hmpy+] [Tf 2 N-] Ionic liquid: a simulation. study. 1. Introduction") ISBN 978-1-84626-081-0 Proceedings of the 2010 International Conference on Application of Mathematics and Physics Volume 1: Advances on Space Weather, Meteorology and Applied Physics Nanjing, P. R. China,
ISBN 978-1-84626-081-0 Proceedings of the 2010 International Conference on Application of Mathematics and Physics Volume 1: Advances on Space Weather, Meteorology and Applied Physics Nanjing, P. R. China,
Atoms & Their Interactions
 Lecture 2 Atoms & Their Interactions Si: the heart of electronic materials Intel, 300mm Si wafer, 200 μm thick and 48-core CPU ( cloud computing on a chip ) Twin Creeks Technologies, San Jose, Si wafer,
Lecture 2 Atoms & Their Interactions Si: the heart of electronic materials Intel, 300mm Si wafer, 200 μm thick and 48-core CPU ( cloud computing on a chip ) Twin Creeks Technologies, San Jose, Si wafer,
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Computer simulation methods (2) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Quantum Chemical Simulations and Descriptors. Dr. Antonio Chana, Dr. Mosè Casalegno
 Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Chapter 2 Experimental sources of intermolecular potentials
 Chapter 2 Experimental sources of intermolecular potentials 2.1 Overview thermodynamical properties: heat of vaporization (Trouton s rule) crystal structures ionic crystals rare gas solids physico-chemical
Chapter 2 Experimental sources of intermolecular potentials 2.1 Overview thermodynamical properties: heat of vaporization (Trouton s rule) crystal structures ionic crystals rare gas solids physico-chemical
CHAPTER 2 INTERATOMIC FORCES. atoms together in a solid?
 CHAPTER 2 INTERATOMIC FORCES What kind of force holds the atoms together in a solid? Interatomic Binding All of the mechanisms which cause bonding between the atoms derive from electrostatic interaction
CHAPTER 2 INTERATOMIC FORCES What kind of force holds the atoms together in a solid? Interatomic Binding All of the mechanisms which cause bonding between the atoms derive from electrostatic interaction
Temperature and Pressure Controls
 Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry room B430, Chemicum 4th floor vesa.hanninen@helsinki.fi September 3, 2013 Introduction and theoretical backround September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry room B430, Chemicum 4th floor vesa.hanninen@helsinki.fi September 3, 2013 Introduction and theoretical backround September
Charge equilibration
 Charge equilibration Taylor expansion of energy of atom A @E E A (Q) =E A0 + Q A + 1 @Q A 0 2 Q2 A @ 2 E @Q 2 A 0 +... The corresponding energy of cation/anion and neutral atom E A (+1) = E A0 + @E @Q
Charge equilibration Taylor expansion of energy of atom A @E E A (Q) =E A0 + Q A + 1 @Q A 0 2 Q2 A @ 2 E @Q 2 A 0 +... The corresponding energy of cation/anion and neutral atom E A (+1) = E A0 + @E @Q
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Condensed matter physics FKA091
 Condensed matter physics FKA091 Ermin Malic Department of Physics Chalmers University of Technology Henrik Johannesson Department of Physics University of Gothenburg Teaching assistants: Roland Jago &
Condensed matter physics FKA091 Ermin Malic Department of Physics Chalmers University of Technology Henrik Johannesson Department of Physics University of Gothenburg Teaching assistants: Roland Jago &
Lattice energy of ionic solids
 1 Lattice energy of ionic solids Interatomic Forces Solids are aggregates of atoms, ions or molecules. The bonding between these particles may be understood in terms of forces that play between them. Attractive
1 Lattice energy of ionic solids Interatomic Forces Solids are aggregates of atoms, ions or molecules. The bonding between these particles may be understood in terms of forces that play between them. Attractive
Ab initio molecular dynamics
 Ab initio molecular dynamics Molecular dynamics Why? allows realistic simulation of equilibrium and transport properties in Nature ensemble averages can be used for statistical mechanics time evolution
Ab initio molecular dynamics Molecular dynamics Why? allows realistic simulation of equilibrium and transport properties in Nature ensemble averages can be used for statistical mechanics time evolution
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012
 Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012 K. Kremer Max Planck Institute for Polymer Research, Mainz Overview Simulations, general considerations
Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012 K. Kremer Max Planck Institute for Polymer Research, Mainz Overview Simulations, general considerations
Molecular Dynamics. A very brief introduction
 Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Chemical bonding in solids from ab-initio Calculations
 Chemical bonding in solids from ab-initio Calculations 1 Prof.P. Ravindran, Department of Physics, Central University of Tamil Nadu, India & Center for Materials Science and Nanotechnology, University
Chemical bonding in solids from ab-initio Calculations 1 Prof.P. Ravindran, Department of Physics, Central University of Tamil Nadu, India & Center for Materials Science and Nanotechnology, University
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Ideal Gas Laws Empirical Gas Laws The Mole Equations of State Dalton's Law The Mole Fraction Extensive and Intensive Variables Graham's Law of
 Ideal Gas Laws Empirical Gas Laws The Mole Equations of State Dalton's Law The Mole Fraction Extensive and Intensive Variables Graham's Law of Effusion The Maxwell-Boltzmann Distribution A Digression on
Ideal Gas Laws Empirical Gas Laws The Mole Equations of State Dalton's Law The Mole Fraction Extensive and Intensive Variables Graham's Law of Effusion The Maxwell-Boltzmann Distribution A Digression on
Interatomic bonding 1
 Interatomic bonding 1 Bonding forces of atoms All forces playing role in bonding are electrostatic Coulomb forces. Nuclei attract electrons, but nuclei repulse each other as well as electrons do. So, bonding
Interatomic bonding 1 Bonding forces of atoms All forces playing role in bonding are electrostatic Coulomb forces. Nuclei attract electrons, but nuclei repulse each other as well as electrons do. So, bonding
Computational Physics. J. M. Thijssen
 Computational Physics J. M. Thijssen Delft University of Technology CAMBRIDGE UNIVERSITY PRESS Contents Preface xi 1 Introduction 1 1.1 Physics and computational physics 1 1.2 Classical mechanics and statistical
Computational Physics J. M. Thijssen Delft University of Technology CAMBRIDGE UNIVERSITY PRESS Contents Preface xi 1 Introduction 1 1.1 Physics and computational physics 1 1.2 Classical mechanics and statistical
7 To solve numerically the equation of motion, we use the velocity Verlet or leap frog algorithm. _ V i n = F i n m i (F.5) For time step, we approxim
 For time step, we approxim") 69 Appendix F Molecular Dynamics F. Introduction In this chapter, we deal with the theories and techniques used in molecular dynamics simulation. The fundamental dynamics equations of any system is the
69 Appendix F Molecular Dynamics F. Introduction In this chapter, we deal with the theories and techniques used in molecular dynamics simulation. The fundamental dynamics equations of any system is the
Thermodynamics of Solids: Harmonic and Quasi-harmonic Approximations
 Thermodynamics of Solids: Harmonic and Quasi-harmonic Approximations, USA, July 9-14, 2017 Alessandro Erba Dipartimento di Chimica, Università di Torino (Italy) alessandro.erba@unito.it 2017 Outline -
Thermodynamics of Solids: Harmonic and Quasi-harmonic Approximations, USA, July 9-14, 2017 Alessandro Erba Dipartimento di Chimica, Università di Torino (Italy) alessandro.erba@unito.it 2017 Outline -
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory.
 Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Molecular mechanics. classical description of molecules. Marcus Elstner and Tomáš Kubař. April 29, 2016
 classical description of molecules April 29, 2016 Chemical bond Conceptual and chemical basis quantum effect solution of the SR numerically expensive (only small molecules can be treated) approximations
classical description of molecules April 29, 2016 Chemical bond Conceptual and chemical basis quantum effect solution of the SR numerically expensive (only small molecules can be treated) approximations
Reactive potentials and applications
 1.021, 3.021, 10.333, 22.00 Introduction to Modeling and Simulation Spring 2011 Part I Continuum and particle methods Reactive potentials and applications Lecture 8 Markus J. Buehler Laboratory for Atomistic
1.021, 3.021, 10.333, 22.00 Introduction to Modeling and Simulation Spring 2011 Part I Continuum and particle methods Reactive potentials and applications Lecture 8 Markus J. Buehler Laboratory for Atomistic
Supplementary Materials
 Supplementary Materials Atomistic Origin of Brittle Failure of Boron Carbide from Large Scale Reactive Dynamics Simulations; Suggestions toward Improved Ductility Qi An and William A. Goddard III * Materials
Supplementary Materials Atomistic Origin of Brittle Failure of Boron Carbide from Large Scale Reactive Dynamics Simulations; Suggestions toward Improved Ductility Qi An and William A. Goddard III * Materials
Lecture 11 - Phonons II - Thermal Prop. Continued
 Phonons II - hermal Properties - Continued (Kittel Ch. 5) Low High Outline Anharmonicity Crucial for hermal expansion other changes with pressure temperature Gruneisen Constant hermal Heat ransport Phonon
Phonons II - hermal Properties - Continued (Kittel Ch. 5) Low High Outline Anharmonicity Crucial for hermal expansion other changes with pressure temperature Gruneisen Constant hermal Heat ransport Phonon
A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane
 A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane Supplementary Information: Simulation Procedure and Physical Property Analysis Simulation Procedure The molecular
A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane Supplementary Information: Simulation Procedure and Physical Property Analysis Simulation Procedure The molecular
Practical Guide to Density Functional Theory (DFT)
") Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
Ionic Bonding. Example: Atomic Radius: Na (r = 0.192nm) Cl (r = 0.099nm) Ionic Radius : Na (r = 0.095nm) Cl (r = 0.181nm)
 Cl (r = 0.099nm) Ionic Radius : Na (r = 0.095nm) Cl (r = 0.181nm)") Ionic Bonding Ion: an atom or molecule that gains or loses electrons (acquires an electrical charge). Atoms form cations (+charge), when they lose electrons, or anions (- charge), when they gain electrons.
Ionic Bonding Ion: an atom or molecule that gains or loses electrons (acquires an electrical charge). Atoms form cations (+charge), when they lose electrons, or anions (- charge), when they gain electrons.
DFT EXERCISES. FELIPE CERVANTES SODI January 2006
 DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
DFT EXERCISES FELIPE CERVANTES SODI January 2006 http://www.csanyi.net/wiki/space/dftexercises Dr. Gábor Csányi 1 Hydrogen atom Place a single H atom in the middle of a largish unit cell (start with a
Chem 3502/4502 Physical Chemistry II (Quantum Mechanics) 3 Credits Spring Semester 2006 Christopher J. Cramer. Lecture 17, March 1, 2006
 3 Credits Spring Semester 2006 Christopher J. Cramer. Lecture 17, March 1, 2006") Chem 3502/4502 Physical Chemistry II (Quantum Mechanics) 3 Credits Spring Semester 2006 Christopher J. Cramer Lecture 17, March 1, 2006 (Some material in this lecture has been adapted from Cramer, C. J.
Chem 3502/4502 Physical Chemistry II (Quantum Mechanics) 3 Credits Spring Semester 2006 Christopher J. Cramer Lecture 17, March 1, 2006 (Some material in this lecture has been adapted from Cramer, C. J.
Materials 218/UCSB: Class III Cohesion in solids van der Waals, ionic, covalent, metallic
 Materials 218/UCSB: Class III Cohesion in solids van der Waals, ionic, covalent, metallic Ram Seshadri (seshadri@mrl.ucsb.edu) Introduction There are four forces in nature. The strong and the weak interactions
Materials 218/UCSB: Class III Cohesion in solids van der Waals, ionic, covalent, metallic Ram Seshadri (seshadri@mrl.ucsb.edu) Introduction There are four forces in nature. The strong and the weak interactions
GEM4 Summer School OpenCourseWare
 GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
GEM4 Summer School OpenCourseWare http://gem4.educommons.net/ http://www.gem4.org/ Lecture: Molecular Mechanics by Ju Li. Given August 9, 2006 during the GEM4 session at MIT in Cambridge, MA. Please use
Simulation of molecular systems by molecular dynamics
 Simulation of molecular systems by molecular dynamics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Dynamics, Label RFCT 2015 November 26, 2015 1 / 35 Introduction
Simulation of molecular systems by molecular dynamics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Dynamics, Label RFCT 2015 November 26, 2015 1 / 35 Introduction
Molecular Mechanics: The Ab Initio Foundation
 Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
Molecular Mechanics: The Ab Initio Foundation Ju Li GEM4 Summer School 2006 Cell and Molecular Mechanics in BioMedicine August 7 18, 2006, MIT, Cambridge, MA, USA 2 Outline Why are electrons quantum? Born-Oppenheimer
1. Thermodynamics 1.1. A macroscopic view of matter
 1. Thermodynamics 1.1. A macroscopic view of matter Intensive: independent of the amount of substance, e.g. temperature,pressure. Extensive: depends on the amount of substance, e.g. internal energy, enthalpy.
1. Thermodynamics 1.1. A macroscopic view of matter Intensive: independent of the amount of substance, e.g. temperature,pressure. Extensive: depends on the amount of substance, e.g. internal energy, enthalpy.
A Study of the Thermal Properties of a One. Dimensional Lennard-Jones System
 A Study of the Thermal Properties of a One Dimensional Lennard-Jones System Abstract In this study, the behavior of a one dimensional (1D) Lennard-Jones (LJ) system is simulated. As part of this research,
A Study of the Thermal Properties of a One Dimensional Lennard-Jones System Abstract In this study, the behavior of a one dimensional (1D) Lennard-Jones (LJ) system is simulated. As part of this research,
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
CHAPTER ELEVEN KINETIC MOLECULAR THEORY OF LIQUIDS AND SOLIDS KINETIC MOLECULAR THEORY OF LIQUIDS AND SOLIDS
 CHAPTER ELEVEN AND LIQUIDS AND SOLIDS KINETIC MOLECULAR THEORY OF LIQUIDS AND SOLIDS Differences between condensed states and gases? KINETIC MOLECULAR THEORY OF LIQUIDS AND SOLIDS Phase Homogeneous part
CHAPTER ELEVEN AND LIQUIDS AND SOLIDS KINETIC MOLECULAR THEORY OF LIQUIDS AND SOLIDS Differences between condensed states and gases? KINETIC MOLECULAR THEORY OF LIQUIDS AND SOLIDS Phase Homogeneous part
1. Introduction to Clusters
 1. Introduction to Clusters 1.1 The Field of Clusters Atomic clusters are aggregates of atoms containing from few to a few thousand atoms. Due to their small size, the properties of the clusters are, in
1. Introduction to Clusters 1.1 The Field of Clusters Atomic clusters are aggregates of atoms containing from few to a few thousand atoms. Due to their small size, the properties of the clusters are, in
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
 lectures accompanying the book: Solid State Physics: An Introduction, by Philip ofmann (2nd edition 2015, ISBN-10: 3527412824, ISBN-13: 978-3527412822, Wiley-VC Berlin. www.philiphofmann.net 1 Bonds between
lectures accompanying the book: Solid State Physics: An Introduction, by Philip ofmann (2nd edition 2015, ISBN-10: 3527412824, ISBN-13: 978-3527412822, Wiley-VC Berlin. www.philiphofmann.net 1 Bonds between
Statistical Mechanics in a Nutshell
 Chapter 2 Statistical Mechanics in a Nutshell Adapted from: Understanding Molecular Simulation Daan Frenkel and Berend Smit Academic Press (2001) pp. 9-22 11 2.1 Introduction In this course, we will treat
Chapter 2 Statistical Mechanics in a Nutshell Adapted from: Understanding Molecular Simulation Daan Frenkel and Berend Smit Academic Press (2001) pp. 9-22 11 2.1 Introduction In this course, we will treat