Ab initio Molecular Dynamics Born Oppenheimer and beyond
|
|
|
- Tracy Leon Robertson
- 5 years ago
- Views:
Transcription
1 Ab initio Molecular Dynamics Born Oppenheimer and beyond
2 Reminder, reliability of MD MD trajectories are chaotic (exponential divergence with respect to initial conditions), BUT... With a good integrator (e.g., Verlet or velocity Verlet) the discrete trajectory, with the same initial and final point as the exact analytical one, can be made infinitely close to the latter (shadow theorem/hypothesis). So: the dynamical information are reliable. What about the ensemble averages? The ergodic theorem/hypothesis tells us that time and ensemble averages are equivalent (at the limit of infinite sampling time). Not true for all systems, though (indeed called non ergodic).
3 Ab initio MD, general formalism Schrödringer equation for a system of N nuclei and n electrons: acts on nuclei acts on electrons
4 Ab initio MD, general formalism Suppose the solutions of the time independent electronic Schrödinger equation are known : correction to the adiabatic eigenvalue of
5 Ab initio MD, Born Oppenheimer approximation If all are negligble: Adiabatic approximation (Born Oppenheimer): Valid in many cases, but, notably, not for electron transfer, photoisomerisation,...
6 Ab initio MD, semi classical approach classical trajectory adiabatic eigenfunctions : probability of finding the system in adiabatic state i at time t
7 Ab initio MD, validity of the BO approximation characteristic length time scale of electronic motion velocity of the nuclei Massay parameter
8 Ab initio MD, semi classical BO approximation The system, when prepared in one adiabatic state i stays there forever Nuclei move according to Newton's equations:
9 Non adiabatic: mean field (Ehrenfest) dynamics The atoms evolve on an effective potential representing an average over the adiabatic states weighted by their state populations Forces: Hellmann Feynman theorem: non adiabatic coupling vectors

10 Ehrenfest dynamics A system that was initially prepared in a pure adiabatic state will be in a mixed state when leaving the region of strong nonadiabatic coupling. The pure adiabatic character of the wavefunction cannot be recovered even in the asymptotic regions of configuration space. The total wavefunction may contain significant contributions from adiabatic states that are energetically inaccessible.
11 Ehrnefest dynamics, violation of microscopic reversibility

12 Surface hopping (fewest switches) Let there be N trajectories: At a later time: Supposing:
13 Surface hopping (fewest switches) Transition selected by using random numbers
14 Surface hopping (fewest switches) Transition from i to k invoked if : Sum of the transition probabilities of the first k states Uniform random number Between 0 and 1
15 Photoisomerization of formaldimine
16 Photoisomerization of formaldimine R P R R
17 Car Parrinello MD, a classical system Suppose a fluid of polarizable molecules E.g., dipolar molecules: permanent + induced dipole moment dipole dipole tensor Induced dipoles follow nuclei adiabatically and is always at its minimum: Iterative solution of (3N) linear equations?
18 Car Parrinello MD, a classical system Extended Lagrangian: Equation of motion for dipoles: Role of M? Adiabadicity: two temperatures?
19 Car Parrinello MD, the idea Starting from minimized Kohn Sham orbitals, define orbital velocities and kinetic energy: units? Lagrange multipliers Equations of motion: Functional derivative. In practice wrt the coefficients of the chosen basis set
20 Car Parrinello MD, the algorithm Initial conditions: Update velocities (Δt/2) Update positions (Δt) Constraint? Initial forces:
21 Car Parrinello MD, the algorithm Iterative solution: Starting from:
22 Car Parrinello MD, the algorithm (summary) Velocities: Positions: Forces update: 5 6
23 Molecular dynamics: summary of extended systems and applications of Born Oppenheimer MD
24 Reminder, reliability of MD MD trajectories are chaotic (exponential divergence with respect to initial conditions), BUT... With a good integrator (e.g., Verlet or velocity Verlet) the discrete trajectory, with the same initial and final point as the exact analytical one, can be made infinitely close to the latter (shadow theorem/hypothesis). So: the dynamical information are reliable. What about the ensemble averages? The ergodic theorem/hypothesis tells us that time and ensemble averages are equivalent (at the limit of infinite sampling time). Not true for all systems, though (indeed called non ergodic).
25 Summary of extended systems: NVT (Nosé) Lagrangian: Conjugated momenta: Hamiltonian Eq. of motion:


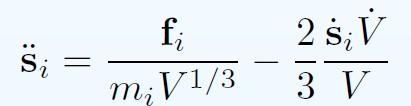

26 Summary of extended systems: NPH (Andersen)

27 Summary of extended systems: NPH (Parrinello Rahman)
28 Summary of extended systems: on the fly optimization Car Parrinello MD Classical example: polarizable fluid
29 Application of plain MD: nature of liquid C Galli et al. Science C atoms 300 fs
30 Application of plain MD: nature of liquid C Galli et al. Phys. Rev. Lett K

31 Application of plain MD: dissociative adsorption Phys. Rev. Lett. 1993

32 Collision Final (400fs) Trajectory 1 Trajectory 2 Trajectory 3 Trajectory 4 Trajectory 5 Kinetic energy: 1 ev
33 Application of plain MD: solvation of DMSO in water Kirchner and Hutter, J. Chem. Phys water molecules K gas phase DMSO Water DMSO
Ab initio molecular dynamics and nuclear quantum effects
 Ab initio molecular dynamics and nuclear quantum effects Luca M. Ghiringhelli Fritz Haber Institute Hands on workshop density functional theory and beyond: First principles simulations of molecules and
Ab initio molecular dynamics and nuclear quantum effects Luca M. Ghiringhelli Fritz Haber Institute Hands on workshop density functional theory and beyond: First principles simulations of molecules and
Ab initio molecular dynamics. Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy. Bangalore, 04 September 2014
 Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Ab initio molecular dynamics: Basic Theory and Advanced Met
 Ab initio molecular dynamics: Basic Theory and Advanced Methods Uni Mainz October 30, 2016 Bio-inspired catalyst for hydrogen production Ab-initio MD simulations are used to learn how the active site
Ab initio molecular dynamics: Basic Theory and Advanced Methods Uni Mainz October 30, 2016 Bio-inspired catalyst for hydrogen production Ab-initio MD simulations are used to learn how the active site
Energy and Forces in DFT
 Energy and Forces in DFT Total Energy as a function of nuclear positions {R} E tot ({R}) = E DF T ({R}) + E II ({R}) (1) where E DF T ({R}) = DFT energy calculated for the ground-state density charge-density
Energy and Forces in DFT Total Energy as a function of nuclear positions {R} E tot ({R}) = E DF T ({R}) + E II ({R}) (1) where E DF T ({R}) = DFT energy calculated for the ground-state density charge-density
MD Thermodynamics. Lecture 12 3/26/18. Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky
 MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
Ab Ini'o Molecular Dynamics (MD) Simula?ons
 Simula?ons") Ab Ini'o Molecular Dynamics (MD) Simula?ons Rick Remsing ICMS, CCDM, Temple University, Philadelphia, PA What are Molecular Dynamics (MD) Simulations? Technique to compute statistical and transport properties
Ab Ini'o Molecular Dynamics (MD) Simula?ons Rick Remsing ICMS, CCDM, Temple University, Philadelphia, PA What are Molecular Dynamics (MD) Simulations? Technique to compute statistical and transport properties
Ab initio molecular dynamics
 Ab initio molecular dynamics Molecular dynamics Why? allows realistic simulation of equilibrium and transport properties in Nature ensemble averages can be used for statistical mechanics time evolution
Ab initio molecular dynamics Molecular dynamics Why? allows realistic simulation of equilibrium and transport properties in Nature ensemble averages can be used for statistical mechanics time evolution
Modeling of Nanostructures and Materials Jacek A. Majewski Summer Semester 2013 Lecture 6 Lecture Modeling of Nanostructures Molecular Dynamics
 Chair of Condensed Matter Physics nstitute of Theoretical Physics Faculty of Physics, Universityof Warsaw Summer Semester 013 Lecture Modeling of Nanostructures and Materials Jacek A. Majewski E-mail:
Chair of Condensed Matter Physics nstitute of Theoretical Physics Faculty of Physics, Universityof Warsaw Summer Semester 013 Lecture Modeling of Nanostructures and Materials Jacek A. Majewski E-mail:
Accelerated Quantum Molecular Dynamics
 Accelerated Quantum Molecular Dynamics Enrique Martinez, Christian Negre, Marc J. Cawkwell, Danny Perez, Arthur F. Voter and Anders M. N. Niklasson Outline Quantum MD Current approaches Challenges Extended
Accelerated Quantum Molecular Dynamics Enrique Martinez, Christian Negre, Marc J. Cawkwell, Danny Perez, Arthur F. Voter and Anders M. N. Niklasson Outline Quantum MD Current approaches Challenges Extended
Electronic Structure Methods for molecular excited states
 Electronic Structure Methods for molecular excited states Summer School on Time dependent Density-Functional Theory: Prospects and Applications Benasque, Spain September 2008 Ivano Tavernelli EPFL CH-1015
Electronic Structure Methods for molecular excited states Summer School on Time dependent Density-Functional Theory: Prospects and Applications Benasque, Spain September 2008 Ivano Tavernelli EPFL CH-1015
Molecular Dynamics Beyond the Born-Oppenheimer Approximation: Mixed Quantum Classical Approaches
 John von Neumann Institute for Computing Molecular Dynamics Beyond the Born-Oppenheimer Approximation: Mixed Quantum Classical Approaches Nikos L. Doltsinis published in Computational Nanoscience: Do It
John von Neumann Institute for Computing Molecular Dynamics Beyond the Born-Oppenheimer Approximation: Mixed Quantum Classical Approaches Nikos L. Doltsinis published in Computational Nanoscience: Do It
Car-Parrinello Molecular Dynamics
 Car-Parrinello Molecular Dynamics Eric J. Bylaska HPCC Group Moral: A man dreams of a miracle and wakes up with loaves of bread Erich Maria Remarque Molecular Dynamics Loop (1) Compute Forces on atoms,
Car-Parrinello Molecular Dynamics Eric J. Bylaska HPCC Group Moral: A man dreams of a miracle and wakes up with loaves of bread Erich Maria Remarque Molecular Dynamics Loop (1) Compute Forces on atoms,
SUPPLEMENTARY INFORMATION
 SUPPLEMENTARY INFORMATION DOI: 10.1038/NPHYS2210 Femtosecond torsional relaxation Theoretical methodology: J. Clark, S. Tretiak, T. Nelson, G. Cirmi & G. Lanzani To model non-adiabatic excited state dynamics
SUPPLEMENTARY INFORMATION DOI: 10.1038/NPHYS2210 Femtosecond torsional relaxation Theoretical methodology: J. Clark, S. Tretiak, T. Nelson, G. Cirmi & G. Lanzani To model non-adiabatic excited state dynamics
Molecular Dynamics. Park City June 2005 Tully
 Molecular Dynamics John Lance Natasa Vinod Xiaosong Dufie Priya Sharani Hongzhi Group: August, 2004 Prelude: Classical Mechanics Newton s equations: F = ma = mq = p Force is the gradient of the potential:
Molecular Dynamics John Lance Natasa Vinod Xiaosong Dufie Priya Sharani Hongzhi Group: August, 2004 Prelude: Classical Mechanics Newton s equations: F = ma = mq = p Force is the gradient of the potential:
IV. Classical Molecular Dynamics
 IV. Classical Molecular Dynamics Basic Assumptions: 1. Born-Oppenheimer Approximation 2. Classical mechanical nuclear motion Unavoidable Additional Approximations: 1. Approximate potential energy surface
IV. Classical Molecular Dynamics Basic Assumptions: 1. Born-Oppenheimer Approximation 2. Classical mechanical nuclear motion Unavoidable Additional Approximations: 1. Approximate potential energy surface
Mixed quantum-classical dynamics. Maurizio Persico. Università di Pisa Dipartimento di Chimica e Chimica Industriale
 Mixed quantum-classical dynamics. Maurizio Persico Università di Pisa Dipartimento di Chimica e Chimica Industriale Outline of this talk. The nuclear coordinates as parameters in the time-dependent Schroedinger
Mixed quantum-classical dynamics. Maurizio Persico Università di Pisa Dipartimento di Chimica e Chimica Industriale Outline of this talk. The nuclear coordinates as parameters in the time-dependent Schroedinger
Ab initio molecular dynamics : BO
 School on First Principles Simulations, JNCASR, 2010 Ab initio molecular dynamics : BO Vardha Srinivasan IISER Bhopal Why ab initio MD Free of parametrization and only fundamental constants required. Bond
School on First Principles Simulations, JNCASR, 2010 Ab initio molecular dynamics : BO Vardha Srinivasan IISER Bhopal Why ab initio MD Free of parametrization and only fundamental constants required. Bond
Conical Intersections. Spiridoula Matsika
 Conical Intersections Spiridoula Matsika The Born-Oppenheimer approximation Energy TS Nuclear coordinate R ν The study of chemical systems is based on the separation of nuclear and electronic motion The
Conical Intersections Spiridoula Matsika The Born-Oppenheimer approximation Energy TS Nuclear coordinate R ν The study of chemical systems is based on the separation of nuclear and electronic motion The
Quantum Molecular Dynamics Basics
 Quantum Molecular Dynamics Basics Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Quantum Molecular Dynamics Basics Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Nonadiabatic Dynamics: Mean-Field and Surface Hopping
 John von Neumann Institute for Computing Nonadiabatic Dynamics: Mean-Field and Surface Hopping Nikos L. Doltsinis published in Quantum Simulations of Complex Many-Body Systems: From Theory to Algorithms,
John von Neumann Institute for Computing Nonadiabatic Dynamics: Mean-Field and Surface Hopping Nikos L. Doltsinis published in Quantum Simulations of Complex Many-Body Systems: From Theory to Algorithms,
23 The Born-Oppenheimer approximation, the Many Electron Hamiltonian and the molecular Schrödinger Equation M I
 23 The Born-Oppenheimer approximation, the Many Electron Hamiltonian and the molecular Schrödinger Equation 1. Now we will write down the Hamiltonian for a molecular system comprising N nuclei and n electrons.
23 The Born-Oppenheimer approximation, the Many Electron Hamiltonian and the molecular Schrödinger Equation 1. Now we will write down the Hamiltonian for a molecular system comprising N nuclei and n electrons.
First-principles Molecular Dynamics Simulations
 First-principles Molecular Dynamics Simulations François Gygi University of California, Davis fgygi@ucdavis.edu http://eslab.ucdavis.edu http://www.quantum-simulation.org MICCoM Computational School, Jul
First-principles Molecular Dynamics Simulations François Gygi University of California, Davis fgygi@ucdavis.edu http://eslab.ucdavis.edu http://www.quantum-simulation.org MICCoM Computational School, Jul
CHEM6085: Density Functional Theory
 Lecture 5 CHEM6085: Density Functional Theory Orbital-free (or pure ) DFT C.-K. Skylaris 1 Consists of three terms The electronic Hamiltonian operator Electronic kinetic energy operator Electron-Electron
Lecture 5 CHEM6085: Density Functional Theory Orbital-free (or pure ) DFT C.-K. Skylaris 1 Consists of three terms The electronic Hamiltonian operator Electronic kinetic energy operator Electron-Electron
Electrochemistry project, Chemistry Department, November Ab-initio Molecular Dynamics Simulation
 Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
Electrochemistry project, Chemistry Department, November 2006 Ab-initio Molecular Dynamics Simulation Outline Introduction Ab-initio concepts Total energy concepts Adsorption energy calculation Project
FIRST PRINCIPLES MOLECULAR DYNAMICS INVOLVING EXCITED STATES AND NONADIABATIC TRANSITIONS
 Journal of Theoretical and Computational Chemistry, Vol., No. 2 (2002) 39 349 c World Scientific Publishing Company FIRST PRINCIPLES MOLECULAR DYNAMICS INVOLVING EXCITED STATES AND NONADIABATIC TRANSITIONS
Journal of Theoretical and Computational Chemistry, Vol., No. 2 (2002) 39 349 c World Scientific Publishing Company FIRST PRINCIPLES MOLECULAR DYNAMICS INVOLVING EXCITED STATES AND NONADIABATIC TRANSITIONS
What is Classical Molecular Dynamics?
 What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
We start with some important background material in classical and quantum mechanics.
 Chapter Basics We start with some important background material in classical and quantum mechanics.. Classical mechanics Lagrangian mechanics Compared to Newtonian mechanics, Lagrangian mechanics has the
Chapter Basics We start with some important background material in classical and quantum mechanics.. Classical mechanics Lagrangian mechanics Compared to Newtonian mechanics, Lagrangian mechanics has the
Density Functional Theory
 Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Density Functional Theory: from theory to Applications
 Density Functional Theory: from theory to Applications Uni Mainz May 27, 2012 Large barrier-activated processes time-dependent bias potential extended Lagrangian formalism Basic idea: during the MD dynamics
Density Functional Theory: from theory to Applications Uni Mainz May 27, 2012 Large barrier-activated processes time-dependent bias potential extended Lagrangian formalism Basic idea: during the MD dynamics
Time reversible Born Oppenheimer molecular dynamics
 Time reversible Born Oppenheimer molecular dynamics Jianfeng Lu Mathematics Department Department of Physics Duke University jianfeng@math.duke.edu KI-Net Conference, CSCAMM, University of Maryland, May
Time reversible Born Oppenheimer molecular dynamics Jianfeng Lu Mathematics Department Department of Physics Duke University jianfeng@math.duke.edu KI-Net Conference, CSCAMM, University of Maryland, May
CHEM6085: Density Functional Theory Lecture 10
 CHEM6085: Density Functional Theory Lecture 10 1) Spin-polarised calculations 2) Geometry optimisation C.-K. Skylaris 1 Unpaired electrons So far we have developed Kohn-Sham DFT for the case of paired
CHEM6085: Density Functional Theory Lecture 10 1) Spin-polarised calculations 2) Geometry optimisation C.-K. Skylaris 1 Unpaired electrons So far we have developed Kohn-Sham DFT for the case of paired
P. W. Atkins and R. S. Friedman. Molecular Quantum Mechanics THIRD EDITION
 P. W. Atkins and R. S. Friedman Molecular Quantum Mechanics THIRD EDITION Oxford New York Tokyo OXFORD UNIVERSITY PRESS 1997 Introduction and orientation 1 Black-body radiation 1 Heat capacities 2 The
P. W. Atkins and R. S. Friedman Molecular Quantum Mechanics THIRD EDITION Oxford New York Tokyo OXFORD UNIVERSITY PRESS 1997 Introduction and orientation 1 Black-body radiation 1 Heat capacities 2 The
Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals
 JOURNAL OF CHEMICAL PHYSICS VOLUME 114, NUMBER 8 JUNE 001 Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals H. Bernhard Schlegel and John M. Millam Department of Chemistry,
JOURNAL OF CHEMICAL PHYSICS VOLUME 114, NUMBER 8 JUNE 001 Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals H. Bernhard Schlegel and John M. Millam Department of Chemistry,
Langevin Dynamics in Constant Pressure Extended Systems
 Langevin Dynamics in Constant Pressure Extended Systems D. Quigley and M.I.J. Probert CMMP 2004 1 Talk Outline Molecular dynamics and ensembles. Existing methods for sampling at NPT. Langevin dynamics
Langevin Dynamics in Constant Pressure Extended Systems D. Quigley and M.I.J. Probert CMMP 2004 1 Talk Outline Molecular dynamics and ensembles. Existing methods for sampling at NPT. Langevin dynamics
Instructor background for the discussion points of Section 2
 Supplementary Information for: Orbitals Some fiction and some facts Jochen Autschbach Department of Chemistry State University of New York at Buffalo Buffalo, NY 14260 3000, USA Instructor background for
Supplementary Information for: Orbitals Some fiction and some facts Jochen Autschbach Department of Chemistry State University of New York at Buffalo Buffalo, NY 14260 3000, USA Instructor background for
Molecular energy levels
 Molecular energy levels Hierarchy of motions and energies in molecules The different types of motion in a molecule (electronic, vibrational, rotational,: : :) take place on different time scales and are
Molecular energy levels Hierarchy of motions and energies in molecules The different types of motion in a molecule (electronic, vibrational, rotational,: : :) take place on different time scales and are
Under evolution for a small time δt the area A(t) = q p evolves into an area
 = q p evolves into an area") Physics 106a, Caltech 6 November, 2018 Lecture 11: Hamiltonian Mechanics II Towards statistical mechanics Phase space volumes are conserved by Hamiltonian dynamics We can use many nearby initial conditions
Physics 106a, Caltech 6 November, 2018 Lecture 11: Hamiltonian Mechanics II Towards statistical mechanics Phase space volumes are conserved by Hamiltonian dynamics We can use many nearby initial conditions
Density Functional Theory
 Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Density Functional Theory March 26, 2009 ? DENSITY FUNCTIONAL THEORY is a method to successfully describe the behavior of atomic and molecular systems and is used for instance for: structural prediction
Time-Dependent Density-Functional Theory
 Summer School on First Principles Calculations for Condensed Matter and Nanoscience August 21 September 3, 2005 Santa Barbara, California Time-Dependent Density-Functional Theory X. Gonze, Université Catholique
Summer School on First Principles Calculations for Condensed Matter and Nanoscience August 21 September 3, 2005 Santa Barbara, California Time-Dependent Density-Functional Theory X. Gonze, Université Catholique
QUANTUM AND THERMAL MOTION IN MOLECULES FROM FIRST-PRINCIPLES
 QUANTUM AND THERMAL MOTION IN MOLECULES FROM FIRST-PRINCIPLES 1 Tapio T. Rantala, Department of Physics, Tampere University of Technology http://www.tut.fi/semiphys CONTENTS MOTIVATION PATH INTEGRAL APPROACH
QUANTUM AND THERMAL MOTION IN MOLECULES FROM FIRST-PRINCIPLES 1 Tapio T. Rantala, Department of Physics, Tampere University of Technology http://www.tut.fi/semiphys CONTENTS MOTIVATION PATH INTEGRAL APPROACH
Reminder from last lecture: potential energy surfaces
 Outline 1 Why Quantum Dynamics? A TDDFT approach Ivano Tavernelli IBM -Research Zurich Summer School of the Max-Planck-EPFL Center for Molecular Nanoscience & Technology 2 Nonadiabatic Bohmian dynamics
Outline 1 Why Quantum Dynamics? A TDDFT approach Ivano Tavernelli IBM -Research Zurich Summer School of the Max-Planck-EPFL Center for Molecular Nanoscience & Technology 2 Nonadiabatic Bohmian dynamics
Classical Molecular Dynamics
 Classical Molecular Dynamics Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, U.K. http://www-users.york.ac.uk/~mijp1 Overview of lecture n Motivation n Types of
Classical Molecular Dynamics Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, U.K. http://www-users.york.ac.uk/~mijp1 Overview of lecture n Motivation n Types of
Project: Vibration of Diatomic Molecules
 Project: Vibration of Diatomic Molecules Objective: Find the vibration energies and the corresponding vibrational wavefunctions of diatomic molecules H 2 and I 2 using the Morse potential. equired Numerical
Project: Vibration of Diatomic Molecules Objective: Find the vibration energies and the corresponding vibrational wavefunctions of diatomic molecules H 2 and I 2 using the Morse potential. equired Numerical
Javier Junquera. Statistical mechanics
 Javier Junquera Statistical mechanics From the microscopic to the macroscopic level: the realm of statistical mechanics Computer simulations Thermodynamic state Generates information at the microscopic
Javier Junquera Statistical mechanics From the microscopic to the macroscopic level: the realm of statistical mechanics Computer simulations Thermodynamic state Generates information at the microscopic
Detailed Balance in Ehrenfest Mixed Quantum-Classical Dynamics
 J. Chem. Theory Comput. 26, 2, 229-235 229 Detailed Balance in Ehrenfest Mixed Quantum-Classical Dynamics Priya V. Parandekar and John C. Tully* Department of Chemistry, Yale UniVersity, P.O. Box 2817,
J. Chem. Theory Comput. 26, 2, 229-235 229 Detailed Balance in Ehrenfest Mixed Quantum-Classical Dynamics Priya V. Parandekar and John C. Tully* Department of Chemistry, Yale UniVersity, P.O. Box 2817,
Exact factorization of the electron-nuclear wave function and the concept of exact forces in MD
 Exact factorization of the electron-nuclear wave function and the concept of exact forces in MD E.K.U. Gross Max-Planck Institute for Microstructure Physics Halle (Saale) OUTLINE Thanks Exact factorisation
Exact factorization of the electron-nuclear wave function and the concept of exact forces in MD E.K.U. Gross Max-Planck Institute for Microstructure Physics Halle (Saale) OUTLINE Thanks Exact factorisation
Chem 442 Review for Exam 2. Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative (3D) components.
 and relative (3D) components.") Chem 44 Review for Exam Hydrogenic atoms: The Coulomb energy between two point charges Ze and e: V r Ze r Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative
Chem 44 Review for Exam Hydrogenic atoms: The Coulomb energy between two point charges Ze and e: V r Ze r Exact separation of the Hamiltonian of a hydrogenic atom into center-of-mass (3D) and relative
III. Kinetic Theory of Gases
 III. Kinetic Theory of Gases III.A General Definitions Kinetic theory studies the macroscopic properties of large numbers of particles, starting from their (classical) equations of motion. Thermodynamics
III. Kinetic Theory of Gases III.A General Definitions Kinetic theory studies the macroscopic properties of large numbers of particles, starting from their (classical) equations of motion. Thermodynamics
7 To solve numerically the equation of motion, we use the velocity Verlet or leap frog algorithm. _ V i n = F i n m i (F.5) For time step, we approxim
 For time step, we approxim") 69 Appendix F Molecular Dynamics F. Introduction In this chapter, we deal with the theories and techniques used in molecular dynamics simulation. The fundamental dynamics equations of any system is the
69 Appendix F Molecular Dynamics F. Introduction In this chapter, we deal with the theories and techniques used in molecular dynamics simulation. The fundamental dynamics equations of any system is the
Ab-initio molecular dynamics: from the basics up to quantum effects Roberto Car Princeton University
 Ab-initio molecular dynamics: from the basics up to quantum effects Roberto Car Princeton University Hands-on Tutorial Workshop on Ab-Initio Molecular Simulations, Fritz- Haber-Institut, Berlin, July 12-21,
Ab-initio molecular dynamics: from the basics up to quantum effects Roberto Car Princeton University Hands-on Tutorial Workshop on Ab-Initio Molecular Simulations, Fritz- Haber-Institut, Berlin, July 12-21,
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
Next generation extended Lagrangian first principles molecular dynamics
 Next generation extended Lagrangian first principles molecular dynamics Anders M. N. Niklasson Theoretical Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545 (Dated: June 1, 017) LA-UR-17-3007
Next generation extended Lagrangian first principles molecular dynamics Anders M. N. Niklasson Theoretical Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545 (Dated: June 1, 017) LA-UR-17-3007
Solid State Theory: Band Structure Methods
 Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
Solid State Theory: Band Structure Methods Lilia Boeri Wed., 11:15-12:45 HS P3 (PH02112) http://itp.tugraz.at/lv/boeri/ele/ Plan of the Lecture: DFT1+2: Hohenberg-Kohn Theorem and Kohn and Sham equations.
Ab initio phonon calculations in mixed systems
 Ab initio phonon calculations in mixed systems Andrei Postnikov apostnik@uos.de Outline: Experiment vs. ab initio theory Ways of theory: linear response and frozen phonon approaches Applications: Be x
Ab initio phonon calculations in mixed systems Andrei Postnikov apostnik@uos.de Outline: Experiment vs. ab initio theory Ways of theory: linear response and frozen phonon approaches Applications: Be x
Temperature and Pressure Controls
 Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Introduction to Hartree-Fock Molecular Orbital Theory
 Introduction to Hartree-Fock Molecular Orbital Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Origins of Mathematical Modeling in Chemistry Plato (ca. 428-347
Introduction to Hartree-Fock Molecular Orbital Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Origins of Mathematical Modeling in Chemistry Plato (ca. 428-347
Lecture 6. Four postulates of quantum mechanics. The eigenvalue equation. Momentum and energy operators. Dirac delta function. Expectation values
 Lecture 6 Four postulates of quantum mechanics The eigenvalue equation Momentum and energy operators Dirac delta function Expectation values Objectives Learn about eigenvalue equations and operators. Learn
Lecture 6 Four postulates of quantum mechanics The eigenvalue equation Momentum and energy operators Dirac delta function Expectation values Objectives Learn about eigenvalue equations and operators. Learn
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride. Dimer. Philip Straughn
 Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Exchange Correlation Functional Investigation of RT-TDDFT on a Sodium Chloride Dimer Philip Straughn Abstract Charge transfer between Na and Cl ions is an important problem in physical chemistry. However,
Distributions of statistical mechanics
 CHAPTER II Distributions of statistical mechanics The purpose of Statistical Mechanics is to explain the thermodynamic properties of macroscopic systems starting from underlying microscopic models possibly
CHAPTER II Distributions of statistical mechanics The purpose of Statistical Mechanics is to explain the thermodynamic properties of macroscopic systems starting from underlying microscopic models possibly
Fast Eigenvalue Solutions
 Fast Eigenvalue Solutions Techniques! Steepest Descent/Conjugate Gradient! Davidson/Lanczos! Carr-Parrinello PDF Files will be available Where HF/DFT calculations spend time Guess ρ Form H Diagonalize
Fast Eigenvalue Solutions Techniques! Steepest Descent/Conjugate Gradient! Davidson/Lanczos! Carr-Parrinello PDF Files will be available Where HF/DFT calculations spend time Guess ρ Form H Diagonalize
G : Statistical Mechanics
 G25.2651: Statistical Mechanics Notes for Lecture 1 Defining statistical mechanics: Statistical Mechanics provies the connection between microscopic motion of individual atoms of matter and macroscopically
G25.2651: Statistical Mechanics Notes for Lecture 1 Defining statistical mechanics: Statistical Mechanics provies the connection between microscopic motion of individual atoms of matter and macroscopically
CHEM3023: Spins, Atoms and Molecules
 CHEM3023: Spins, Atoms and Molecules Lecture 3 The Born-Oppenheimer approximation C.-K. Skylaris Learning outcomes Separate molecular Hamiltonians to electronic and nuclear parts according to the Born-Oppenheimer
CHEM3023: Spins, Atoms and Molecules Lecture 3 The Born-Oppenheimer approximation C.-K. Skylaris Learning outcomes Separate molecular Hamiltonians to electronic and nuclear parts according to the Born-Oppenheimer
Quantum Physics III (8.06) Spring 2007 FINAL EXAMINATION Monday May 21, 9:00 am You have 3 hours.
 Spring 2007 FINAL EXAMINATION Monday May 21, 9:00 am You have 3 hours.") Quantum Physics III (8.06) Spring 2007 FINAL EXAMINATION Monday May 21, 9:00 am You have 3 hours. There are 10 problems, totalling 180 points. Do all problems. Answer all problems in the white books provided.
Quantum Physics III (8.06) Spring 2007 FINAL EXAMINATION Monday May 21, 9:00 am You have 3 hours. There are 10 problems, totalling 180 points. Do all problems. Answer all problems in the white books provided.
Calculating Vibrational Spectra from Molecular Dynamics
 Calculating Vibrational Spectra from Molecular Dynamics A Simulating a Trajectory with Wannier Centers To calculate IR spectra from Molecular Dynamics, it is necessary to have dipole information for the
Calculating Vibrational Spectra from Molecular Dynamics A Simulating a Trajectory with Wannier Centers To calculate IR spectra from Molecular Dynamics, it is necessary to have dipole information for the
Time-dependent density functional theory (TDDFT)
") Advanced Workshop on High-Performance & High-Throughput Materials Simulations using Quantum ESPRESSO ICTP, Trieste, Italy, January 16 to 27, 2017 Time-dependent density functional theory (TDDFT) Ralph
Advanced Workshop on High-Performance & High-Throughput Materials Simulations using Quantum ESPRESSO ICTP, Trieste, Italy, January 16 to 27, 2017 Time-dependent density functional theory (TDDFT) Ralph
Srinivasan S. Iyengar Department of Chemistry and Henry Eyring Center for Theoretical Chemistry, University of Utah, Salt Lake City, Utah
 JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 8 DECEMBER 001 Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. II. Generalizations based on mass-weighting, idempotency,
JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 8 DECEMBER 001 Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. II. Generalizations based on mass-weighting, idempotency,
Ab initio Molecular Dynamics
 Ab initio Molecular Dynamics Jürg Hutter Department of Chemistry, University of Zurich Overview Equations of motion and Integrators General Lagrangian for AIMD Car Parrinello MD and Born Oppenheimer MD
Ab initio Molecular Dynamics Jürg Hutter Department of Chemistry, University of Zurich Overview Equations of motion and Integrators General Lagrangian for AIMD Car Parrinello MD and Born Oppenheimer MD
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić
 Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Introduction to Density Functional Theory with Applications to Graphene Branislav K. Nikolić Department of Physics and Astronomy, University of Delaware, Newark, DE 19716, U.S.A. http://wiki.physics.udel.edu/phys824
Wavepacket Correlation Function Approach for Nonadiabatic Reactions: Quasi-Jahn-Teller Model
 Wavepacket Correlation for Nonadiabatic Reactions Bull. Korean Chem. Soc. 04, Vol. 35, No. 4 06 http://dx.doi.org/0.50/bkcs.04.35.4.06 Wavepacket Correlation Function Approach for Nonadiabatic Reactions:
Wavepacket Correlation for Nonadiabatic Reactions Bull. Korean Chem. Soc. 04, Vol. 35, No. 4 06 http://dx.doi.org/0.50/bkcs.04.35.4.06 Wavepacket Correlation Function Approach for Nonadiabatic Reactions:
LETTERS. Photodissociation, Recombination, and Electron Transfer in Cluster Ions: A Nonadiabatic Molecular Dynamics Study of I 2- (CO 2 ) n
 n") Copyright 1997 by the American Chemical Society VOLUME 101, NUMBER 44, OCTOBER 30, 1997 LETTERS Photodissociation, Recombination, and Electron Transfer in Cluster Ions: A Nonadiabatic Molecular Dynamics
Copyright 1997 by the American Chemical Society VOLUME 101, NUMBER 44, OCTOBER 30, 1997 LETTERS Photodissociation, Recombination, and Electron Transfer in Cluster Ions: A Nonadiabatic Molecular Dynamics
Density Functional Theory for Electrons in Materials
 Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
Density Functional Theory for Electrons in Materials Richard M. Martin Department of Physics and Materials Research Laboratory University of Illinois at Urbana-Champaign 1 Density Functional Theory for
1 Setting the stage: why ab initio molecular dynamics?
 1 Setting the stage: why ab initio molecular dynamics? Classical molecular dynamics using predefined potentials, force fields, either based on empirical data or on independent electronic structure calculations,
1 Setting the stage: why ab initio molecular dynamics? Classical molecular dynamics using predefined potentials, force fields, either based on empirical data or on independent electronic structure calculations,
Introduction to Electronic Structure Theory
 Introduction to Electronic Structure Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology June 2002 Last Revised: June 2003 1 Introduction The purpose of these
Introduction to Electronic Structure Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology June 2002 Last Revised: June 2003 1 Introduction The purpose of these
Mixing Quantum and Classical Mechanics: A Partially Miscible Solution
 Mixing Quantum and Classical Mechanics: A Partially Miscible Solution R. Kapral S. Nielsen A. Sergi D. Mac Kernan G. Ciccotti quantum dynamics in a classical condensed phase environment how to simulate
Mixing Quantum and Classical Mechanics: A Partially Miscible Solution R. Kapral S. Nielsen A. Sergi D. Mac Kernan G. Ciccotti quantum dynamics in a classical condensed phase environment how to simulate
Time-dependent density functional theory (TDDFT)
") 04/05/16 Hands-on workshop and Humboldt-Kolleg: Density-Functional Theory and Beyond - Basic Principles and Modern Insights Isfahan University of Technology, Isfahan, Iran, May 2 to 13, 2016 Time-dependent
04/05/16 Hands-on workshop and Humboldt-Kolleg: Density-Functional Theory and Beyond - Basic Principles and Modern Insights Isfahan University of Technology, Isfahan, Iran, May 2 to 13, 2016 Time-dependent
LECTURE 6 : BASICS FOR MOLECULAR SIMULATIONS - Historical perspective - Skimming over Statistical Mechanics - General idea of Molecular Dynamics -
 LECTURE 6 : BASICS FOR MOLECULAR SIMULATIONS - Historical perspective - Skimming over Statistical Mechanics - General idea of Molecular Dynamics - Force calculations, structure of MD, equations of motion
LECTURE 6 : BASICS FOR MOLECULAR SIMULATIONS - Historical perspective - Skimming over Statistical Mechanics - General idea of Molecular Dynamics - Force calculations, structure of MD, equations of motion
Chem 442 Review of Spectroscopy
 Chem 44 Review of Spectroscopy General spectroscopy Wavelength (nm), frequency (s -1 ), wavenumber (cm -1 ) Frequency (s -1 ): n= c l Wavenumbers (cm -1 ): n =1 l Chart of photon energies and spectroscopies
Chem 44 Review of Spectroscopy General spectroscopy Wavelength (nm), frequency (s -1 ), wavenumber (cm -1 ) Frequency (s -1 ): n= c l Wavenumbers (cm -1 ): n =1 l Chart of photon energies and spectroscopies
Molecular Bonding. Molecular Schrödinger equation. r - nuclei s - electrons. M j = mass of j th nucleus m 0 = mass of electron
 Molecular onding Molecular Schrödinger equation r - nuclei s - electrons 1 1 W V r s j i j1 M j m i1 M j = mass of j th nucleus m = mass of electron j i Laplace operator for nuclei Laplace operator for
Molecular onding Molecular Schrödinger equation r - nuclei s - electrons 1 1 W V r s j i j1 M j m i1 M j = mass of j th nucleus m = mass of electron j i Laplace operator for nuclei Laplace operator for
Problems and Multiple Choice Questions
 Problems and Multiple Choice Questions 1. A momentum operator in one dimension is 2. A position operator in 3 dimensions is 3. A kinetic energy operator in 1 dimension is 4. If two operator commute, a)
Problems and Multiple Choice Questions 1. A momentum operator in one dimension is 2. A position operator in 3 dimensions is 3. A kinetic energy operator in 1 dimension is 4. If two operator commute, a)
Oslo node. Highly accurate calculations benchmarking and extrapolations
 Oslo node Highly accurate calculations benchmarking and extrapolations Torgeir Ruden, with A. Halkier, P. Jørgensen, J. Olsen, W. Klopper, J. Gauss, P. Taylor Explicitly correlated methods Pål Dahle, collaboration
Oslo node Highly accurate calculations benchmarking and extrapolations Torgeir Ruden, with A. Halkier, P. Jørgensen, J. Olsen, W. Klopper, J. Gauss, P. Taylor Explicitly correlated methods Pål Dahle, collaboration
CHEM6085: Density Functional Theory
 Lecture 11 CHEM6085: Density Functional Theory DFT for periodic crystalline solids C.-K. Skylaris 1 Electron in a one-dimensional periodic box (in atomic units) Schrödinger equation Energy eigenvalues
Lecture 11 CHEM6085: Density Functional Theory DFT for periodic crystalline solids C.-K. Skylaris 1 Electron in a one-dimensional periodic box (in atomic units) Schrödinger equation Energy eigenvalues
PolyCEID: towards a better description of non-adiabatic molecular processes by Correlated Electron-Ion Dynamics
 PolyCEID: towards a better description of non-adiabatic molecular processes by Correlated Electron-Ion Dynamics Lorenzo Stella, R. Miranda, A.P. Horsfield, A.J. Fisher London Centre for Nanotechnology
PolyCEID: towards a better description of non-adiabatic molecular processes by Correlated Electron-Ion Dynamics Lorenzo Stella, R. Miranda, A.P. Horsfield, A.J. Fisher London Centre for Nanotechnology
Intro to ab initio methods
 Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Nonadiabatic Molecular Dynamics with Kohn-Sham DFT: Modeling Nanoscale Materials. Oleg Prezhdo
 Nonadiabatic Molecular Dynamics with Kohn-Sham DFT: Modeling Nanoscale Materials Oleg Prezhdo CSCAMM Mar 11, 2010 Outline Nonadiabatic MD with Kohn-Sham DFT Advantages & Validity Quantum Backreaction &
Nonadiabatic Molecular Dynamics with Kohn-Sham DFT: Modeling Nanoscale Materials Oleg Prezhdo CSCAMM Mar 11, 2010 Outline Nonadiabatic MD with Kohn-Sham DFT Advantages & Validity Quantum Backreaction &
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Non-adiabatic dynamics with external laser fields
 January 18, 21 1 MQCD Mixed Quantum-Classical Dynamics Quantum Dynamics with Trajectories 2 Radiation Field Interaction Hamiltonian 3 NAC NAC in DFT 4 NAC computation Sternheimer Implementation Nearly
January 18, 21 1 MQCD Mixed Quantum-Classical Dynamics Quantum Dynamics with Trajectories 2 Radiation Field Interaction Hamiltonian 3 NAC NAC in DFT 4 NAC computation Sternheimer Implementation Nearly
CHE3935. Lecture 4 Quantum Mechanical Simulation Methods Continued
 CHE3935 Lecture 4 Quantum Mechanical Simulation Methods Continued 1 OUTLINE Review Introduction to CPMD MD and ensembles The functionals of density functional theory Return to ab initio methods Binding
CHE3935 Lecture 4 Quantum Mechanical Simulation Methods Continued 1 OUTLINE Review Introduction to CPMD MD and ensembles The functionals of density functional theory Return to ab initio methods Binding
Errors in electron - molecule collision calculations (at low energies)
") e - Errors in electron - molecule collision calculations (at low energies) Jonathan Tennyson University College London Outer region Inner region IAEA May 2013 Electron processes: at low impact energies
e - Errors in electron - molecule collision calculations (at low energies) Jonathan Tennyson University College London Outer region Inner region IAEA May 2013 Electron processes: at low impact energies
The electronic structure of materials 1
 Quantum mechanics 2 - Lecture 9 December 18, 2013 1 An overview 2 Literature Contents 1 An overview 2 Literature Electronic ground state Ground state cohesive energy equilibrium crystal structure phase
Quantum mechanics 2 - Lecture 9 December 18, 2013 1 An overview 2 Literature Contents 1 An overview 2 Literature Electronic ground state Ground state cohesive energy equilibrium crystal structure phase
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
On the Asymptotic Convergence. of the Transient and Steady State Fluctuation Theorems. Gary Ayton and Denis J. Evans. Research School Of Chemistry
 1 On the Asymptotic Convergence of the Transient and Steady State Fluctuation Theorems. Gary Ayton and Denis J. Evans Research School Of Chemistry Australian National University Canberra, ACT 0200 Australia
1 On the Asymptotic Convergence of the Transient and Steady State Fluctuation Theorems. Gary Ayton and Denis J. Evans Research School Of Chemistry Australian National University Canberra, ACT 0200 Australia
Density Functional Theory. Martin Lüders Daresbury Laboratory
 Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Semiclassical limit and longtime asymptotics of the central spin problem. Gang Chen Doron Bergman Leon Balents
 Semiclassical limit and longtime asymptotics of the central spin problem Gang Chen Doron Bergman Leon Balents Trieste, June 2007 Outline The problem electron-nuclear interactions in a quantum dot Experiments
Semiclassical limit and longtime asymptotics of the central spin problem Gang Chen Doron Bergman Leon Balents Trieste, June 2007 Outline The problem electron-nuclear interactions in a quantum dot Experiments
Theoretical Photochemistry WiSe 2017/18
 Theoretical Photochemistry WiSe 2017/18 Lecture 7 Irene Burghardt (burghardt@chemie.uni-frankfurt.de) http://www.theochem.uni-frankfurt.de/teaching/ Theoretical Photochemistry 1 Topics 1. Photophysical
Theoretical Photochemistry WiSe 2017/18 Lecture 7 Irene Burghardt (burghardt@chemie.uni-frankfurt.de) http://www.theochem.uni-frankfurt.de/teaching/ Theoretical Photochemistry 1 Topics 1. Photophysical
Set the initial conditions r i. Update neighborlist. Get new forces F i
 v Set the initial conditions r i ( t 0 ), v i ( t 0 ) Update neighborlist Quantum mechanical models Get new forces F i ( r i ) Solve the equations of motion numerically over time step Δt : r i ( t n )
v Set the initial conditions r i ( t 0 ), v i ( t 0 ) Update neighborlist Quantum mechanical models Get new forces F i ( r i ) Solve the equations of motion numerically over time step Δt : r i ( t n )
Time part of the equation can be separated by substituting independent equation
 Lecture 9 Schrödinger Equation in 3D and Angular Momentum Operator In this section we will construct 3D Schrödinger equation and we give some simple examples. In this course we will consider problems where
Lecture 9 Schrödinger Equation in 3D and Angular Momentum Operator In this section we will construct 3D Schrödinger equation and we give some simple examples. In this course we will consider problems where
Resonant enhanced electron impact dissociation of molecules
 Journal of Physics: Conference Series Resonant enhanced electron impact dissociation of molecules Recent citations - An R-matrix study of singlet and triplet continuum states of N 2 Duncan A Little and
Journal of Physics: Conference Series Resonant enhanced electron impact dissociation of molecules Recent citations - An R-matrix study of singlet and triplet continuum states of N 2 Duncan A Little and
Optimization Methods via Simulation
 Optimization Methods via Simulation Optimization problems are very important in science, engineering, industry,. Examples: Traveling salesman problem Circuit-board design Car-Parrinello ab initio MD Protein
Optimization Methods via Simulation Optimization problems are very important in science, engineering, industry,. Examples: Traveling salesman problem Circuit-board design Car-Parrinello ab initio MD Protein
Selected Publications of Prof. Dr. Wenjian Liu
 Selected Publications of Prof. Dr. Wenjian Liu College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China 1 Fundamentals of relativistic molecular quantum mechanics 1. Handbook
Selected Publications of Prof. Dr. Wenjian Liu College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China 1 Fundamentals of relativistic molecular quantum mechanics 1. Handbook