Density Functional Theory: from theory to Applications
|
|
|
- Beryl Howard
- 5 years ago
- Views:
Transcription
1 Density Functional Theory: from theory to Applications Uni Mainz January 17, 2011
2 CP-PAW/COSMO Interface
 and the environment which is not neglected but treated at lower level of accuracy (MM). 1 1 See pioneering work of Warshel and Levitt, JMB 1976, 103, 227.")
3 Mixed quantum/ classical molecular dynamics (QM/MM) Quantum mechanics is computationally expensive Idea: subdivide a complex system in a small, relevant part (QM) and the environment which is not neglected but treated at lower level of accuracy (MM). 1 1 See pioneering work of Warshel and Levitt, JMB 1976, 103, 227.
 are in the enzyme s active site (with amino")
4 Metallo-beta-lactamase enzyme. Two zinc ions (purple spheres) are in the enzyme s active site (with amino acids coordinating the metals represented as sticks).
5 : both QM and MM are based on particle representation. The full Hamiltonian is: E QM/MM = E QM ({R α }) + E MM ({R I }) + E QM MM ({R α }, {R I }) (1) E QM ({R α }) is the Kohn-Sham Hamiltonian He KS, e.g. in a plane wave / pseudopotential representation. E MM ({R I }) are force field energy expressions: short-range bonded interaction with fixed topology + non-bonded interaction (electrostic and van der Waals)
6 MM: Bonds and bends
7 MM: Urey-Bradley and Dihedral
8 MM: Improper (Out of plane motion)
9 MM: Non-bonded Outline Steric interactions are usually described by standard Lenard-Jones interaction, but different functional forms are also possible Electrostatics: V (r ij ) = q iq j ɛr ij (2)
. J.W. Ponder and D.A. Case. Force fields for protein simulations. Adv. Prot. Chem. 66, 27-85 (2003).")
10 Biomolecular simulations: AMBER D.A. Case, T.E. Cheatham, III, T. Darden, H. Gohlke, R. Luo, K.M. Merz, Jr., A. Onufriev, C. Simmerling, B. Wang and R. Woods. The Amber biomolecular simulation programs. J. Computat. Chem. 26, (2005). J.W. Ponder and D.A. Case. Force fields for protein simulations. Adv. Prot. Chem. 66, (2003).
11 Biomolecular simulations: CHARMM CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations, J. Comp. Chem. 4, (1983), by B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. CHARMM: The Energy Function and Its Parameterization with an Overview of the Program, in The Encyclopedia of Computational Chemistry, 1, , P. v. R. Schleyer et al., editors (John Wiley & Sons: Chichester, 1998), by A. D. MacKerell, Jr., B. Brooks,C. L. Brooks, III, L. Nilsson, B. Roux, Y. Won, and M. Karplus.
12 OPLS (Optimized Potentials for Liquid Simulations) Jorgensen WL, Tirado-Rives J (1988). The OPLS Force Field for Proteins. Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J. Am. Chem. Soc. 110: Jorgensen WL, Maxwell DS, Tirado-Rives J (1996). Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 118 (45):
13 Class II force fields Greater transferability: Application of empirical force field parameters to molecules not explicitly included during the parameter optimization. Example: Merck Molecular Force Field (MMFF), a Class II force field designed to be a transferable force field for pharmaceutical compounds that accurately treats conformational energetics and non-bonded interactions. This would, ideally, produce a force field that was adequate for both gas phase and condensed phase calculations.
14 Class II force fields: MMFF
15 E QM MM ({R α }, {R I }) E QM MM = E QM MM b E QM MM nb = E QM MM es + E QM MM nb (3) + E QM MM steric (4) generally follows the model used in E MM (Lenard-Jones-type potentials). E QM MM steric For the Ees QM MM there different coupling schemes: mechanical embedding: no influence of MM charges on the QM system electrostatic embedding: electrostatic interaction between mm charges and charge density of the QM system polarized embedding: MM polarization due to the QM system is also included
16 Technical issues connected with the electrostic coupling Straightforward numerical evaluation of the electrostatic coupling term: = q I E QM MM es I MM n(r) dr (5) r R I is prohibitive. Indeed it would involve a number of operations which scales as N g (number of grid points) times N MM (number of MM atoms). Electron spill out or charge leakage
17 Outline Pioneering qm/mm work by Bloch and coworkers 2. Motivation: studying chemical reaction of complex system and computing free energy profile using thermodynamics integration. The instaneous electronic density of the QM system is mapped onto a set of Gaussian smearing functions tied to the QM sites. The corresponding Lagrangian is: L QM/MM = L QM CP + LMM + L QM MM (6) = 1 2 MαṘ2 α + µ φi φ KS i Ψ 0 H e ({R α }) Ψ 0 (7) α i + i,j + I Λ i,j ( φ i φ j δ ij ) (8) 1 2 M I Ṙ2 I + E MM ({R I }) + E QM MM ({R I }, {R α } (9) 2 J. Chem. Phys. 103, 7422
18 Outline Separate Nose -Hoover thermostats for the QM/MM regions Decoupling scheme between periodic images The continuous charge density is compressed to a linear superposition of of atom-centered spherical Gaussian functions: n G (r) = α q α G(r R α ) (10) Gaussian parameters are determined imposing that n G (r) reproduce the multiple moments of n(r). In the limit of infinitesimally small width the Gaussian density becomes: n pc (r) = q α δ(r R α ) (11) α The electrostatic coupling is obtained between these effective QM charges and the MM charges.


19 Handling bond cuts: the hydrogen capping
20 A multiple timestep scheme Relevant dynamical changes in the QM and MM systems typically occur on quite disparate time scales. Accessible time-scale for QM dynamics is the ps time scale, whereas large amplitude conformational changes in large biomolecules occurs on the ns timescale. Multiple time step scheme oversampling artificially decreasing the MM masses, in this case the generated dynamics is fictious.
21 The Full Hamiltonian QM/MM coupling. The QM/MM interface 3 is tailored to study dynamics of complex biomolecular systems and chemical reactions. The non-bonded part of the total energy is written as: E QM MM nb = Ees QM MM + E QM MM = n(r) q I r R I dr + I MM steric (12) I MM α QM v vdw ( R α R I (13) ) Short range electrostatic: the spill out problem. Ees QM MM = q I n(r)vi eff ( r R I )dr (14) v eff I (r I ) = I MM rci m ri m r m+1 ci r m+1 I 1 r I (r I ) (15) where m = 4 and R c,i covalent radius for the I th atom is a reasonable choice. 3 Laio, VandeVondele, Rothlisberger, J. Chem. Phys. 116, 6941 (2002). Laio, VandeVondele, Rothlisberger, J. Phys. Density Chem. Functional B 106, Theory: 7300,(2002) from theory to Applications
22 Long Range Electrostatic The Coulombic electrostatic field is included exactly only for a set of MM atoms in the vicinity of the QM system. The electrostatic field on the MM atoms that do not belong to the NN set is calculated by a multi-polar expansion
23 Bonded Interactions Two option have been employed: Hydrogen capping Carbon monovalent pseudopotentials
24 CPMD/GROMS: validation QM water in MM (SPC) water Pair correlation functions for qm/mm compared with continuous line with circles (SPC water) continuous line with diamonds (full QM) Continuous line: r c for oxygen and hydrogen are 0.8 and 0.4 Å, respectively; dashed line: r c for oxygen and hydrogen are 0.8 and 0.8 Å; dotted line: r c for oxygen and hydrogen are 1.4 and 0.8 Å. 1) QM oxygen-mm hydrogen. 2) QM hydrogen-mm oxygen. 3) QM oxygen-mm oxygen.
25 CPMD/GROMS: validation Fictitious electronic kinetic energy for a QM water in a box of classical SPC water at 300 K and normal density. The time step is 6 a.u. Continuous line: no modification of the Coulomb potential. Dotted line: modified Coulomb potential. (r =0.8 Åfor oxygen Marialoreand Sulpizi r =0.4Density ÅforFunctional hydrogen). Theory: from theory to Applications
26 Hydrogen-water oxygen pair correlation function for a ammonium ion in a box of water. Continuous line: full quantum result Dashed line: QM/MM result. The quantum ammonium ion is solvated in a box of SPC water.
27 This approach 4 is implemented in CP2K 5. Which is a GPWs code, suing a mixed Gaussian and plane wave basis set for the expansion of the wavefunction and of the charge density. This approach is based on the use of a multi-grid technique in conjunction with a Gaussian expansion of the electrostatic potential (GEEP) 4 JCTC 2005, 1,
28 The exact Coulomb potential is replaced by which is the exact potential energy function originated by a Gaussian charge distribution and has the desired property of tending to 1/r at large distances and going smoothly to a constant for small r. This potential can be written as sum of Gaussian functions with different cutoffs according to

29 The advantage of this decomposition scheme is that grids of different spacing can be used to represent the different contributions to v a (r, r a ). Sharp Gaussians require fine grids, while coarser grids are necessary for the smoothest components. Gaussians can be truncated beyond a certain threshold value, which makes the collocation of the Gaussians on the grid very efficient.
30 Gaussian will be represented on the same number of grid points irrespective of its width. (e.g. a mesh of 25 X 25 x 25 suffices for an optimal Gaussian representation). The Gaussian can be considered a compact domain function, i.e., it is zero beyond a certain distance. Thus only MM atoms embedded into the QM box, or close to it, will contribute to the finest grid levels. The speed up is abut 2 order of magnitude
31 CP-PAW/COSMO Interface CP-PAW/COSMO: the environment is treated with a conductor-like screening model homogeneous and inert solvent treated as dielectric continuum: suitable if environment is not involved in the ongoing chemistry the essential aspect of this approach is to treat the surface charges at the cavity boundaries as fictious dynamical variables: L QM/MM = L QM CP + LMM + L COSMO (16) L COSMO = 1 2 M Q I I 2 + Ges COSMO ({Q I }, {R α }) I G steric ({R α }) I k(1 Θ I )Q 2 I (17) where the G steric = σ 0 + σ 1 A (18) with A surface area of the cavity and σ i, empirical, fixed parameters.


32 CP-PAW/COSMO Interface COSMO surface of a pentaacrylate molecule (red = negative, green = positive equilibrium layer).
33 CP-PAW/COSMO Interface A bonded term between the QM and MM parts, E QM MM b does not exists in this approach. The fictious variables Q I, with associated inertia parameters M Q I, are discretized, scaled surface charges, located at the center of segments s I. Switching functions are introduced to switch off the charges of the segments which are not exposed to the solvent. Last term in eq:17 is a penalty function introduced to keep the instantaneous switched off charges from blowing-up during the dynamics
34 CP-PAW/COSMO Interface The COSMO free energy can be written down in terms of of electrostatic solute-solvent, solvent-solvent, and self energy contributions: Ges COSMO = n(r) Q I Θ I I V r s I dr + 1 Q I Θ I Q J Θ J (19) f s I s J I <J + c QI 2Θ2 I (20) f ai I G steric = σ 0 + σ 1 a I Θ I (21) f is a constant screening factor, c is a geometry-dependent constant and V is the cavity volume. Spill-out is prevented using the Gaussian model density as described for the CP-PAW/AMBER. I

35 A QM/MM application: reaction mechanism of caspase Caspase-3 is a cysteine protease involved in the programmed cell-death (apoptosis). Idea: study the enzymatic reaction in the protein environment. Calculate free energy barrier associated with the proposed mechanism 6. 6 Sulpizi et al, Proteins, 52(2): 212 (2003).
36
37

38
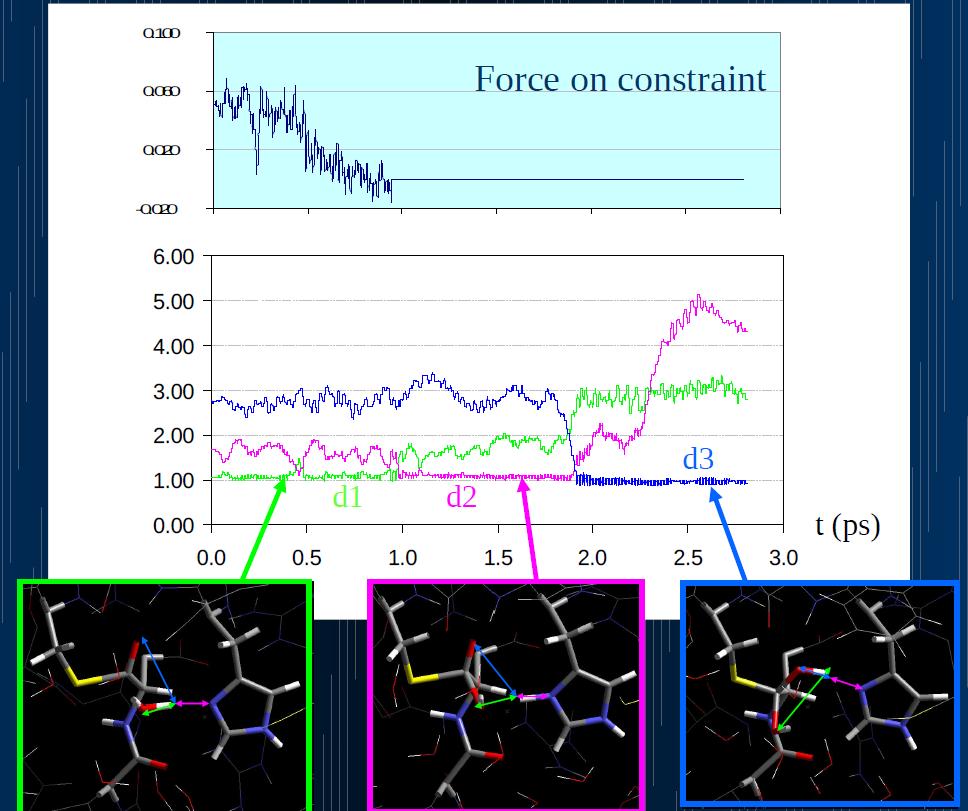
39

40 For our calculation a F 20 kcal/mol is compatible with the 1

41 The free energy barrier associated with this step is only 5 kcal/mol, so the previous step results to be the rate determing one.
42 Importance of the environment No secondary structure elements which are relevant to active site electric field (as i.e. in papain and cathepsins) Protein structure seems to act as geometrical constraint which reduces entropy of reaction provides proper conformation to catalytic dyad
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Multiscale Materials Modeling
 Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model. John Dood Hope College
 Water Model. John Dood Hope College") Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model John Dood Hope College What are MD simulations? Model and predict the structure and dynamics of large macromolecules.
Developing Monovalent Ion Parameters for the Optimal Point Charge (OPC) Water Model John Dood Hope College What are MD simulations? Model and predict the structure and dynamics of large macromolecules.
k θ (θ θ 0 ) 2 angles r i j r i j
 2 angles r i j r i j") 1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D.
 3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
Computer simulation methods (2) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Chapter 11 Molecular Mechanics
 Chapter 11 Molecular Mechanics Molecular Mechanics uses an analytical, differentiable, and relatively simple potential energy function, (R), for describing the interactions between a set of atoms specified
Chapter 11 Molecular Mechanics Molecular Mechanics uses an analytical, differentiable, and relatively simple potential energy function, (R), for describing the interactions between a set of atoms specified
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015,
 Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation
 Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
The Molecular Dynamics Method
 H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
Computational Molecular Biophysics. Computational Biophysics, GRS Jülich SS 2013
 Computational Molecular Biophysics Computational Biophysics, GRS Jülich SS 2013 Computational Considerations Number of terms for different energy contributions / E el ~N ~N ~N ~N 2 Computational cost for
Computational Molecular Biophysics Computational Biophysics, GRS Jülich SS 2013 Computational Considerations Number of terms for different energy contributions / E el ~N ~N ~N ~N 2 Computational cost for
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Subject of the Lecture:
 Subject of the Lecture: Conceptual basis for the development of force fields. Implementation/validation Water - a worked example Extensions - combining molecular mechanics and quantum mechanics (QM/MM)
Subject of the Lecture: Conceptual basis for the development of force fields. Implementation/validation Water - a worked example Extensions - combining molecular mechanics and quantum mechanics (QM/MM)
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia
 University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
Biomolecular modeling I
 2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
2016, December 6 Biomolecular structure Structural elements of life Biomolecules proteins, nucleic acids, lipids, carbohydrates... Biomolecular structure Biomolecules biomolecular complexes aggregates...
MODIFIED PROXIMITY CRITERIA FOR THE ANALYSIS OF THE SOLVATION OF A POLYFUNCTIONAL SOLUTE.
 Molecular Simulation 1988, Vol. 1 pp.327-332 c 1988 Gordon and Breach Science Publishers S.A. MODIFIED PROXIMITY CRITERIA FOR THE ANALYSIS OF THE SOLVATION OF A POLYFUNCTIONAL SOLUTE. MIHALY MEZEI Department
Molecular Simulation 1988, Vol. 1 pp.327-332 c 1988 Gordon and Breach Science Publishers S.A. MODIFIED PROXIMITY CRITERIA FOR THE ANALYSIS OF THE SOLVATION OF A POLYFUNCTIONAL SOLUTE. MIHALY MEZEI Department
Hyeyoung Shin a, Tod A. Pascal ab, William A. Goddard III abc*, and Hyungjun Kim a* Korea
 The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
Accounting for Solvation in Quantum Chemistry. Comp chem spring school 2014 CSC, Finland
 Accounting for Solvation in Quantum Chemistry Comp chem spring school 2014 CSC, Finland In solution or in a vacuum? Solvent description is important when: Polar solvent: electrostatic stabilization Solvent
Accounting for Solvation in Quantum Chemistry Comp chem spring school 2014 CSC, Finland In solution or in a vacuum? Solvent description is important when: Polar solvent: electrostatic stabilization Solvent
Scuola di Chimica Computazionale
 Societa Chimica Italiana Gruppo Interdivisionale di Chimica Computazionale Scuola di Chimica Computazionale Introduzione, per Esercizi, all Uso del Calcolatore in Chimica Organica e Biologica Modellistica
Societa Chimica Italiana Gruppo Interdivisionale di Chimica Computazionale Scuola di Chimica Computazionale Introduzione, per Esercizi, all Uso del Calcolatore in Chimica Organica e Biologica Modellistica
Density Functional Theory: from theory to Applications
 Density Functional Theory: from theory to Applications Uni Mainz May 27, 2012 Large barrier-activated processes time-dependent bias potential extended Lagrangian formalism Basic idea: during the MD dynamics
Density Functional Theory: from theory to Applications Uni Mainz May 27, 2012 Large barrier-activated processes time-dependent bias potential extended Lagrangian formalism Basic idea: during the MD dynamics
Reactive Empirical Force Fields
 Reactive Empirical Force Fields Jason Quenneville jasonq@lanl.gov X-1: Solid Mechanics, EOS and Materials Properties Applied Physics Division Los Alamos National Laboratory Timothy C. Germann, Los Alamos
Reactive Empirical Force Fields Jason Quenneville jasonq@lanl.gov X-1: Solid Mechanics, EOS and Materials Properties Applied Physics Division Los Alamos National Laboratory Timothy C. Germann, Los Alamos
The Dominant Interaction Between Peptide and Urea is Electrostatic in Nature: A Molecular Dynamics Simulation Study
 Dror Tobi 1 Ron Elber 1,2 Devarajan Thirumalai 3 1 Department of Biological Chemistry, The Hebrew University, Jerusalem 91904, Israel 2 Department of Computer Science, Cornell University, Ithaca, NY 14853
Dror Tobi 1 Ron Elber 1,2 Devarajan Thirumalai 3 1 Department of Biological Chemistry, The Hebrew University, Jerusalem 91904, Israel 2 Department of Computer Science, Cornell University, Ithaca, NY 14853
Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions Van der Waals Interactions
Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions Van der Waals Interactions
Biomolecular modeling I
 2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
2015, December 15 Biomolecular simulation Elementary body atom Each atom x, y, z coordinates A protein is a set of coordinates. (Gromacs, A. P. Heiner) Usually one molecule/complex of interest (e.g. protein,
Ab initio molecular dynamics. Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy. Bangalore, 04 September 2014
 Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
Ab initio molecular dynamics Simone Piccinin CNR-IOM DEMOCRITOS Trieste, Italy Bangalore, 04 September 2014 What is MD? 1) Liquid 4) Dye/TiO2/electrolyte 2) Liquids 3) Solvated protein 5) Solid to liquid
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro. Karel Berka, Ph.D.
 4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
An Informal AMBER Small Molecule Force Field :
 An Informal AMBER Small Molecule Force Field : parm@frosst Credit Christopher Bayly (1992-2010) initiated, contributed and lead the efforts Daniel McKay (1997-2010) and Jean-François Truchon (2002-2010)
An Informal AMBER Small Molecule Force Field : parm@frosst Credit Christopher Bayly (1992-2010) initiated, contributed and lead the efforts Daniel McKay (1997-2010) and Jean-François Truchon (2002-2010)
Molecular Dynamics Simulation of a Nanoconfined Water Film
 Molecular Dynamics Simulation of a Nanoconfined Water Film Kyle Lindquist, Shu-Han Chao May 7, 2013 1 Introduction The behavior of water confined in nano-scale environment is of interest in many applications.
Molecular Dynamics Simulation of a Nanoconfined Water Film Kyle Lindquist, Shu-Han Chao May 7, 2013 1 Introduction The behavior of water confined in nano-scale environment is of interest in many applications.
COSMO-RS Theory. The Basics
 Theory The Basics From µ to properties Property µ 1 µ 2 activity coefficient vapor pressure Infinite dilution Gas phase Pure compound Pure bulk compound Partition coefficient Phase 1 Phase 2 Liquid-liquid
Theory The Basics From µ to properties Property µ 1 µ 2 activity coefficient vapor pressure Infinite dilution Gas phase Pure compound Pure bulk compound Partition coefficient Phase 1 Phase 2 Liquid-liquid
Molecular Simulation II. Classical Mechanical Treatment
 Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
QM/MM Theory and Examples with ORCA
 QM/MM Theory and Examples with ORCA Marius Retegan Max Planck Institute for Chemical Energy Conversion Stiftstr. 34-36 Mülheim an der Ruhr QM/MM: an historical overview 1976: Warshell and Levitt Theoretical
QM/MM Theory and Examples with ORCA Marius Retegan Max Planck Institute for Chemical Energy Conversion Stiftstr. 34-36 Mülheim an der Ruhr QM/MM: an historical overview 1976: Warshell and Levitt Theoretical
Supporting Information
 alladium monophosphine d(h 3 ): does it really exist in solution? ietro Vidossich*, Gregori Ujaque,* and Agustí Lledós* Supporting Information Contents: 1. Schemes S1 and S2 2. Computational details 3.
alladium monophosphine d(h 3 ): does it really exist in solution? ietro Vidossich*, Gregori Ujaque,* and Agustí Lledós* Supporting Information Contents: 1. Schemes S1 and S2 2. Computational details 3.
74 these states cannot be reliably obtained from experiments. In addition, the barriers between the local minima can also not be obtained reliably fro
 73 Chapter 5 Development of Adiabatic Force Field for Polyvinyl Chloride (PVC) and Chlorinated PVC (CPVC) 5.1 Introduction Chlorinated polyvinyl chloride has become an important specialty polymer due to
73 Chapter 5 Development of Adiabatic Force Field for Polyvinyl Chloride (PVC) and Chlorinated PVC (CPVC) 5.1 Introduction Chlorinated polyvinyl chloride has become an important specialty polymer due to
Modeling in Solution. Important Observations. Energy Concepts
 Modeling in Solution 1 Important Observations Various electrostatic effects for a solvated molecule are often less important than for an isolated gaseous molecule when the molecule is dissolved in a solvent
Modeling in Solution 1 Important Observations Various electrostatic effects for a solvated molecule are often less important than for an isolated gaseous molecule when the molecule is dissolved in a solvent
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water?
 Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Supporting Information
 Supporting Information Constant ph molecular dynamics reveals ph-modulated binding of two small-molecule BACE1 inhibitors Christopher R. Ellis 1,, Cheng-Chieh Tsai 1,, Xinjun Hou 2, and Jana Shen 1, 1
Supporting Information Constant ph molecular dynamics reveals ph-modulated binding of two small-molecule BACE1 inhibitors Christopher R. Ellis 1,, Cheng-Chieh Tsai 1,, Xinjun Hou 2, and Jana Shen 1, 1
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Molecular Modelling. part of Bioinformatik von RNA- und Proteinstrukturen. Sonja Prohaska. Leipzig, SS Computational EvoDevo University Leipzig
 part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
Semi Empirical Force Fields and Their Limitations. Potential Energy Surface (PES)
") Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
Semi Empirical Force Fields and Their Limitations Ioan Kosztin Beckman Institute University of Illinois at Urbana-Champaign Potential Energy Surface (PES) Schrödinger equation: H T Ψ( r, = E Ψ( r, H =
Development of Molecular Dynamics Simulation System for Large-Scale Supra-Biomolecules, PABIOS (PArallel BIOmolecular Simulator)
") Development of Molecular Dynamics Simulation System for Large-Scale Supra-Biomolecules, PABIOS (PArallel BIOmolecular Simulator) Group Representative Hisashi Ishida Japan Atomic Energy Research Institute
Development of Molecular Dynamics Simulation System for Large-Scale Supra-Biomolecules, PABIOS (PArallel BIOmolecular Simulator) Group Representative Hisashi Ishida Japan Atomic Energy Research Institute
Supporting Information
 Supporting Information ph-responsive self-assembly of polysaccharide through a rugged energy landscape Brian H. Morrow, Gregory F. Payne, and Jana Shen Department of Pharmaceutical Sciences, School of
Supporting Information ph-responsive self-assembly of polysaccharide through a rugged energy landscape Brian H. Morrow, Gregory F. Payne, and Jana Shen Department of Pharmaceutical Sciences, School of
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
 A Nobel Prize for Molecular Dynamics and QM/MM What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential
A Nobel Prize for Molecular Dynamics and QM/MM What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential
Aqueous solutions. Solubility of different compounds in water
 Aqueous solutions Solubility of different compounds in water The dissolution of molecules into water (in any solvent actually) causes a volume change of the solution; the size of this volume change is
Aqueous solutions Solubility of different compounds in water The dissolution of molecules into water (in any solvent actually) causes a volume change of the solution; the size of this volume change is
A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane
 A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane Supplementary Information: Simulation Procedure and Physical Property Analysis Simulation Procedure The molecular
A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane Supplementary Information: Simulation Procedure and Physical Property Analysis Simulation Procedure The molecular
A new extension of QM/MM methods: the adaptive buffered-force QM/MM method
 A new extension of QM/MM methods: the adaptive buffered-force QM/MM method Letif Mones Engineering Department, University of Cambridge lam81@cam.ac.uk Overview Basic concept of the QM/MM methods MM vs.
A new extension of QM/MM methods: the adaptive buffered-force QM/MM method Letif Mones Engineering Department, University of Cambridge lam81@cam.ac.uk Overview Basic concept of the QM/MM methods MM vs.
Molecular Dynamics Simulations. Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia
 Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Molecular Dynamics Simulations Dr. Noelia Faginas Lago Dipartimento di Chimica,Biologia e Biotecnologie Università di Perugia 1 An Introduction to Molecular Dynamics Simulations Macroscopic properties
Density functional theory in solution: Implementing an implicit solvent model for CASTEP and ONETEP
 Density functional theory in solution: Implementing an implicit solvent model for CASTEP and ONETEP, November 2016 James C. Womack University of Southampton, Southampton, UK Overview Background The model
Density functional theory in solution: Implementing an implicit solvent model for CASTEP and ONETEP, November 2016 James C. Womack University of Southampton, Southampton, UK Overview Background The model
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Applications of the Spring. Force: molecules
 Applications of the Spring Atoms interact via the coulomb force Force: molecules When atoms are far apart, they are attractive When atoms are too close, they are repulsive Atoms in a molecule display relative
Applications of the Spring Atoms interact via the coulomb force Force: molecules When atoms are far apart, they are attractive When atoms are too close, they are repulsive Atoms in a molecule display relative
Supplementary Information
 Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Force Fields for MD simulations
 Force Fields for MD simulations Topology/parameter files Where do the numbers an MD code uses come from? ow to make topology files for ligands, cofactors, special amino acids, ow to obtain/develop missing
Force Fields for MD simulations Topology/parameter files Where do the numbers an MD code uses come from? ow to make topology files for ligands, cofactors, special amino acids, ow to obtain/develop missing
Molecular Dynamics Simulation of a Biomolecule with High Speed, Low Power and Accuracy Using GPU-Accelerated TSUBAME2.
 APSIPA ASC 2011 Xi an Molecular Dynamics Simulation of a Biomolecule with High Speed, Low Power and Accuracy Using GPU-Accelerated TSUBAME2.0 Supercomputer Shiqiao Du, Takuro Udagawa, Toshio Endo and Masakazu
APSIPA ASC 2011 Xi an Molecular Dynamics Simulation of a Biomolecule with High Speed, Low Power and Accuracy Using GPU-Accelerated TSUBAME2.0 Supercomputer Shiqiao Du, Takuro Udagawa, Toshio Endo and Masakazu
Gromacs Workshop Spring CSC
 Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
Towards fast and accurate binding affinity. prediction with pmemdgti: an efficient. implementation of GPU-accelerated. Thermodynamic Integration
 Towards fast and accurate binding affinity prediction with pmemdgti: an efficient implementation of GPU-accelerated Thermodynamic Integration Tai-Sung Lee,, Yuan Hu, Brad Sherborne, Zhuyan Guo, and Darrin
Towards fast and accurate binding affinity prediction with pmemdgti: an efficient implementation of GPU-accelerated Thermodynamic Integration Tai-Sung Lee,, Yuan Hu, Brad Sherborne, Zhuyan Guo, and Darrin
A generalized Born formalism for heterogeneous dielectric environments: Application to the implicit modeling of biological membranes
 THE JOURNAL OF CHEMICAL PHYSICS 122, 124706 2005 A generalized Born formalism for heterogeneous dielectric environments: Application to the implicit modeling of biological membranes Seiichiro Tanizaki
THE JOURNAL OF CHEMICAL PHYSICS 122, 124706 2005 A generalized Born formalism for heterogeneous dielectric environments: Application to the implicit modeling of biological membranes Seiichiro Tanizaki
Water models in classical simulations
 Water models in classical simulations Maria Fyta Institut für Computerphysik, Universität Stuttgart Stuttgart, Germany Water transparent, odorless, tasteless and ubiquitous really simple: two H atoms attached
Water models in classical simulations Maria Fyta Institut für Computerphysik, Universität Stuttgart Stuttgart, Germany Water transparent, odorless, tasteless and ubiquitous really simple: two H atoms attached
What is Classical Molecular Dynamics?
 What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
Dominant Paths in Protein Folding
 Dominant Paths in Protein Folding Henri Orland SPhT, CEA-Saclay France work in collaboration with P. Faccioli, F. Pederiva, M. Sega University of Trento Henri Orland Annecy meeting 2006 Outline Basic notions
Dominant Paths in Protein Folding Henri Orland SPhT, CEA-Saclay France work in collaboration with P. Faccioli, F. Pederiva, M. Sega University of Trento Henri Orland Annecy meeting 2006 Outline Basic notions
Helix Folding Simulations with Various Initial Conformations
 1 796 Biophysical Journal Volume 66 June 1994 1796-1803 Helix Folding Simulations with Various Initial Conformations Shen-Shu Sung Research Institute, The Cleveland Clinic Foundation, Cleveland Ohio 44195
1 796 Biophysical Journal Volume 66 June 1994 1796-1803 Helix Folding Simulations with Various Initial Conformations Shen-Shu Sung Research Institute, The Cleveland Clinic Foundation, Cleveland Ohio 44195
Statistical Mechanics for Proteins
 The Partition Function From Q all relevant thermodynamic properties can be obtained by differentiation of the free energy F: = kt q p E q pd d h T V Q ), ( exp 1! 1 ),, ( 3 3 3 ),, ( ln ),, ( T V Q kt
The Partition Function From Q all relevant thermodynamic properties can be obtained by differentiation of the free energy F: = kt q p E q pd d h T V Q ), ( exp 1! 1 ),, ( 3 3 3 ),, ( ln ),, ( T V Q kt
A Hamiltonian electrostatic coupling scheme for hybrid Car Parrinello molecular dynamics simulations
 JOURNAL OF CHEMICAL PHYSICS VOLUME 116, NUMBER 16 22 APRIL 2002 A Hamiltonian electrostatic coupling scheme for hybrid Car Parrinello molecular dynamics simulations Alessandro Laio, Joost VandeVondele,
JOURNAL OF CHEMICAL PHYSICS VOLUME 116, NUMBER 16 22 APRIL 2002 A Hamiltonian electrostatic coupling scheme for hybrid Car Parrinello molecular dynamics simulations Alessandro Laio, Joost VandeVondele,
Molecular Dynamics Investigation of the ω-current in the Kv1.2 Voltage Sensor Domains
 Molecular Dynamics Investigation of the ω-current in the Kv1.2 Voltage Sensor Domains Fatemeh Khalili-Araghi, Emad Tajkhorshid, Benoît Roux, and Klaus Schulten Department of Physics, Department of Biochemistry,
Molecular Dynamics Investigation of the ω-current in the Kv1.2 Voltage Sensor Domains Fatemeh Khalili-Araghi, Emad Tajkhorshid, Benoît Roux, and Klaus Schulten Department of Physics, Department of Biochemistry,
Coupling the Level-Set Method with Variational Implicit Solvent Modeling of Molecular Solvation
 Coupling the Level-Set Method with Variational Implicit Solvent Modeling of Molecular Solvation Bo Li Math Dept & CTBP, UCSD Li-Tien Cheng (Math, UCSD) Zhongming Wang (Math & Biochem, UCSD) Yang Xie (MAE,
Coupling the Level-Set Method with Variational Implicit Solvent Modeling of Molecular Solvation Bo Li Math Dept & CTBP, UCSD Li-Tien Cheng (Math, UCSD) Zhongming Wang (Math & Biochem, UCSD) Yang Xie (MAE,
Practical Guide to Density Functional Theory (DFT)
") Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
Practical Guide to Density Functional Theory (DFT) Brad Malone, Sadas Shankar Quick recap of where we left off last time BD Malone, S Shankar Therefore there is a direct one-to-one correspondence between
Improved Resolution of Tertiary Structure Elasticity in Muscle Protein
 Improved Resolution of Tertiary Structure Elasticity in Muscle Protein Jen Hsin and Klaus Schulten* Department of Physics and Beckman Institute, University of Illinois at Urbana-Champaign, Urbana, Illinois
Improved Resolution of Tertiary Structure Elasticity in Muscle Protein Jen Hsin and Klaus Schulten* Department of Physics and Beckman Institute, University of Illinois at Urbana-Champaign, Urbana, Illinois
Performance Comparison of Generalized Born and Poisson Methods in the Calculation of Electrostatic Solvation Energies for Protein Structures
 Performance Comparison of Generalized Born and Poisson Methods in the Calculation of Electrostatic Solvation Energies for Protein Structures MICHAEL FEIG, 1 ALEXEY ONUFRIEV, 1 MICHAEL S. LEE, 2 WONPIL
Performance Comparison of Generalized Born and Poisson Methods in the Calculation of Electrostatic Solvation Energies for Protein Structures MICHAEL FEIG, 1 ALEXEY ONUFRIEV, 1 MICHAEL S. LEE, 2 WONPIL
Supporting Information for. Ab Initio Metadynamics Study of VO + 2 /VO2+ Redox Reaction Mechanism at the Graphite. Edge Water Interface
 Supporting Information for Ab Initio Metadynamics Study of VO + 2 /VO2+ Redox Reaction Mechanism at the Graphite Edge Water Interface Zhen Jiang, Konstantin Klyukin, and Vitaly Alexandrov,, Department
Supporting Information for Ab Initio Metadynamics Study of VO + 2 /VO2+ Redox Reaction Mechanism at the Graphite Edge Water Interface Zhen Jiang, Konstantin Klyukin, and Vitaly Alexandrov,, Department
The change in free energy on transferring an ion from a medium of low dielectric constantε1 to one of high dielectric constant ε2:
 The Born Energy of an Ion The free energy density of an electric field E arising from a charge is ½(ε 0 ε E 2 ) per unit volume Integrating the energy density of an ion over all of space = Born energy:
The Born Energy of an Ion The free energy density of an electric field E arising from a charge is ½(ε 0 ε E 2 ) per unit volume Integrating the energy density of an ion over all of space = Born energy:
Chem 1140; Molecular Modeling
 P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
Universal Repulsive Contribution to the. Solvent-Induced Interaction Between Sizable, Curved Hydrophobes: Supporting Information
 Universal Repulsive Contribution to the Solvent-Induced Interaction Between Sizable, Curved Hydrophobes: Supporting Information B. Shadrack Jabes, Dusan Bratko, and Alenka Luzar Department of Chemistry,
Universal Repulsive Contribution to the Solvent-Induced Interaction Between Sizable, Curved Hydrophobes: Supporting Information B. Shadrack Jabes, Dusan Bratko, and Alenka Luzar Department of Chemistry,
Supporting Material for. Microscopic origin of gating current fluctuations in a potassium channel voltage sensor
 Supporting Material for Microscopic origin of gating current fluctuations in a potassium channel voltage sensor J. Alfredo Freites, * Eric V. Schow, * Stephen H. White, and Douglas J. Tobias * * Department
Supporting Material for Microscopic origin of gating current fluctuations in a potassium channel voltage sensor J. Alfredo Freites, * Eric V. Schow, * Stephen H. White, and Douglas J. Tobias * * Department
QM/MM: what have we learned, where are we, and where do we go from here?
 Theor Chem Acc (2007) 117: 185 199 DOI 10.1007/s00214-006-0143-z FEATURE ARTICLE Hai Lin Donald G. Truhlar QM/MM: what have we learned, where are we, and where do we go from here? Received: 25 May 2005
Theor Chem Acc (2007) 117: 185 199 DOI 10.1007/s00214-006-0143-z FEATURE ARTICLE Hai Lin Donald G. Truhlar QM/MM: what have we learned, where are we, and where do we go from here? Received: 25 May 2005
Applications of Molecular Dynamics
 June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
 Title Super- and subcritical hydration of Thermodynamics of hydration Author(s) Matubayasi, N; Nakahara, M Citation JOURNAL OF CHEMICAL PHYSICS (2000), 8109 Issue Date 2000-05-08 URL http://hdl.handle.net/2433/50350
Title Super- and subcritical hydration of Thermodynamics of hydration Author(s) Matubayasi, N; Nakahara, M Citation JOURNAL OF CHEMICAL PHYSICS (2000), 8109 Issue Date 2000-05-08 URL http://hdl.handle.net/2433/50350
Atomic and molecular interaction forces in biology
 Atomic and molecular interaction forces in biology 1 Outline Types of interactions relevant to biology Van der Waals interactions H-bond interactions Some properties of water Hydrophobic effect 2 Types
Atomic and molecular interaction forces in biology 1 Outline Types of interactions relevant to biology Van der Waals interactions H-bond interactions Some properties of water Hydrophobic effect 2 Types
Molecular Mechanics. Yohann Moreau. November 26, 2015
 Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Specific Ion Solvtion in Ethylene Carbonate and Propylene Carbonate
 Specific Ion Solvtion in Ethylene Carbonate and Propylene Carbonate A. Arslanargin, A. Powers, S. Rick, T. Pollard, T. Beck Univ Cincinnati Chemistry Support: NSF, OSC TSRC 2016 November 2, 2016 A. Arslanargin,
Specific Ion Solvtion in Ethylene Carbonate and Propylene Carbonate A. Arslanargin, A. Powers, S. Rick, T. Pollard, T. Beck Univ Cincinnati Chemistry Support: NSF, OSC TSRC 2016 November 2, 2016 A. Arslanargin,
T6.2 Molecular Mechanics
 T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
Force Fields in Molecular Mechanics
 Force Fields in Molecular Mechanics Rajarshi Guha (9915607) and Rajesh Sardar (9915610) March 21, 2001 1 Introduction With the advent of computers chemists have realized the utility of carrying out simulations
Force Fields in Molecular Mechanics Rajarshi Guha (9915607) and Rajesh Sardar (9915610) March 21, 2001 1 Introduction With the advent of computers chemists have realized the utility of carrying out simulations
Dielectric polarization of 2-pyrrolidinone molecules in benzene solution - a quantum-chemical study
 Dielectric polarization of 2-pyrrolidinone molecules in benzene solution - a quantum-chemical study L. Gorb* ), J. Jadżyn $) and K. W. Wojciechowski #) Institute of Molecular Physics, Polish Academy of
Dielectric polarization of 2-pyrrolidinone molecules in benzene solution - a quantum-chemical study L. Gorb* ), J. Jadżyn $) and K. W. Wojciechowski #) Institute of Molecular Physics, Polish Academy of
D. R. Berard, D. Wei. Centre de Recherche en Calcul Applique, 5160 Boulevard Decarie, Bureau 400, D. R. Salahub
 Towards a density functional treatment of chemical reactions in complex media D. R. Berard, D. Wei Centre de Recherche en Calcul Applique, 5160 Boulevard Decarie, Bureau 400, Montreal, Quebec, Canada H3X
Towards a density functional treatment of chemical reactions in complex media D. R. Berard, D. Wei Centre de Recherche en Calcul Applique, 5160 Boulevard Decarie, Bureau 400, Montreal, Quebec, Canada H3X
SUPPLEMENTARY INFORMATION. Very Low Temperature Membrane Free Desalination. by Directional Solvent Extraction
 SUPPLEMENTARY INFORMATION Very Low Temperature Membrane Free Desalination by Directional Solvent Extraction Anurag Bajpayee, Tengfei Luo, Andrew J. Muto, and Gang Chen Department of Mechanical Engineering,
SUPPLEMENTARY INFORMATION Very Low Temperature Membrane Free Desalination by Directional Solvent Extraction Anurag Bajpayee, Tengfei Luo, Andrew J. Muto, and Gang Chen Department of Mechanical Engineering,
Biomolecules are dynamic no single structure is a perfect model
 Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
3. Solutions W = N!/(N A!N B!) (3.1) Using Stirling s approximation ln(n!) = NlnN N: ΔS mix = k (N A lnn + N B lnn N A lnn A N B lnn B ) (3.
 (3.1) Using Stirling s approximation ln(n!) = NlnN N: ΔS mix = k (N A lnn + N B lnn N A lnn A N B lnn B ) (3.") 3. Solutions Many biological processes occur between molecules in aqueous solution. In addition, many protein and nucleic acid molecules adopt three-dimensional structure ( fold ) in aqueous solution.
3. Solutions Many biological processes occur between molecules in aqueous solution. In addition, many protein and nucleic acid molecules adopt three-dimensional structure ( fold ) in aqueous solution.