Density Functional Theory: from theory to Applications
|
|
|
- Raymond Watts
- 5 years ago
- Views:
Transcription
1 Density Functional Theory: from theory to Applications Uni Mainz May 27, 2012
2
3 Large barrier-activated processes time-dependent bias potential extended Lagrangian formalism Basic idea: during the MD dynamics preventing the system revisiting area in the configuration space where it has been in the past using a history dependent bias potential - Laio A and Parrinello M 2002 Proc. Natl Acad. Sci. USA Iannuzzi M, Laio A and Parrinello M 2003 Phys. Rev. Lett Ensing B, De Vivo M, Liu Z, Moore P, and Klein M, Acc. Chem. Res. 2006, 39, Laio A and Gervasio FL, 2008 Rep. Prog. Phys
4 Filling a 1D model potential with hills, starting in the middle well. After 20 hills, the system escapes to the well on the left and so forth.

5
6 It is crucial to introduce a small set of (scaled and dimensionless) collective variables S = {S 1,..., A α,..., S Ns } defined in terms of the Cartesian coordinates of the nuclei R = {R 1,..., R I,..., R N }. N s N reduction of dimensionality makes possible the filling of minima with a manageable number of Gaussian. Collective variables must include all relevant slow motions.
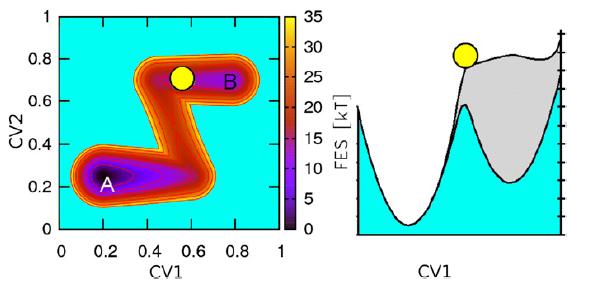
7
8 The extended Lagrangian is: N s L = L CP + α=1 1 2 µs αṡ 2 α(t) N s α=1 1 2 k α [S α (R(t)) s α (t)] 2 + V (t, [s]) (1) where {s α } are the fictious variables; third term is a restraining potential that keeps the instantaneous dynamic variables close to the collective variables S α. The time dependent potential is: t V (t, [s]) = W (t )exp [ ((s(t) s(t )) 2 ] 2( s ) 2 dt (2) t 0
9 The time dependent potential: t V (t, [s]) = W (t )exp [ ((s(t) s(t )) 2 ] 2( s ) 2 dt (3) t 0 can be expressed in a discrete form as: imax V (t, [s]) V (t, {s(t i )}) = W (t i )exp [ ((s(t) s(t )) 2 ] 2( s ) 2 i=0 exp [ [(s(t i+1) s(t i )) (s(t) s(t i ))] 2 ] 2 s(t i+1 ) s(t i ) 4 (4) (5) s t is the meta-timestep ( s t t) s is the width of the Gaussian orthogonal to the direction of motion in the s-space s (t i ) = s(t i+1 ) s(t i ) is adapted to the surface topology W (t i ) is the height of the Gaussian can be fixed or adaptive.
10 the associated Euler-Lagrange equations are: d L = L dt s α s α (6) d L CP = L CP dt Ṙ I R I (7) d L CP = L CP dt φ φ i i (8) the trajectory that are generated do not reflect physical real time dynamics the fictious dynamics is used to explore the free energy landscape
11 The adiabatic decoupling of the collective variable dynamics from the auxiliary electronic wave function dynamics requires additional attention to avoid deviation from the Born- Oppenheimer ground state. The meta timestep t s (dropping rate of the Gaussian) must be larger than the ab initio molecular dynamics time-step t. Typically t s = t.
12 Metadynamics is used to map the free energy profile: where P(s) = 1 Z α It can be shown that: F(s) = K B TlnP(s) (9) [ [δ(s α (R) s α )] exp E KS ] (R) dr (10) K B T F(s) = lim t V (t, {s(t i )}) (11) and the associated error can be estimated as: W s ɛ s t (12)
13 The error W s ɛ s t does not depend on µ s α and on k α. Recipe to choose metadynamics parameters: preliminary run without dropping Gaussians to estimate the fluctuations of the collective variables.


14 The metadynamics method can be used with different Hamiltonian, e.g. in conjunction with classical force-field based MD.

15 The initial configuration corresponds to a C7ax conformation. As the history-dependent potential is added, the fluctuations of ψ increase, until the system fills the free energy basin to reach a C7eq conformation. After approximately 0.8 ns, the added potential has mostly filled the two energy basins and the CV starts to show a diffusive behavior, visiting repeatedly the two basins.
16 Accumulated profiles at 0.8 ns intervals. It can be seen that after 2.4 ns the profile has reached an almost complete convergence.
17 In conclusion, a metadynamics run requires Equilibrate the system by normal molecular dynamics Choose the collective variables, taking into account (i) the performance of metadynamics is optimal with two or three variables. One variable is practically equivalent to thermodynamic integration. Choose the metadynamics parameters, keeping in mind that: (i) width(s) chosen by monitoring the value of standard deviation the CV(s) in a finite temperature run. (ii) height W and the time t s are not independent: the accuracy is determined by their ratio. Check the convergence of the results: the history-dependent potentials evaluated at different times must be approximately similar.
18 In the Parrinello-Rahman method the box is allowed to change its shape in order to comply with a new structure. Often the system has to cross a significant barrier in order to transform from one structure to another. One possibility: overpressurize the system close to the point of mechanical instability. PR can be combined with metadynamics to explore the Gibss free energy landscape 6 independent components of h now represent collective coordinates R. Martonak, A. Laio, and M. Parrinello, Phys. Rev. Lett. 90, (2003)

19 The tight-binding parametrization captures some of the main features of the Si phase diagram and provides a convenient test model. A supercell of 216 atoms is used and only the Γ point of the Brillouin zone is considered. At P=1 atm the equilibrium structure is the diamond, the two high-pressure phases are the β-tin (metastable) and simple hexagonal (SH).

20 At metastep 35 there is a phase transition: the system had made a transition to the SH structure, whose parameters are a = 2.61Å and c = 2.48Å.
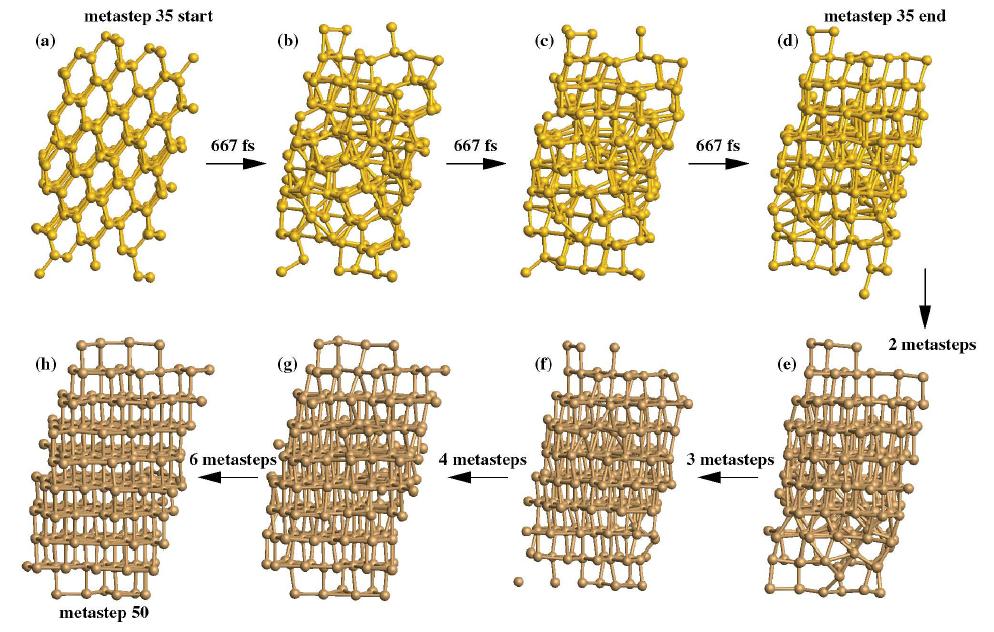
21
Crossing the barriers - simulations of activated processes
 Crossing the barriers - simulations of activated processes Mgr. Ján Hreha for 6 th Student Colloquium and School on Mathematical Physics Faculty of Mathematics, Physics and Informatics Comenius University
Crossing the barriers - simulations of activated processes Mgr. Ján Hreha for 6 th Student Colloquium and School on Mathematical Physics Faculty of Mathematics, Physics and Informatics Comenius University
Performing Metadynamics Simulations Using NAMD
 Performing Metadynamics Simulations Using NAMD Author: Zhaleh Ghaemi Contents 1 Introduction 2 1.1 Goals of this tutorial.......................... 2 2 Standard metadynamics simulations 3 2.1 Free energy
Performing Metadynamics Simulations Using NAMD Author: Zhaleh Ghaemi Contents 1 Introduction 2 1.1 Goals of this tutorial.......................... 2 2 Standard metadynamics simulations 3 2.1 Free energy
Ab initio molecular dynamics
 Ab initio molecular dynamics Kari Laasonen, Physical Chemistry, Aalto University, Espoo, Finland (Atte Sillanpää, Jaakko Saukkoriipi, Giorgio Lanzani, University of Oulu) Computational chemistry is a field
Ab initio molecular dynamics Kari Laasonen, Physical Chemistry, Aalto University, Espoo, Finland (Atte Sillanpää, Jaakko Saukkoriipi, Giorgio Lanzani, University of Oulu) Computational chemistry is a field
GROMACS Implementation of Metadynamics in Essential Coordinates
 GROMACS implementation of metadynamics in essential coordinates 1 GROMACS Implementation of Metadynamics in Essential Coordinates Contact: Vojtěch Spiwok Centre for Glycomics Chemical Department, Slovak
GROMACS implementation of metadynamics in essential coordinates 1 GROMACS Implementation of Metadynamics in Essential Coordinates Contact: Vojtěch Spiwok Centre for Glycomics Chemical Department, Slovak
arxiv:cond-mat/ v1 [cond-mat.stat-mech] 2 Feb 2005
![arxiv:cond-mat/ v1 [cond-mat.stat-mech] 2 Feb 2005](/thumbs/83/87052497.jpg "arxiv:cond-mat/ v1 [cond-mat.stat-mech] 2 Feb 2005") Topological defects and bulk melting of hexagonal ice Davide Donadio, Paolo Raiteri, and Michele Parrinello Computational Science, Department of Chemistry and Applied Biosciences, ETH Zürich, USI Campus,
Topological defects and bulk melting of hexagonal ice Davide Donadio, Paolo Raiteri, and Michele Parrinello Computational Science, Department of Chemistry and Applied Biosciences, ETH Zürich, USI Campus,
Computing free energy: Thermodynamic perturbation and beyond
 Computing free energy: Thermodynamic perturbation and beyond Extending the scale Length (m) 1 10 3 Potential Energy Surface: {Ri} 10 6 (3N+1) dimensional 10 9 E Thermodynamics: p, T, V, N continuum ls
Computing free energy: Thermodynamic perturbation and beyond Extending the scale Length (m) 1 10 3 Potential Energy Surface: {Ri} 10 6 (3N+1) dimensional 10 9 E Thermodynamics: p, T, V, N continuum ls
Supporting Information for. Ab Initio Metadynamics Study of VO + 2 /VO2+ Redox Reaction Mechanism at the Graphite. Edge Water Interface
 Supporting Information for Ab Initio Metadynamics Study of VO + 2 /VO2+ Redox Reaction Mechanism at the Graphite Edge Water Interface Zhen Jiang, Konstantin Klyukin, and Vitaly Alexandrov,, Department
Supporting Information for Ab Initio Metadynamics Study of VO + 2 /VO2+ Redox Reaction Mechanism at the Graphite Edge Water Interface Zhen Jiang, Konstantin Klyukin, and Vitaly Alexandrov,, Department
Accelerated Quantum Molecular Dynamics
 Accelerated Quantum Molecular Dynamics Enrique Martinez, Christian Negre, Marc J. Cawkwell, Danny Perez, Arthur F. Voter and Anders M. N. Niklasson Outline Quantum MD Current approaches Challenges Extended
Accelerated Quantum Molecular Dynamics Enrique Martinez, Christian Negre, Marc J. Cawkwell, Danny Perez, Arthur F. Voter and Anders M. N. Niklasson Outline Quantum MD Current approaches Challenges Extended
Ab initio molecular dynamics. Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy. Bangalore, 04 September 2014
 Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Ab Ini'o Molecular Dynamics (MD) Simula?ons
 Simula?ons") Ab Ini'o Molecular Dynamics (MD) Simula?ons Rick Remsing ICMS, CCDM, Temple University, Philadelphia, PA What are Molecular Dynamics (MD) Simulations? Technique to compute statistical and transport properties
Ab Ini'o Molecular Dynamics (MD) Simula?ons Rick Remsing ICMS, CCDM, Temple University, Philadelphia, PA What are Molecular Dynamics (MD) Simulations? Technique to compute statistical and transport properties
Ab initio molecular dynamics: Basic Theory and Advanced Met
 Ab initio molecular dynamics: Basic Theory and Advanced Methods Uni Mainz October 30, 2016 Bio-inspired catalyst for hydrogen production Ab-initio MD simulations are used to learn how the active site
Ab initio molecular dynamics: Basic Theory and Advanced Methods Uni Mainz October 30, 2016 Bio-inspired catalyst for hydrogen production Ab-initio MD simulations are used to learn how the active site
MD Thermodynamics. Lecture 12 3/26/18. Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky
 MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
Free energy calculations and the potential of mean force
 Free energy calculations and the potential of mean force IMA Workshop on Classical and Quantum Approaches in Molecular Modeling Mark Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Science
Free energy calculations and the potential of mean force IMA Workshop on Classical and Quantum Approaches in Molecular Modeling Mark Tuckerman Dept. of Chemistry and Courant Institute of Mathematical Science
Computing free energies with PLUMED 2.0
 Computing free energies with PLUMED 2.0 Davide Branduardi Formerly at MPI Biophysics, Frankfurt a.m. TyOutline of the talk Relevance of free energy computation Free-energy from histograms: issues Tackling
Computing free energies with PLUMED 2.0 Davide Branduardi Formerly at MPI Biophysics, Frankfurt a.m. TyOutline of the talk Relevance of free energy computation Free-energy from histograms: issues Tackling
Bacterial Outer Membrane Porins as Electrostatic Nanosieves: Exploring Transport Rules of Small Polar Molecules
 Bacterial Outer Membrane Porins as Electrostatic Nanosieves: Exploring Transport Rules of Small Polar Molecules Harsha Bajaj, Silvia Acosta Gutiérrez, Igor Bodrenko, Giuliano Malloci, Mariano Andrea Scorciapino,
Bacterial Outer Membrane Porins as Electrostatic Nanosieves: Exploring Transport Rules of Small Polar Molecules Harsha Bajaj, Silvia Acosta Gutiérrez, Igor Bodrenko, Giuliano Malloci, Mariano Andrea Scorciapino,
Representing High-Dimensional Potential-Energy Surfaces by Artificial Neural Networks
 Energy Landscapes, Chemnitz 200 Representing High-Dimensional Potential-Energy Surfaces by Artificial Neural Networks Jörg Behler Lehrstuhl für Theoretische Chemie Ruhr-Universität Bochum D-44780 Bochum,
Energy Landscapes, Chemnitz 200 Representing High-Dimensional Potential-Energy Surfaces by Artificial Neural Networks Jörg Behler Lehrstuhl für Theoretische Chemie Ruhr-Universität Bochum D-44780 Bochum,
Energy and Forces in DFT
 Energy and Forces in DFT Total Energy as a function of nuclear positions {R} E tot ({R}) = E DF T ({R}) + E II ({R}) (1) where E DF T ({R}) = DFT energy calculated for the ground-state density charge-density
Energy and Forces in DFT Total Energy as a function of nuclear positions {R} E tot ({R}) = E DF T ({R}) + E II ({R}) (1) where E DF T ({R}) = DFT energy calculated for the ground-state density charge-density
Supporting Information
 Supporting Information The effect of strong acid functional groups on electrode rise potential in capacitive mixing by double layer expansion Marta C. Hatzell, a Muralikrishna Raju, b Valerie J. Watson,
Supporting Information The effect of strong acid functional groups on electrode rise potential in capacitive mixing by double layer expansion Marta C. Hatzell, a Muralikrishna Raju, b Valerie J. Watson,
Supporting Information
 1 Supporting Information 2 3 4 Hydrothermal Decomposition of Amino Acids and Origins of Prebiotic Meteoritic Organic Compounds 5 6 7 Fabio Pietrucci 1, José C. Aponte 2,3,*, Richard Starr 2,4, Andrea Pérez-Villa
1 Supporting Information 2 3 4 Hydrothermal Decomposition of Amino Acids and Origins of Prebiotic Meteoritic Organic Compounds 5 6 7 Fabio Pietrucci 1, José C. Aponte 2,3,*, Richard Starr 2,4, Andrea Pérez-Villa
Ab initio Molecular Dynamics Born Oppenheimer and beyond
 Ab initio Molecular Dynamics Born Oppenheimer and beyond Reminder, reliability of MD MD trajectories are chaotic (exponential divergence with respect to initial conditions), BUT... With a good integrator
Ab initio Molecular Dynamics Born Oppenheimer and beyond Reminder, reliability of MD MD trajectories are chaotic (exponential divergence with respect to initial conditions), BUT... With a good integrator
Sampling the free energy surfaces of collective variables
 Sampling the free energy surfaces of collective variables Jérôme Hénin Enhanced Sampling and Free-Energy Calculations Urbana, 12 September 2018 Please interrupt! struct bioinform phys chem theoretical
Sampling the free energy surfaces of collective variables Jérôme Hénin Enhanced Sampling and Free-Energy Calculations Urbana, 12 September 2018 Please interrupt! struct bioinform phys chem theoretical
Density Functional Theory: from theory to Applications
 Density Functional Theory: from theory to Applications Uni Mainz May 14, 2012 All electrons vs pseudopotentials Classes of Basis-set Condensed phase: Bloch s th and PBC Hamann-Schlüter-Chiang pseudopotentials
Density Functional Theory: from theory to Applications Uni Mainz May 14, 2012 All electrons vs pseudopotentials Classes of Basis-set Condensed phase: Bloch s th and PBC Hamann-Schlüter-Chiang pseudopotentials
Characterization of the free-energy landscapes of proteins by NMR-guided metadynamics
 Characterization of the free-energy landscapes of proteins by NMR-guided metadynamics B Results and Discussion BIOPHYSICS ND COMPUTTIONL BIOLOGY SI Text Inset B χ χ Inset SI Text Inset C Folding of GB3
Characterization of the free-energy landscapes of proteins by NMR-guided metadynamics B Results and Discussion BIOPHYSICS ND COMPUTTIONL BIOLOGY SI Text Inset B χ χ Inset SI Text Inset C Folding of GB3
Supporting Information for. Dynamics Study"
 Supporting Information for "CO 2 Adsorption and Reactivity on Rutile TiO 2 (110) in Water: An Ab Initio Molecular Dynamics Study" Konstantin Klyukin and Vitaly Alexandrov,, Department of Chemical and Biomolecular
Supporting Information for "CO 2 Adsorption and Reactivity on Rutile TiO 2 (110) in Water: An Ab Initio Molecular Dynamics Study" Konstantin Klyukin and Vitaly Alexandrov,, Department of Chemical and Biomolecular
Ab initio molecular dynamics
 Ab initio molecular dynamics Molecular dynamics Why? allows realistic simulation of equilibrium and transport properties in Nature ensemble averages can be used for statistical mechanics time evolution
Ab initio molecular dynamics Molecular dynamics Why? allows realistic simulation of equilibrium and transport properties in Nature ensemble averages can be used for statistical mechanics time evolution
Molecular dynamics (MD) and the Monte Carlo simulation
 and the Monte Carlo simulation") Escaping free-energy minima Alessandro Laio and Michele Parrinello* Centro Svizzero di Calcolo Scientifico, Via Cantonale, CH-6928 Manno, Switzerland; and Department of Chemistry, Eidgenössische Technische
Escaping free-energy minima Alessandro Laio and Michele Parrinello* Centro Svizzero di Calcolo Scientifico, Via Cantonale, CH-6928 Manno, Switzerland; and Department of Chemistry, Eidgenössische Technische
Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC
 Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC Sergey V. Churakov sergey.churakov@psi.ch Paul Scherrer Institute Switzerland Cement Phase Composition C-S-H H Solid Solution Model
Structure of Cement Phases from ab initio Modeling Crystalline C-S-HC Sergey V. Churakov sergey.churakov@psi.ch Paul Scherrer Institute Switzerland Cement Phase Composition C-S-H H Solid Solution Model
Ab initio molecular dynamics and nuclear quantum effects
 Ab initio molecular dynamics and nuclear quantum effects Luca M. Ghiringhelli Fritz Haber Institute Hands on workshop density functional theory and beyond: First principles simulations of molecules and
Ab initio molecular dynamics and nuclear quantum effects Luca M. Ghiringhelli Fritz Haber Institute Hands on workshop density functional theory and beyond: First principles simulations of molecules and
What is Classical Molecular Dynamics?
 What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
Supporting Information
 Prebiotic NH 3 formation: Insights from simulations András Stirling 1, Tamás Rozgonyi 2, Matthias Krack 3, Marco Bernasconi 4 1 Institute of Organic Chemistry, Research Centre for Natural Sciences of the
Prebiotic NH 3 formation: Insights from simulations András Stirling 1, Tamás Rozgonyi 2, Matthias Krack 3, Marco Bernasconi 4 1 Institute of Organic Chemistry, Research Centre for Natural Sciences of the
arxiv: v2 [physics.comp-ph] 4 Feb 2016
![arxiv: v2 [physics.comp-ph] 4 Feb 2016](/thumbs/76/73896299.jpg "arxiv: v2 [physics.comp-ph] 4 Feb 2016") Sampling Free Energy Surfaces as Slices by Combining Umbrella Sampling and Metadynamics Shalini Awasthi, Venkat Kapil and Nisanth N. Nair 1 arxiv:1507.06764v2 [physics.comp-ph] 4 Feb 2016 Department of
Sampling Free Energy Surfaces as Slices by Combining Umbrella Sampling and Metadynamics Shalini Awasthi, Venkat Kapil and Nisanth N. Nair 1 arxiv:1507.06764v2 [physics.comp-ph] 4 Feb 2016 Department of
Importance Sampling in Monte Carlo Simulation of Rare Transition Events
 Importance Sampling in Monte Carlo Simulation of Rare Transition Events Wei Cai Lecture 1. August 1, 25 1 Motivation: time scale limit and rare events Atomistic simulations such as Molecular Dynamics (MD)
Importance Sampling in Monte Carlo Simulation of Rare Transition Events Wei Cai Lecture 1. August 1, 25 1 Motivation: time scale limit and rare events Atomistic simulations such as Molecular Dynamics (MD)
Metadynamics-biased ab initio Molecular Dynamics Study of
 Metadynamics-biased ab initio Molecular Dynamics Study of Heterogeneous CO 2 Reduction via Surface Frustrated Lewis Pairs Mireille Ghoussoub 1, Shwetank Yadav 1, Kulbir Kaur Ghuman 1, Geoffrey A. Ozin
Metadynamics-biased ab initio Molecular Dynamics Study of Heterogeneous CO 2 Reduction via Surface Frustrated Lewis Pairs Mireille Ghoussoub 1, Shwetank Yadav 1, Kulbir Kaur Ghuman 1, Geoffrey A. Ozin
Determining Energy Barriers and Selectivities of a Multi-Pathway System With Infrequent Metadynamics
 Determining Energy Barriers and Selectivities of a Multi-Pathway System With Infrequent Metadynamics Christopher D. Fu 1, Luiz F.L. Oliveira 1 and Jim Pfaendtner 1,a) 1 Department of Chemical Engineering,
Determining Energy Barriers and Selectivities of a Multi-Pathway System With Infrequent Metadynamics Christopher D. Fu 1, Luiz F.L. Oliveira 1 and Jim Pfaendtner 1,a) 1 Department of Chemical Engineering,
Controlling fluctuations
 Controlling fluctuations Michele Parrinello Department of Chemistry and Applied Biosciences ETH Zurich and ICS, Università della Svizzera Italiana, Lugano, Switzerland Today s menu Introduction Fluctuations
Controlling fluctuations Michele Parrinello Department of Chemistry and Applied Biosciences ETH Zurich and ICS, Università della Svizzera Italiana, Lugano, Switzerland Today s menu Introduction Fluctuations
Unraveling the degradation of artificial amide bonds in Nylon oligomer hydrolase: From induced-fit to acylation processes
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Supporting Information for Unraveling the degradation of artificial amide bonds
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2015 Supporting Information for Unraveling the degradation of artificial amide bonds
Metadynamics. Day 2, Lecture 3 James Dama
 Metadynamics Day 2, Lecture 3 James Dama Metadynamics The bare bones of metadynamics Bias away from previously visited configuraaons In a reduced space of collecave variables At a sequenaally decreasing
Metadynamics Day 2, Lecture 3 James Dama Metadynamics The bare bones of metadynamics Bias away from previously visited configuraaons In a reduced space of collecave variables At a sequenaally decreasing
The Potential Energy Surface (PES) Preamble to the Basic Force Field Chem 4021/8021 Video II.i
 Preamble to the Basic Force Field Chem 4021/8021 Video II.i") The Potential Energy Surface (PES) Preamble to the Basic Force Field Chem 4021/8021 Video II.i The Potential Energy Surface Captures the idea that each structure that is, geometry has associated with it
The Potential Energy Surface (PES) Preamble to the Basic Force Field Chem 4021/8021 Video II.i The Potential Energy Surface Captures the idea that each structure that is, geometry has associated with it
Importance Sampling in Monte Carlo Simulation of Rare Transition Events
 Importance Sampling in Monte Carlo Simulation of Rare Transition Events Wei Cai Lecture 1. August 1, 25 1 Motivation: time scale limit and rare events Atomistic simulations such as Molecular Dynamics (MD)
Importance Sampling in Monte Carlo Simulation of Rare Transition Events Wei Cai Lecture 1. August 1, 25 1 Motivation: time scale limit and rare events Atomistic simulations such as Molecular Dynamics (MD)
Making the best of a bad situation: a multiscale approach to free energy calculation
 Making the best of a bad situation: arxiv:1901.04455v2 [physics.comp-ph] 15 Jan 2019 a multiscale approach to free energy calculation Michele Invernizzi,, and Michele Parrinello, Department of Physics,
Making the best of a bad situation: arxiv:1901.04455v2 [physics.comp-ph] 15 Jan 2019 a multiscale approach to free energy calculation Michele Invernizzi,, and Michele Parrinello, Department of Physics,
Lecture 14: Advanced Conformational Sampling
 Lecture 14: Advanced Conformational Sampling Dr. Ronald M. Levy ronlevy@temple.edu Multidimensional Rough Energy Landscapes MD ~ ns, conformational motion in macromolecules ~µs to sec Interconversions
Lecture 14: Advanced Conformational Sampling Dr. Ronald M. Levy ronlevy@temple.edu Multidimensional Rough Energy Landscapes MD ~ ns, conformational motion in macromolecules ~µs to sec Interconversions
Quantum Molecular Dynamics Basics
 Quantum Molecular Dynamics Basics Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Quantum Molecular Dynamics Basics Aiichiro Nakano Collaboratory for Advanced Computing & Simulations Depts. of Computer Science, Physics & Astronomy, Chemical Engineering & Materials Science, and Biological
Kinetic Monte Carlo: from transition probabilities to transition rates
 Kinetic Monte Carlo: from transition probabilities to transition rates With MD we can only reproduce the dynamics of the system for 100 ns. Slow thermallyactivated processes, such as diffusion, cannot
Kinetic Monte Carlo: from transition probabilities to transition rates With MD we can only reproduce the dynamics of the system for 100 ns. Slow thermallyactivated processes, such as diffusion, cannot
Temperature and Pressure Controls
 Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Molecular Dynamics. Park City June 2005 Tully
 Molecular Dynamics John Lance Natasa Vinod Xiaosong Dufie Priya Sharani Hongzhi Group: August, 2004 Prelude: Classical Mechanics Newton s equations: F = ma = mq = p Force is the gradient of the potential:
Molecular Dynamics John Lance Natasa Vinod Xiaosong Dufie Priya Sharani Hongzhi Group: August, 2004 Prelude: Classical Mechanics Newton s equations: F = ma = mq = p Force is the gradient of the potential:
Density Functional Theory: from theory to Applications
 Density Functional Theory: from theory to Applications Uni Mainz January 17, 2011 CP-PAW/COSMO Interface Mixed quantum/ classical molecular dynamics (QM/MM) Quantum mechanics is computationally expensive
Density Functional Theory: from theory to Applications Uni Mainz January 17, 2011 CP-PAW/COSMO Interface Mixed quantum/ classical molecular dynamics (QM/MM) Quantum mechanics is computationally expensive
Metadynamics with adaptive Gaussians
 Metadynamics with adaptive Gaussians Davide Branduardi Theoretical Molecular Biophysics Group, Max Planck Institute for Biophysics, Max-von-Laue strasse 5, 60438, Frankfurt am Main, Germany Giovanni Bussi
Metadynamics with adaptive Gaussians Davide Branduardi Theoretical Molecular Biophysics Group, Max Planck Institute for Biophysics, Max-von-Laue strasse 5, 60438, Frankfurt am Main, Germany Giovanni Bussi
Au-C Au-Au. g(r) r/a. Supplementary Figures
 r/a. Supplementary Figures") g(r) Supplementary Figures 60 50 40 30 20 10 0 Au-C Au-Au 2 4 r/a 6 8 Supplementary Figure 1 Radial bond distributions for Au-C and Au-Au bond. The zero density regime between the first two peaks in g
g(r) Supplementary Figures 60 50 40 30 20 10 0 Au-C Au-Au 2 4 r/a 6 8 Supplementary Figure 1 Radial bond distributions for Au-C and Au-Au bond. The zero density regime between the first two peaks in g
Current address: Department of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong,
 Hydrolysis of Cisplatin - A Metadynamics Study Supporting Information Justin Kai-Chi Lau a and Bernd Ensing* b Department of Chemistry and Applied Bioscience, ETH Zurich, USI Campus, Computational Science,
Hydrolysis of Cisplatin - A Metadynamics Study Supporting Information Justin Kai-Chi Lau a and Bernd Ensing* b Department of Chemistry and Applied Bioscience, ETH Zurich, USI Campus, Computational Science,
Optimization Methods via Simulation
 Optimization Methods via Simulation Optimization problems are very important in science, engineering, industry,. Examples: Traveling salesman problem Circuit-board design Car-Parrinello ab initio MD Protein
Optimization Methods via Simulation Optimization problems are very important in science, engineering, industry,. Examples: Traveling salesman problem Circuit-board design Car-Parrinello ab initio MD Protein
Introduction to Geometry Optimization. Computational Chemistry lab 2009
 Introduction to Geometry Optimization Computational Chemistry lab 9 Determination of the molecule configuration H H Diatomic molecule determine the interatomic distance H O H Triatomic molecule determine
Introduction to Geometry Optimization Computational Chemistry lab 9 Determination of the molecule configuration H H Diatomic molecule determine the interatomic distance H O H Triatomic molecule determine
PHYSICAL REVIEW LETTERS
 PHYSICAL REVIEW LETTERS VOLUME 86 28 MAY 21 NUMBER 22 Mathematical Analysis of Coupled Parallel Simulations Michael R. Shirts and Vijay S. Pande Department of Chemistry, Stanford University, Stanford,
PHYSICAL REVIEW LETTERS VOLUME 86 28 MAY 21 NUMBER 22 Mathematical Analysis of Coupled Parallel Simulations Michael R. Shirts and Vijay S. Pande Department of Chemistry, Stanford University, Stanford,
Ab initio molecular dynamics : BO
 School on First Principles Simulations, JNCASR, 2010 Ab initio molecular dynamics : BO Vardha Srinivasan IISER Bhopal Why ab initio MD Free of parametrization and only fundamental constants required. Bond
School on First Principles Simulations, JNCASR, 2010 Ab initio molecular dynamics : BO Vardha Srinivasan IISER Bhopal Why ab initio MD Free of parametrization and only fundamental constants required. Bond
Multi-Ensemble Markov Models and TRAM. Fabian Paul 21-Feb-2018
 Multi-Ensemble Markov Models and TRAM Fabian Paul 21-Feb-2018 Outline Free energies Simulation types Boltzmann reweighting Umbrella sampling multi-temperature simulation accelerated MD Analysis methods
Multi-Ensemble Markov Models and TRAM Fabian Paul 21-Feb-2018 Outline Free energies Simulation types Boltzmann reweighting Umbrella sampling multi-temperature simulation accelerated MD Analysis methods
Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering (REST2)
") pubs.acs.org/jpcb Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering (REST2) Lingle Wang, Richard A. Friesner, and B. J. Berne* Department of Chemistry,
pubs.acs.org/jpcb Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering (REST2) Lingle Wang, Richard A. Friesner, and B. J. Berne* Department of Chemistry,
University of Zurich. Proton transfer in imidazole-based molecular crystals. Zurich Open Repository and Archive. Iannuzzi, M.
 University of Zurich Zurich Open Repository and Archive Winterthurerstr. 190 CH-8057 Zurich http://www.zora.uzh.ch Year: 2006 Proton transfer in imidazole-based molecular crystals Iannuzzi, M Iannuzzi,
University of Zurich Zurich Open Repository and Archive Winterthurerstr. 190 CH-8057 Zurich http://www.zora.uzh.ch Year: 2006 Proton transfer in imidazole-based molecular crystals Iannuzzi, M Iannuzzi,
A self-activated mechanism for nucleic acid polymerization catalyzed by DNA/RNA polymerases
 A self-activated mechanism for nucleic acid polymerization catalyzed by DNA/RNA polymerases Vito Genna 1,2,, Pietro Vidossich 2, Emiliano Ippoliti 2, Paolo Carloni 2* and Marco De Vivo 1,2* 1 Laboratory
A self-activated mechanism for nucleic acid polymerization catalyzed by DNA/RNA polymerases Vito Genna 1,2,, Pietro Vidossich 2, Emiliano Ippoliti 2, Paolo Carloni 2* and Marco De Vivo 1,2* 1 Laboratory
Advanced sampling. fluids of strongly orientation-dependent interactions (e.g., dipoles, hydrogen bonds)
") Advanced sampling ChE210D Today's lecture: methods for facilitating equilibration and sampling in complex, frustrated, or slow-evolving systems Difficult-to-simulate systems Practically speaking, one is
Advanced sampling ChE210D Today's lecture: methods for facilitating equilibration and sampling in complex, frustrated, or slow-evolving systems Difficult-to-simulate systems Practically speaking, one is
Supplementary Information
 Pt II as proton shuttle during C H bond activation in the Shilov process Pietro Vidossich,* a Gregori Ujaque, a Agustí Lledós a a Departament de Química, Universitat Autònoma de Barcelona, 08193 Cerdanyola
Pt II as proton shuttle during C H bond activation in the Shilov process Pietro Vidossich,* a Gregori Ujaque, a Agustí Lledós a a Departament de Química, Universitat Autònoma de Barcelona, 08193 Cerdanyola
Quantum (Path Integral) Molecular Dynamics
 Molecular Dynamics") Quantum (Path Integral) Molecular Dynamics Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, U.K. http://www-users.york.ac.uk/~mijp1 Overview of lecture n Background
Quantum (Path Integral) Molecular Dynamics Matt Probert Condensed Matter Dynamics Group Department of Physics, University of York, U.K. http://www-users.york.ac.uk/~mijp1 Overview of lecture n Background
The PLUMED plugin and free energy methods in electronic-structure-based molecular dynamics
 The PLUMED plugin and free energy methods in electronic-structure-based molecular dynamics Davide Branduardi, Theoretical Molecular Biophysics Group Max Planck for Biophysics, Frankfurt am Main, Germany
The PLUMED plugin and free energy methods in electronic-structure-based molecular dynamics Davide Branduardi, Theoretical Molecular Biophysics Group Max Planck for Biophysics, Frankfurt am Main, Germany
Major Concepts Lecture #11 Rigoberto Hernandez. TST & Transport 1
 Major Concepts Onsager s Regression Hypothesis Relaxation of a perturbation Regression of fluctuations Fluctuation-Dissipation Theorem Proof of FDT & relation to Onsager s Regression Hypothesis Response
Major Concepts Onsager s Regression Hypothesis Relaxation of a perturbation Regression of fluctuations Fluctuation-Dissipation Theorem Proof of FDT & relation to Onsager s Regression Hypothesis Response
Energy Landscapes and Accelerated Molecular- Dynamical Techniques for the Study of Protein Folding
 Energy Landscapes and Accelerated Molecular- Dynamical Techniques for the Study of Protein Folding John K. Prentice Boulder, CO BioMed Seminar University of New Mexico Physics and Astronomy Department
Energy Landscapes and Accelerated Molecular- Dynamical Techniques for the Study of Protein Folding John K. Prentice Boulder, CO BioMed Seminar University of New Mexico Physics and Astronomy Department
Spinor Bose gases lecture outline
 Spinor Bose gases lecture outline 1. Basic properties 2. Magnetic order of spinor Bose-Einstein condensates 3. Imaging spin textures 4. Spin-mixing dynamics 5. Magnetic excitations We re here Coupling
Spinor Bose gases lecture outline 1. Basic properties 2. Magnetic order of spinor Bose-Einstein condensates 3. Imaging spin textures 4. Spin-mixing dynamics 5. Magnetic excitations We re here Coupling
Clusters and Percolation
 Chapter 6 Clusters and Percolation c 2012 by W. Klein, Harvey Gould, and Jan Tobochnik 5 November 2012 6.1 Introduction In this chapter we continue our investigation of nucleation near the spinodal. We
Chapter 6 Clusters and Percolation c 2012 by W. Klein, Harvey Gould, and Jan Tobochnik 5 November 2012 6.1 Introduction In this chapter we continue our investigation of nucleation near the spinodal. We
Testing the transition state theory in stochastic dynamics of a. genetic switch
 Testing the transition state theory in stochastic dynamics of a genetic switch Tomohiro Ushikubo 1, Wataru Inoue, Mitsumasa Yoda 1 1,, 3, and Masaki Sasai 1 Department of Computational Science and Engineering,
Testing the transition state theory in stochastic dynamics of a genetic switch Tomohiro Ushikubo 1, Wataru Inoue, Mitsumasa Yoda 1 1,, 3, and Masaki Sasai 1 Department of Computational Science and Engineering,
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
Introduction to DFTB. Marcus Elstner. July 28, 2006
 Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Selected Publications of Prof. Dr. Wenjian Liu
 Selected Publications of Prof. Dr. Wenjian Liu College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China 1 Fundamentals of relativistic molecular quantum mechanics 1. Handbook
Selected Publications of Prof. Dr. Wenjian Liu College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China 1 Fundamentals of relativistic molecular quantum mechanics 1. Handbook
Supplemental Material for Global Langevin model of multidimensional biomolecular dynamics
 Supplemental Material for Global Langevin model of multidimensional biomolecular dynamics Norbert Schaudinnus, Benjamin Lickert, Mithun Biswas and Gerhard Stock Biomolecular Dynamics, Institute of Physics,
Supplemental Material for Global Langevin model of multidimensional biomolecular dynamics Norbert Schaudinnus, Benjamin Lickert, Mithun Biswas and Gerhard Stock Biomolecular Dynamics, Institute of Physics,
New Insights in Hybrid Inflation
 Dr. Sébastien Clesse TU Munich, T70 group: Theoretical Physics of the Early Universe Excellence Cluster Universe Based on S.C., B. Garbrecht, Y. Zhu, Non-gaussianities and curvature perturbations in hybrid
Dr. Sébastien Clesse TU Munich, T70 group: Theoretical Physics of the Early Universe Excellence Cluster Universe Based on S.C., B. Garbrecht, Y. Zhu, Non-gaussianities and curvature perturbations in hybrid
7 To solve numerically the equation of motion, we use the velocity Verlet or leap frog algorithm. _ V i n = F i n m i (F.5) For time step, we approxim
 For time step, we approxim") 69 Appendix F Molecular Dynamics F. Introduction In this chapter, we deal with the theories and techniques used in molecular dynamics simulation. The fundamental dynamics equations of any system is the
69 Appendix F Molecular Dynamics F. Introduction In this chapter, we deal with the theories and techniques used in molecular dynamics simulation. The fundamental dynamics equations of any system is the
Key concepts in Density Functional Theory (I) Silvana Botti
 Silvana Botti") From the many body problem to the Kohn-Sham scheme European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address: Centre
From the many body problem to the Kohn-Sham scheme European Theoretical Spectroscopy Facility (ETSF) CNRS - Laboratoire des Solides Irradiés Ecole Polytechnique, Palaiseau - France Temporary Address: Centre
Gibb s free energy change with temperature in a single component system
 Gibb s free energy change with temperature in a single component system An isolated system always tries to maximize the entropy. That means the system is stable when it has maximum possible entropy. Instead
Gibb s free energy change with temperature in a single component system An isolated system always tries to maximize the entropy. That means the system is stable when it has maximum possible entropy. Instead
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
METAGUI A VMD EXTENSION TO ANALYZE AND VISUALIZE METADYNAMICS SIMULATIONS
 METAGUI A VMD EXTENSION TO ANALYZE AND VISUALIZE METADYNAMICS SIMULATIONS Alessandro Laio SISSA & DEMOCRITOS, Trieste Coworkers: Xevi Biarnes Fabio Pietrucci Fabrizio Marinelli Metadynamics (Laio A. and
METAGUI A VMD EXTENSION TO ANALYZE AND VISUALIZE METADYNAMICS SIMULATIONS Alessandro Laio SISSA & DEMOCRITOS, Trieste Coworkers: Xevi Biarnes Fabio Pietrucci Fabrizio Marinelli Metadynamics (Laio A. and
COARSE-GRAINING AND THERMODYNAMICS IN FAR-FROM-EQUILIBRIUM SYSTEMS
 Vol. 44 (2013) ACTA PHYSICA POLONICA B No 5 COARSE-GRAINING AND THERMODYNAMICS IN FAR-FROM-EQUILIBRIUM SYSTEMS J. Miguel Rubí, A. Pérez-Madrid Departament de Física Fonamental, Facultat de Física, Universitat
Vol. 44 (2013) ACTA PHYSICA POLONICA B No 5 COARSE-GRAINING AND THERMODYNAMICS IN FAR-FROM-EQUILIBRIUM SYSTEMS J. Miguel Rubí, A. Pérez-Madrid Departament de Física Fonamental, Facultat de Física, Universitat
Molecular Dynamics Simulations of Fusion Materials: Challenges and Opportunities (Recent Developments)
") Molecular Dynamics Simulations of Fusion Materials: Challenges and Opportunities (Recent Developments) Fei Gao gaofeium@umich.edu Limitations of MD Time scales Length scales (PBC help a lot) Accuracy of
Molecular Dynamics Simulations of Fusion Materials: Challenges and Opportunities (Recent Developments) Fei Gao gaofeium@umich.edu Limitations of MD Time scales Length scales (PBC help a lot) Accuracy of
arxiv: v1 [q-bio.bm] 9 Jul 2007
![arxiv: v1 [q-bio.bm] 9 Jul 2007](/thumbs/74/69979420.jpg "arxiv: v1 [q-bio.bm] 9 Jul 2007") Exploring the Protein G Helix Free Energy Surface by Solute Tempering Metadynamics Carlo Camilloni, 1, 2 Davide Provasi, 1, 2 Guido Tiana, 1, 2, and Ricardo A. Broglia 1,2,3 1 Department of Physics, University
Exploring the Protein G Helix Free Energy Surface by Solute Tempering Metadynamics Carlo Camilloni, 1, 2 Davide Provasi, 1, 2 Guido Tiana, 1, 2, and Ricardo A. Broglia 1,2,3 1 Department of Physics, University
Ab initio phonon calculations in mixed systems
 Ab initio phonon calculations in mixed systems Andrei Postnikov apostnik@uos.de Outline: Experiment vs. ab initio theory Ways of theory: linear response and frozen phonon approaches Applications: Be x
Ab initio phonon calculations in mixed systems Andrei Postnikov apostnik@uos.de Outline: Experiment vs. ab initio theory Ways of theory: linear response and frozen phonon approaches Applications: Be x
Car-Parrinello Molecular Dynamics
 Car-Parrinello Molecular Dynamics Eric J. Bylaska HPCC Group Moral: A man dreams of a miracle and wakes up with loaves of bread Erich Maria Remarque Molecular Dynamics Loop (1) Compute Forces on atoms,
Car-Parrinello Molecular Dynamics Eric J. Bylaska HPCC Group Moral: A man dreams of a miracle and wakes up with loaves of bread Erich Maria Remarque Molecular Dynamics Loop (1) Compute Forces on atoms,
Testing Convergence of Different Free-Energy Methods in a Simple Analytical System with Hidden Barriers
 computation Article Testing Convergence of Different Free-Energy Methods in a Simple Analytical System with Hidden Barriers S. Alexis Paz 1, ID and Cameron F. Abrams 3, * ID 1 Departamento de Química Teórica
computation Article Testing Convergence of Different Free-Energy Methods in a Simple Analytical System with Hidden Barriers S. Alexis Paz 1, ID and Cameron F. Abrams 3, * ID 1 Departamento de Química Teórica
Anisotropy of Earth s D layer and stacking faults in MgSiO 3 post-perovskite
 1 Anisotropy of Earth s D layer and stacking faults in MgSiO 3 post-perovskite Artem R. Oganov 1*, Roman Martoňák 2, Alessandro Laio 2, Paolo Raiteri 2, Michele Parrinello 2 1 Laboratory of Crystallography,
1 Anisotropy of Earth s D layer and stacking faults in MgSiO 3 post-perovskite Artem R. Oganov 1*, Roman Martoňák 2, Alessandro Laio 2, Paolo Raiteri 2, Michele Parrinello 2 1 Laboratory of Crystallography,
Rare Event Simulations
 Rare Event Simulations Homogeneous nucleation is a rare event (e.g. Liquid Solid) Crystallization requires supercooling (µ solid < µ liquid ) Crystal nucleus 2r Free - energy gain r 3 4! 3! GBulk = ñr!
Rare Event Simulations Homogeneous nucleation is a rare event (e.g. Liquid Solid) Crystallization requires supercooling (µ solid < µ liquid ) Crystal nucleus 2r Free - energy gain r 3 4! 3! GBulk = ñr!
Liquid to Glass Transition
 Landscape Approach to Glass Transition and Relaxation (Lecture # 3, March 30) Liquid to Glass Transition Instructor: Prabhat Gupta The Ohio State University (gupta.3@osu.edu) PKGupta(OSU) Landscape #3
Landscape Approach to Glass Transition and Relaxation (Lecture # 3, March 30) Liquid to Glass Transition Instructor: Prabhat Gupta The Ohio State University (gupta.3@osu.edu) PKGupta(OSU) Landscape #3
Molecular energy levels
 Molecular energy levels Hierarchy of motions and energies in molecules The different types of motion in a molecule (electronic, vibrational, rotational,: : :) take place on different time scales and are
Molecular energy levels Hierarchy of motions and energies in molecules The different types of motion in a molecule (electronic, vibrational, rotational,: : :) take place on different time scales and are
Introduction to Numerical Simulations and High Performance Computing: From Materials Science to Biochemistry
 Introduction to Numerical Simulations and High Performance Computing: From Materials Science to Biochemistry Mauro Boero Institut de Physique et Chimie des Matériaux de Strasbourg University of Strasbourg
Introduction to Numerical Simulations and High Performance Computing: From Materials Science to Biochemistry Mauro Boero Institut de Physique et Chimie des Matériaux de Strasbourg University of Strasbourg
Diffusion in multicomponent solids. Anton Van der Ven Department of Materials Science and Engineering University of Michigan Ann Arbor, MI
 Diffusion in multicomponent solids nton Van der Ven Department of Materials Science and Engineering University of Michigan nn rbor, MI Coarse graining time Diffusion in a crystal Two levels of time coarse
Diffusion in multicomponent solids nton Van der Ven Department of Materials Science and Engineering University of Michigan nn rbor, MI Coarse graining time Diffusion in a crystal Two levels of time coarse
Hill climbing: Simulated annealing and Tabu search
 Hill climbing: Simulated annealing and Tabu search Heuristic algorithms Giovanni Righini University of Milan Department of Computer Science (Crema) Hill climbing Instead of repeating local search, it is
Hill climbing: Simulated annealing and Tabu search Heuristic algorithms Giovanni Righini University of Milan Department of Computer Science (Crema) Hill climbing Instead of repeating local search, it is
IV. Classical Molecular Dynamics
 IV. Classical Molecular Dynamics Basic Assumptions: 1. Born-Oppenheimer Approximation 2. Classical mechanical nuclear motion Unavoidable Additional Approximations: 1. Approximate potential energy surface
IV. Classical Molecular Dynamics Basic Assumptions: 1. Born-Oppenheimer Approximation 2. Classical mechanical nuclear motion Unavoidable Additional Approximations: 1. Approximate potential energy surface
Vibrational Spectroscopy & Intramolecular Vibrational Redistribution (IVR)
") Vibrational Spectroscopy & Intramolecular Vibrational Redistribution (IVR) 1 The Role of Vibrational Energy in Chemical Reactions Unimolecular reactions: The Rice-Rampsberger-Kassel-Marcus (RRKM) theory
Vibrational Spectroscopy & Intramolecular Vibrational Redistribution (IVR) 1 The Role of Vibrational Energy in Chemical Reactions Unimolecular reactions: The Rice-Rampsberger-Kassel-Marcus (RRKM) theory
MARTINI simulation details
 S1 Appendix MARTINI simulation details MARTINI simulation initialization and equilibration In this section, we describe the initialization of simulations from Main Text section Residue-based coarsegrained
S1 Appendix MARTINI simulation details MARTINI simulation initialization and equilibration In this section, we describe the initialization of simulations from Main Text section Residue-based coarsegrained
TDDFT II. Alberto Castro
 Problem set Alberto Castro Institute for Biocomputation and Physics of Complex Systems (BIFI) and Zaragoza Scientific Center for Advanced Modeling (ZCAM), University of Zaragoza, Spain ELK Summit, Lausanne
Problem set Alberto Castro Institute for Biocomputation and Physics of Complex Systems (BIFI) and Zaragoza Scientific Center for Advanced Modeling (ZCAM), University of Zaragoza, Spain ELK Summit, Lausanne
CHEM3023: Spins, Atoms and Molecules
 CHEM3023: Spins, Atoms and Molecules Lecture 3 The Born-Oppenheimer approximation C.-K. Skylaris Learning outcomes Separate molecular Hamiltonians to electronic and nuclear parts according to the Born-Oppenheimer
CHEM3023: Spins, Atoms and Molecules Lecture 3 The Born-Oppenheimer approximation C.-K. Skylaris Learning outcomes Separate molecular Hamiltonians to electronic and nuclear parts according to the Born-Oppenheimer
Ab Initio Molecular Dynamic
 Ab Initio Molecular Dynamic Paul Fleurat-Lessard AtoSim Master / RFCT Ab Initio Molecular Dynamic p.1/67 Summary I. Introduction II. Classical Molecular Dynamic III. Ab Initio Molecular Dynamic (AIMD)
Ab Initio Molecular Dynamic Paul Fleurat-Lessard AtoSim Master / RFCT Ab Initio Molecular Dynamic p.1/67 Summary I. Introduction II. Classical Molecular Dynamic III. Ab Initio Molecular Dynamic (AIMD)
Essential dynamics sampling of proteins. Tuorial 6 Neva Bešker
 Essential dynamics sampling of proteins Tuorial 6 Neva Bešker Relevant time scale Why we need enhanced sampling? Interconvertion between basins is infrequent at the roomtemperature: kinetics and thermodynamics
Essential dynamics sampling of proteins Tuorial 6 Neva Bešker Relevant time scale Why we need enhanced sampling? Interconvertion between basins is infrequent at the roomtemperature: kinetics and thermodynamics
3.320: Lecture 19 (4/14/05) Free Energies and physical Coarse-graining. ,T) + < σ > dµ
 Free Energies and physical Coarse-graining. ,T) + < σ > dµ") 3.320: Lecture 19 (4/14/05) F(µ,T) = F(µ ref,t) + < σ > dµ µ µ ref Free Energies and physical Coarse-graining T S(T) = S(T ref ) + T T ref C V T dt Non-Boltzmann sampling and Umbrella sampling Simple
3.320: Lecture 19 (4/14/05) F(µ,T) = F(µ ref,t) + < σ > dµ µ µ ref Free Energies and physical Coarse-graining T S(T) = S(T ref ) + T T ref C V T dt Non-Boltzmann sampling and Umbrella sampling Simple
Temperature and Pressure Controls
 Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Ensembles Temperature and Pressure Controls 1. (E, V, N) microcanonical (constant energy) 2. (T, V, N) canonical, constant volume 3. (T, P N) constant pressure 4. (T, V, µ) grand canonical #2, 3 or 4 are
Limitations of temperature replica exchange (T-REMD) for protein folding simulations
 for protein folding simulations") Limitations of temperature replica exchange (T-REMD) for protein folding simulations Jed W. Pitera, William C. Swope IBM Research pitera@us.ibm.com Anomalies in protein folding kinetic thermodynamic 322K
Limitations of temperature replica exchange (T-REMD) for protein folding simulations Jed W. Pitera, William C. Swope IBM Research pitera@us.ibm.com Anomalies in protein folding kinetic thermodynamic 322K
Free energy calculations using molecular dynamics simulations. Anna Johansson
 Free energy calculations using molecular dynamics simulations Anna Johansson 2007-03-13 Outline Introduction to concepts Why is free energy important? Calculating free energy using MD Thermodynamical Integration
Free energy calculations using molecular dynamics simulations Anna Johansson 2007-03-13 Outline Introduction to concepts Why is free energy important? Calculating free energy using MD Thermodynamical Integration