Computational Molecular Biophysics. Computational Biophysics, GRS Jülich SS 2013
|
|
|
- Claude Stephen Skinner
- 6 years ago
- Views:
Transcription
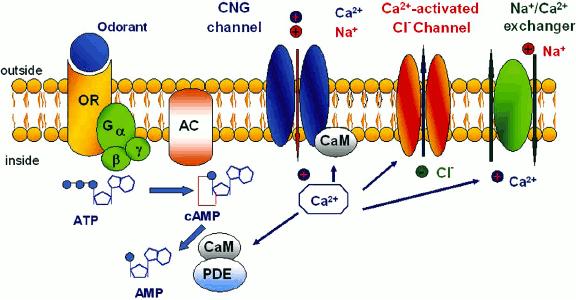
1 Computational Molecular Biophysics Computational Biophysics, GRS Jülich SS 2013
2 Computational Considerations Number of terms for different energy contributions / E el ~N ~N ~N ~N 2 Computational cost for non-bonded interactions Number of atoms: 302 ~96% ~98.8% ~99.88% 2
3 Non-Bonded Interactions Approaches for reducing computational costs Spherical cutoffs Suitable for van der Waals interactions (~1/r 6 ) Particle Mesh Ewald Preferred method in biological simulations Fast multipole schemes Continuum solvation methods 3
4 Non-Bonded Interactions Computational Bottleneck Pairwise calculation ~N 2 (N = number of atoms) Cutoff-Schemes Neglect long range interactions beyond some distance Poor approximation for highly charged systems 4
5 Non-Bonded Interactions Cutoff Schemes Applied to either energy or force function of the non-bonded potential. Truncation Abruptly defines values to be zero at the cutoff b. Do no alter values for distances r < b. Switching Begin to change values at a nonzero value a < b. Leave values unchanged for r < a. Shift functions Alter functions more gradually for all r < b. Atom-based cutoffs Group-based cutoffs: Distances between group centers. 5
6 Non-Bonded Interactions Cutoff Schemes Guidelines Short-range energies and forces should be altered as minimally as possible (while satisfying other criteria). Energies should be altered gradually rather than abruptly to avoid the introduction of artificial minima (where the potential energy and gradient values are suddenly zero). Avoid introducing large forces around the cutoff region. Should alter the function in a way to approximately conserve energy. 6

7 Cutoff Schemes Altered potential Non-Bonded Interactions Errors in the forces Formula appear on the following 2 slides 7
8 Non-Bonded Interactions Cutoff Schemes Distance-dependent shift or switch function: Truncation: Discontinuity Fails to conserve energy Potential switch 8 Potential shift CHARMM: electrostatics S 1 : group-based pot. shift S 2 : atom-based pot. shift
9 Non-Bonded Interactions Cutoff Schemes Potential shift: CHARMM van der Waals shift 9
10 Periodic Boundary Conditions PBC Unit cell Infinitely many images in space 10
11 Periodic Boundary Conditions Minimum Image Convention Only coordinates of the unit cell need to be recorded and propagated. As an atom leaves the unit cell, an image enters to replace it conserving the total number of particles. Cell size: solvated biomolecule with at least 10 Å layer of water. Each atom acts only with the closest periodic image of the other N-1 particles. Spherical cutoff: distance at most L/2, where L is the dimension of the side of the box. 11
12 Ewald sum Ewald sum and crystallography Reciprocal term The reciprocal lattice is an orthogonal system related to the orthogonal system associated with the atoms in the unit cell (real-space lattice). This lattice is used to express scattering vector in crystals. Ewald summation: two terms arise for the computation of the electrostatic energy: Atom pairs in the direct lattice: Direct calculation Atom pairs corresponding to interactions with images of the unit cell atoms: Fourier transformation Fast Fourier transforms (FFT) Ewald sum: evaluate the electrostatic energy corresponding to the reciprocal lattice. Electron density distribution Electrostatic energy in terms of charge density rather than point charges to make the infinite sum convergent. 12

13 13 Tricks from X-Ray Crystallography
14 14 Tricks from X-Ray Crystallography

15 15 Tricks from X-Ray Crystallography

16 Ewald Summation Coulomb energy in periodic domains (Without constants) The sum is conditionally convergent since the terms decay of 1/ n like a harmonic series. 16
17 Ewald Summation Idea: Splitting into short-range and long-range contributions, which converts a conditionally convergent sum into two terms that are rapidly and absolutely converging 17 Short range Embedding each point charge in a screening potential: Gaussian with opposite sign: outside this range (width of the Gaussian) the net charge is zero => interaction between screened point charges is shortranged and can be evaluated directly in real space. Long range To recover the original point charge interaction, the effect of the screening potentials must be subtracted again. This compensating term is an interaction between Gaussian charge distributions evaluated efficiently in reciprocal space.
18 Ewald Summation Idea: Splitting into short-range and long-range contributions Calculated in reciprocal (Fourier) space. Calculated in real space. 18
19 Ewald Summation Ewald sum Convert the sum into an expression involving a sum of two absolutely and rapidly converging series in direct and reciprocal space. Conversion is accomplished by representing each point charge as a Gaussian charge density, producing an exponentially decaying function. This Gaussian transformation must be counteracted by an analogous subtraction to leave the net result of an effective point charge (delta function charge). This cancelling distribution is summed in the reciprocal space and transformed back to real space because it is a smoothly-varying periodic function which can be represented by a rapidly convergent Fourier series. 19 Short-ranged with singularity at origin long-ranged smooth
20 Ewald Summation Error function Complementary error function Screening Gaussians centered at each point charge (parameterized by ß) Instead of the cumulative point charge density described by a sum of delta functions 20 a sum of localized densities is used to define the total Gaussian screening charge density

21 Ewald Summation Real space sum Given in terms of the complementary error function using Poisson s equation for the electrostatic potential, due to the Gaussian charge cloud followed by integration. Solution of the Poisson equation leads to Thus, the sum for the Coulomb energy is converted into the pair of terms 21
22 Ewald Summation 22 Ewald sum complexity Optimization of ß (Gaussian width) As ß increases, the real space sum converges more rapidly, but the recipropcal sum more slowly. Both sums are truncated in practice A sufficiently large ß yields an accurate direct space sum with an appropriate truncation resulting in ~N rather than ~N 2. The reciprocal space sum still requires ~N 2 work. In total ~N 3/2. Mesh interpolation Trigonometric function values in the Fourier series used to represent the reciprocal space term can be evaluated through a smooth interpolation of the potential over a regular grid. Particle-Mesh Ewald (PME): ~N(log N). Variations Particle-Particle Particle Mesh Ewald (P 3 M): Long range forces in large systems are split into a short-range, rapidly varying part (direct particle-particle sum) and a smooth, slowly varying remainder (particle-mesh).
23 Ewald Summation Resulting Ewald Summation E real : real-space sum of electrostatic energy due to the point charges screened by oppositely charged Gaussians. E recipe : associated cancelling term summed in reciprocal space using smooth interpolation of Fourier-series values. 23
24 Ewald Summation Resulting Ewald Summation E cor, self : Subtracts the self-interaction term (each point charge and its corresponding charge cloud). E cor, ex : Subtracts the Coulomb contribution from the nonbonded energies included in the Coulomb energy since they are separately accounted for in bonded, bond-angle, and possibly dihedral-angle terms. E cor, ε : Accounts for non-infinite dielectric medium 24
25 Ewald Summation Practical implementation Gaussian width Real space term should acquire the desired accuracy tolerance 10 / L (L ranges roughly from 60 to 100 Å). Grid size and accuracy Coarse interpolation grid: 1-2 Å (moderate accuracy, 10-4 relative force error) Finer grid: -0.5 Å (very high accuracy, relative force error) 25
26 Fast Multipole Moment (FMM) Splitting into near- and far field Near field: exact calculation Far field: dividing space into boxes Interaction between all molecules in one box with all molecules in another is approximated as interactions between multipoles located at the box centers. The further away from each other two boxes are, the larger the boxes can be for a given accuracy, thereby reducing the formal N 2 scaling to linear scaling. Cross-over point when FMM method become faster than PME: ~ 10 5 atoms. Works best when particles are relatively uniformly distributed. Maximum error is significantly larger than for Ewald methods. No requirement of periodicity, thus applicable to large non-periodic systems. 26 Pairwise interactions Multipole interactions
 27 O(M")
27 Naive Multipol- Method U AB Fast Multipole Moment (FMM) U AB FMM Method U AC1 U AC2 U AC3 U AC4 1/4 U AB U AC O(M 2 ) 27 O(M 1 )
28 Reducing computational costs Long range interaction - Ewald summation method Scaling: O(N 3/2 ) - Particle mesh Ewald techniques O(N log N) - Fast multipole methods O(N) N = number of atoms 28

29 Force Fields Parameterization Non-bonded and torsion parameters affect performance most, stretching and bending are less important. Aim to reproduce exp. structures exp. thermodynamics QM derived properties 29
30 Force Fields Parameterization Parameter set and functional forms are self-consistent, thus parameters are not transferable from one force field to another. Force constants may differ by almost a factor of two, but this is of course related to the stiffness of the bond. The associated energy contributions are small regardless of the force constant, i.e. the minimum energy geometry is insensitive to the exact value of the force constant. 30
31 Force Fields Class I / Harmonic / Diagonal / Classical FF Designed to treat large systems such as proteins or DNA Simple functional form (only harmonic functions for bonded interactions, no cross terms, Lennard-Jones potential for van der Waals interactions). CHARMM, AMBER, OPLS, GROMOS, Class II / Second generation FF Reproducing small to medium sized molecules with high accuracy. Cross terms, cubic or quartic expansions for bond and angle potentials. Reproduce also vibrational frequencies MMFF94 (Merck), MM2/MM3/MM4 (Allinger, organic molecules), Class III Refinements allowing parameters to depend on neighboring atom types e.g. for modeling hyperconjugation including electronic polarization effects. 31
32 Force Fields Class I potential energy function 32
33 Force Fields Class II potential energy function 33
34 Polarizable Force Fields (Class III) Based on electronic structure theory X-Pol: Explicit polarization theory, fragment based electronic structure method (J. Gao) Based on induced dipoles CFF/ind, ENZYMIX DFF90 (P. Th. van Duijnen) PIPF (J. Gao) Based on point charges PFF: Polarizable force field (R. A. Friesner) CHARMM PFF (S. Patel, C. L. Brooks III) CHARMM PFF: classical Drude oscillators (A. MacKerell, B. Roux) AMBER PFF (J. Caldwell) Based on distributed multipoles AMOEBA (P. Ren, J. W. Ponder) Based on density Gaussian electrostatic model (GEM) (T. Darden) Kim-Gordon approach (J. Hutter) 34
35 Simplified Force Fields United atom force fields Do not consider hydrogen atoms explicitly Option for AMBER, CHARMM, GROMOS, DREIDING Reduce the effort by a factor ~2-3. CH 2 atom Coarse-grained FF Atom groups as single particles (pseudo atoms or beads) E.g. amino acid: 1-2 beads on the backbone, 0-3 beads for side chain. MARTINI 35

36 Coarse-grained Force Fields MARTINI force field: Amino acid representation 1-4 beads Coarse-grained representation of all amino acids. Different colors represent different particle types. 36 Luca Monticelli; Senthil K. Kandasamy; Xavier Periole; Ronald G. Larson; D. Peter Tieleman; Siewert-Jan Marrink; J. Chem. Theory Comput. 2008, 4,

37 Coarse-grained Force Fields MARTINI force field 37 Luca Monticelli; Senthil K. Kandasamy; Xavier Periole; Ronald G. Larson; D. Peter Tieleman; Siewert-Jan Marrink; J. Chem. Theory Comput. 2008, 4,

38 Reactive Force Fields Empirical Valence Bond (EVB) A. Warshel Chemical reactions, e.g. enzymatic catalysis, proton transfer, Multi-state EVB: G. A. Voth Y. Peng, G. A. Voth, Biochemica et Biophysica Acta Bioenergetics 2012, 1817,
39 Knowledge-Based Force Fields Knowledge-based FF as alternative to physics-based FF Energy functions derived from databases of know protein structures Probabilities are calculated that residues appear in specific configurations and that residues appear together in a particular relative geometry. Probabilities are converted into an effective potential energy using the Boltzmann equation. c 1 : amino acid type, c 2 : structure descriptor of the same or neighboring residue, p: relative occurrence in the data base. Pro: Any behavior seen in protein structures can be modeled. Con: Phenomenological functions cannot predict behavior not present in the training data set. Examples: Go models (P. Wolynes), Rosetta (D. Baker) 39
40 40 Force Fields
41 41 Force Fields
42 Force Fields Class I: AMBER (Assisted Model Building and Energy Refinement) ff94, ff96, ff98 proteins ff94 biased towards helices, ff96 favoring extended conformations too much f99 proteins, RNA, DNA good nonbonded interactions, overstabilization of α helices ff99sb proteins, RNA, DNA fixed overstabilization of helices ff03 proteins, RNA, DNA partial charges of f99 reparameterized, better balance between α helix and ß sheet ff99sb-ildn ff99sb-ildn-nmr 42 Improved side-chain torsional potentials for Ile, Leu, Asp, and Asn. NMR-based torsional modifications ff03*, f99sb* proteins, RNA, DNA modified backbone potential for a better balance between helix and coil GAFF any molecule general Amber force field, used for ligands GLYCAM sugars carb
43 Force Fields Class I: CHARMM (Chemistry at HARvard Molecular Mechanics) CHARMM19 proteins, lipids united atom CHARMM22 proteins all-atom, parameterized for TIP3P (adapted for CHARMM), though often used with implicit solvent, tends to favor π helices. CHARMM22-CMAP proteins CMAP backbone dihedral angle corrections CHARMM27 RNA, DNA, lipids, sugars based on CHARMM22/ CMAP 43
44 Force Fields Class I: GROMOS (GROningen MOlecular Simulation package) OPLS (Optimized Potentials for Liquid Simulations) GROMOS87, GROMOS96, GROMOS05, GROMOS11 alkanes, proteins, sugars united atom Parameter sets: 54, 53,45, 43, traditionally used with SPC water OPLS-UA proteins united atom, TIP3P/TIP4P water models, optimized to fit exp. properties of liquids. OPLS-AA proteins all atom, TIP3P/TIP4P water models, optimized to fit exp. properties of liquids. OPLS-2001, OPLS-2005 proteins, sugars based on OPLS-AA 44
45 Force Fields Class III: Polarizable force fields CHARMM-FQ CHARMM-CHEQ proteins CHARMM-Drude proteins Drude model fluctuating charge model or charge equilibration model 45

46 Force Field Improvement Lindorff-Larsen et al. PLOS-ONE 2012, 7, e ff99sb*-ildn and CHARMM22* perform consistently well in reproducing the experimental data in the test set. 46
47 Force Field Deficiencies Deficiencies / Need for improvement Determination of partial charges Improvement of electrostatic potentials (e.g. use of distributed multipole analysis) Methods for solvent representation. Resolve conflicting results by different models and potentials. Van der Waals parameter are strongly environment dependent Inclusion of polarization. 47
48 App1: Derivatives of MM Energy Functions Derivatives for forces 48
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
Computer simulation methods (2) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Non-bonded interactions
 speeding up the number-crunching continued Marcus Elstner and Tomáš Kubař December 3, 2013 why care? number of individual pair-wise interactions bonded interactions proportional to N: O(N) non-bonded interactions
speeding up the number-crunching continued Marcus Elstner and Tomáš Kubař December 3, 2013 why care? number of individual pair-wise interactions bonded interactions proportional to N: O(N) non-bonded interactions
Non-bonded interactions
 speeding up the number-crunching Marcus Elstner and Tomáš Kubař May 8, 2015 why care? key to understand biomolecular structure and function binding of a ligand efficiency of a reaction color of a chromophore
speeding up the number-crunching Marcus Elstner and Tomáš Kubař May 8, 2015 why care? key to understand biomolecular structure and function binding of a ligand efficiency of a reaction color of a chromophore
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Lecture 12: Solvation Models: Molecular Mechanics Modeling of Hydration Effects
 Statistical Thermodynamics Lecture 12: Solvation Models: Molecular Mechanics Modeling of Hydration Effects Dr. Ronald M. Levy ronlevy@temple.edu Bare Molecular Mechanics Atomistic Force Fields: torsion
Statistical Thermodynamics Lecture 12: Solvation Models: Molecular Mechanics Modeling of Hydration Effects Dr. Ronald M. Levy ronlevy@temple.edu Bare Molecular Mechanics Atomistic Force Fields: torsion
Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia
 University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
University of Groningen Peptide folding in non-aqueous environments investigated with molecular dynamics simulations Soto Becerra, Patricia IMPORTANT NOTE: You are advised to consult the publisher's version
Molecular Mechanics. Yohann Moreau. November 26, 2015
 Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Multiscale Materials Modeling
 Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015,
 Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Subject of the Lecture:
 Subject of the Lecture: Conceptual basis for the development of force fields. Implementation/validation Water - a worked example Extensions - combining molecular mechanics and quantum mechanics (QM/MM)
Subject of the Lecture: Conceptual basis for the development of force fields. Implementation/validation Water - a worked example Extensions - combining molecular mechanics and quantum mechanics (QM/MM)
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Theoretical and Computational Biophysics Group, Beckman Institute Departments of Biochemistry and Pharmacology,
Water models in classical simulations
 Water models in classical simulations Maria Fyta Institut für Computerphysik, Universität Stuttgart Stuttgart, Germany Water transparent, odorless, tasteless and ubiquitous really simple: two H atoms attached
Water models in classical simulations Maria Fyta Institut für Computerphysik, Universität Stuttgart Stuttgart, Germany Water transparent, odorless, tasteless and ubiquitous really simple: two H atoms attached
Hyeyoung Shin a, Tod A. Pascal ab, William A. Goddard III abc*, and Hyungjun Kim a* Korea
 The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
Gromacs Workshop Spring CSC
 Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
Gromacs Workshop Spring 2007 @ CSC Erik Lindahl Center for Biomembrane Research Stockholm University, Sweden David van der Spoel Dept. Cell & Molecular Biology Uppsala University, Sweden Berk Hess Max-Planck-Institut
Force fields in computer simulation of soft nanomaterials
 Lecture 2 Part B Force fields in computer simulation of soft nanomaterials Recommended reading: Leach Chapter 4 1 Force Field Methods Force field methods are simulation methods that use classical force
Lecture 2 Part B Force fields in computer simulation of soft nanomaterials Recommended reading: Leach Chapter 4 1 Force Field Methods Force field methods are simulation methods that use classical force
MOLECULAR DYNAMICS SIMULATIONS OF BIOMOLECULES: Long-Range Electrostatic Effects
 Annu. Rev. Biophys. Biomol. Struct. 1999. 28:155 79 Copyright c 1999 by Annual Reviews. All rights reserved MOLECULAR DYNAMICS SIMULATIONS OF BIOMOLECULES: Long-Range Electrostatic Effects Celeste Sagui
Annu. Rev. Biophys. Biomol. Struct. 1999. 28:155 79 Copyright c 1999 by Annual Reviews. All rights reserved MOLECULAR DYNAMICS SIMULATIONS OF BIOMOLECULES: Long-Range Electrostatic Effects Celeste Sagui
Applications of Molecular Dynamics
 June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
June 4, 0 Molecular Modeling and Simulation Applications of Molecular Dynamics Agricultural Bioinformatics Research Unit, Graduate School of Agricultural and Life Sciences, The University of Tokyo Tohru
Molecular Dynamics. A very brief introduction
 Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
An introduction to Molecular Dynamics. EMBO, June 2016
 An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
An introduction to Molecular Dynamics EMBO, June 2016 What is MD? everything that living things do can be understood in terms of the jiggling and wiggling of atoms. The Feynman Lectures in Physics vol.
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Computational protein design
 Computational protein design There are astronomically large number of amino acid sequences that needs to be considered for a protein of moderate size e.g. if mutating 10 residues, 20^10 = 10 trillion sequences
Computational protein design There are astronomically large number of amino acid sequences that needs to be considered for a protein of moderate size e.g. if mutating 10 residues, 20^10 = 10 trillion sequences
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular Simulation II. Classical Mechanical Treatment
 Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation II Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Principles of Physical Biochemistry
 Principles of Physical Biochemistry Kensal E. van Hold e W. Curtis Johnso n P. Shing Ho Preface x i PART 1 MACROMOLECULAR STRUCTURE AND DYNAMICS 1 1 Biological Macromolecules 2 1.1 General Principles
Principles of Physical Biochemistry Kensal E. van Hold e W. Curtis Johnso n P. Shing Ho Preface x i PART 1 MACROMOLECULAR STRUCTURE AND DYNAMICS 1 1 Biological Macromolecules 2 1.1 General Principles
Molecular Modelling. part of Bioinformatik von RNA- und Proteinstrukturen. Sonja Prohaska. Leipzig, SS Computational EvoDevo University Leipzig
 part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland
 Dr. Adrian Mulholland") Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Example questions for Molecular modelling (Level 4) Dr. Adrian Mulholland 1) Question. Two methods which are widely used for the optimization of molecular geometies are the Steepest descents and Newton-Raphson
Analysis of the simulation
 Analysis of the simulation Marcus Elstner and Tomáš Kubař January 7, 2014 Thermodynamic properties time averages of thermodynamic quantites correspond to ensemble averages (ergodic theorem) some quantities
Analysis of the simulation Marcus Elstner and Tomáš Kubař January 7, 2014 Thermodynamic properties time averages of thermodynamic quantites correspond to ensemble averages (ergodic theorem) some quantities
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations
 Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Density Functional Theory: from theory to Applications
 Density Functional Theory: from theory to Applications Uni Mainz January 17, 2011 CP-PAW/COSMO Interface Mixed quantum/ classical molecular dynamics (QM/MM) Quantum mechanics is computationally expensive
Density Functional Theory: from theory to Applications Uni Mainz January 17, 2011 CP-PAW/COSMO Interface Mixed quantum/ classical molecular dynamics (QM/MM) Quantum mechanics is computationally expensive
A Fast N-Body Solver for the Poisson(-Boltzmann) Equation
 Equation") A Fast N-Body Solver for the Poisson(-Boltzmann) Equation Robert D. Skeel Departments of Computer Science (and Mathematics) Purdue University http://bionum.cs.purdue.edu/2008december.pdf 1 Thesis Poisson(-Boltzmann)
A Fast N-Body Solver for the Poisson(-Boltzmann) Equation Robert D. Skeel Departments of Computer Science (and Mathematics) Purdue University http://bionum.cs.purdue.edu/2008december.pdf 1 Thesis Poisson(-Boltzmann)
Coarse-Grained Models!
 Coarse-Grained Models! Large and complex molecules (e.g. long polymers) can not be simulated on the all-atom level! Requires coarse-graining of the model! Coarse-grained models are usually also particles
Coarse-Grained Models! Large and complex molecules (e.g. long polymers) can not be simulated on the all-atom level! Requires coarse-graining of the model! Coarse-grained models are usually also particles
Neighbor Tables Long-Range Potentials
 Neighbor Tables Long-Range Potentials Today we learn how we can handle long range potentials. Neighbor tables Long-range potential Ewald sums MSE485/PHY466/CSE485 1 Periodic distances Minimum Image Convention:
Neighbor Tables Long-Range Potentials Today we learn how we can handle long range potentials. Neighbor tables Long-range potential Ewald sums MSE485/PHY466/CSE485 1 Periodic distances Minimum Image Convention:
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Using Molecular Dynamics to Compute Properties CHEM 430
 Using Molecular Dynamics to Compute Properties CHEM 43 Heat Capacity and Energy Fluctuations Running an MD Simulation Equilibration Phase Before data-collection and results can be analyzed the system
Using Molecular Dynamics to Compute Properties CHEM 43 Heat Capacity and Energy Fluctuations Running an MD Simulation Equilibration Phase Before data-collection and results can be analyzed the system
Atomistic Modeling of Small-Angle Scattering Data Using SASSIE-web
 Course Introduction Atomistic Modeling of Small-Angle Scattering Data Using SASSIE-web September 21-23, 2016 Advanced Photon Source Argonne National Laboratory, Argonne, IL ccpsas.org Scatters & Simulators
Course Introduction Atomistic Modeling of Small-Angle Scattering Data Using SASSIE-web September 21-23, 2016 Advanced Photon Source Argonne National Laboratory, Argonne, IL ccpsas.org Scatters & Simulators
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water?
 Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Ruhong Zhou 1 and Bruce J. Berne 2 1 IBM Thomas J. Watson Research Center; and 2 Department of Chemistry,
Simulations of bulk phases. Periodic boundaries. Cubic boxes
 Simulations of bulk phases ChE210D Today's lecture: considerations for setting up and running simulations of bulk, isotropic phases (e.g., liquids and gases) Periodic boundaries Cubic boxes In simulations
Simulations of bulk phases ChE210D Today's lecture: considerations for setting up and running simulations of bulk, isotropic phases (e.g., liquids and gases) Periodic boundaries Cubic boxes In simulations
Optimizing GROMACS for parallel performance
 Optimizing GROMACS for parallel performance Outline 1. Why optimize? Performance status quo 2. GROMACS as a black box. (PME) 3. How does GROMACS spend its time? (MPE) 4. What you can do What I want to
Optimizing GROMACS for parallel performance Outline 1. Why optimize? Performance status quo 2. GROMACS as a black box. (PME) 3. How does GROMACS spend its time? (MPE) 4. What you can do What I want to
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation
 Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro. Karel Berka, Ph.D.
 4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
4 th Advanced in silico Drug Design KFC/ADD Molecular Modelling Intro Karel Berka, Ph.D. UP Olomouc, 21.1.-25.1. 2019 Motto A theory is something nobody believes, except the person who made it An experiment
Scuola di Chimica Computazionale
 Societa Chimica Italiana Gruppo Interdivisionale di Chimica Computazionale Scuola di Chimica Computazionale Introduzione, per Esercizi, all Uso del Calcolatore in Chimica Organica e Biologica Modellistica
Societa Chimica Italiana Gruppo Interdivisionale di Chimica Computazionale Scuola di Chimica Computazionale Introduzione, per Esercizi, all Uso del Calcolatore in Chimica Organica e Biologica Modellistica
The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii
 And the Basic Force Field Chem 4021/8021 Video II.iii") The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii Fundamental Points About Which to Be Thinking It s clear the PES is useful, so how can I construct it for an arbitrary
The Potential Energy Surface (PES) And the Basic Force Field Chem 4021/8021 Video II.iii Fundamental Points About Which to Be Thinking It s clear the PES is useful, so how can I construct it for an arbitrary
Contents. xiii. Preface v
 Contents Preface Chapter 1 Biological Macromolecules 1.1 General PrincipIes 1.1.1 Macrornolecules 1.2 1.1.2 Configuration and Conformation Molecular lnteractions in Macromolecular Structures 1.2.1 Weak
Contents Preface Chapter 1 Biological Macromolecules 1.1 General PrincipIes 1.1.1 Macrornolecules 1.2 1.1.2 Configuration and Conformation Molecular lnteractions in Macromolecular Structures 1.2.1 Weak
Advanced Molecular Molecular Dynamics
 Advanced Molecular Molecular Dynamics Technical details May 12, 2014 Integration of harmonic oscillator r m period = 2 k k and the temperature T determine the sampling of x (here T is related with v 0
Advanced Molecular Molecular Dynamics Technical details May 12, 2014 Integration of harmonic oscillator r m period = 2 k k and the temperature T determine the sampling of x (here T is related with v 0
Biomolecular modeling II
 2015, December 16 System boundary and the solvent Biomolecule in solution typical MD simulations molecular system in aqueous solution task make the system as small as possible (reduce cost) straightforward
2015, December 16 System boundary and the solvent Biomolecule in solution typical MD simulations molecular system in aqueous solution task make the system as small as possible (reduce cost) straightforward
k θ (θ θ 0 ) 2 angles r i j r i j
 2 angles r i j r i j") 1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
Course Introduction. SASSIE CCP-SAS Workshop. January 23-25, ISIS Neutron and Muon Source Rutherford Appleton Laboratory UK
 Course Introduction SASSIE CCP-SAS Workshop January 23-25, 2017 ISIS Neutron and Muon Source Rutherford Appleton Laboratory UK ccpsas.org Scatters & Simulators Develop a community of users and developers
Course Introduction SASSIE CCP-SAS Workshop January 23-25, 2017 ISIS Neutron and Muon Source Rutherford Appleton Laboratory UK ccpsas.org Scatters & Simulators Develop a community of users and developers
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
SUPPLEMENTAL MATERIAL
 SUPPLEMENTAL MATERIAL Systematic Coarse-Grained Modeling of Complexation between Small Interfering RNA and Polycations Zonghui Wei 1 and Erik Luijten 1,2,3,4,a) 1 Graduate Program in Applied Physics, Northwestern
SUPPLEMENTAL MATERIAL Systematic Coarse-Grained Modeling of Complexation between Small Interfering RNA and Polycations Zonghui Wei 1 and Erik Luijten 1,2,3,4,a) 1 Graduate Program in Applied Physics, Northwestern
Introduction to Classical Molecular Dynamics. Giovanni Chillemi HPC department, CINECA
 Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
Introduction to Classical Molecular Dynamics Giovanni Chillemi g.chillemi@cineca.it HPC department, CINECA MD ingredients Coordinates Velocities Force field Topology MD Trajectories Input parameters Analysis
T6.2 Molecular Mechanics
 T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
T6.2 Molecular Mechanics We have seen that Benson group additivities are capable of giving heats of formation of molecules with accuracies comparable to those of the best ab initio procedures. However,
Biochemistry 530: Introduction to Structural Biology. Autumn Quarter 2014 BIOC 530
 Biochemistry 530: Introduction to Structural Biology Autumn Quarter 2014 Course Information Course Description Graduate-level discussion of the structure, function, and chemistry of proteins and nucleic
Biochemistry 530: Introduction to Structural Biology Autumn Quarter 2014 Course Information Course Description Graduate-level discussion of the structure, function, and chemistry of proteins and nucleic
Force Fields for Classical Molecular Dynamics simulations of Biomolecules. Emad Tajkhorshid
 Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Force Fields for Classical Molecular Dynamics simulations of Biomolecules Emad Tajkhorshid Beckman Institute Departments of Biochemistry Center for Biophysics and Computational Biology University of Illinois
Intermolecular Forces and Monte-Carlo Integration 열역학특수연구
 Intermolecular Forces and Monte-Carlo Integration 열역학특수연구 2003.3.28 Source of the lecture note. J.M.Prausnitz and others, Molecular Thermodynamics of Fluid Phase Equiliria Atkins, Physical Chemistry Lecture
Intermolecular Forces and Monte-Carlo Integration 열역학특수연구 2003.3.28 Source of the lecture note. J.M.Prausnitz and others, Molecular Thermodynamics of Fluid Phase Equiliria Atkins, Physical Chemistry Lecture
Coarse-Grained Peptide Modeling Using a Systematic Multiscale Approach
 Biophysical Journal Volume 92 June 2007 4289 4303 4289 Coarse-Grained Peptide Modeling Using a Systematic Multiscale Approach Jian Zhou, Ian F. Thorpe, Sergey Izvekov, and Gregory A. Voth Center for Biophysical
Biophysical Journal Volume 92 June 2007 4289 4303 4289 Coarse-Grained Peptide Modeling Using a Systematic Multiscale Approach Jian Zhou, Ian F. Thorpe, Sergey Izvekov, and Gregory A. Voth Center for Biophysical
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Protein Structures. 11/19/2002 Lecture 24 1
 Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D.
 3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
3rd Advanced in silico Drug Design KFC/ADD Molecular mechanics intro Karel Berka, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. Thierry Langer, Ph.D. Jana Vrbková, Ph.D. UP Olomouc, 23.1.-26.1. 2018
ATOMIC BONDING Atomic Bonding
 ATOMIC BONDING Atomic Bonding Primary Bonds Secondary Bonds Ionic Covalent Metallic van der Waals 1. IONIC BONDING q 11 Na & 17 Cl These two ions are attracted to eachother by the electrostatic force developed
ATOMIC BONDING Atomic Bonding Primary Bonds Secondary Bonds Ionic Covalent Metallic van der Waals 1. IONIC BONDING q 11 Na & 17 Cl These two ions are attracted to eachother by the electrostatic force developed
An Informal AMBER Small Molecule Force Field :
 An Informal AMBER Small Molecule Force Field : parm@frosst Credit Christopher Bayly (1992-2010) initiated, contributed and lead the efforts Daniel McKay (1997-2010) and Jean-François Truchon (2002-2010)
An Informal AMBER Small Molecule Force Field : parm@frosst Credit Christopher Bayly (1992-2010) initiated, contributed and lead the efforts Daniel McKay (1997-2010) and Jean-François Truchon (2002-2010)
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Proteins polymer molecules, folded in complex structures. Konstantin Popov Department of Biochemistry and Biophysics
 Proteins polymer molecules, folded in complex structures Konstantin Popov Department of Biochemistry and Biophysics Outline General aspects of polymer theory Size and persistent length of ideal linear
Proteins polymer molecules, folded in complex structures Konstantin Popov Department of Biochemistry and Biophysics Outline General aspects of polymer theory Size and persistent length of ideal linear
Modeling in Solution. Important Observations. Energy Concepts
 Modeling in Solution 1 Important Observations Various electrostatic effects for a solvated molecule are often less important than for an isolated gaseous molecule when the molecule is dissolved in a solvent
Modeling in Solution 1 Important Observations Various electrostatic effects for a solvated molecule are often less important than for an isolated gaseous molecule when the molecule is dissolved in a solvent
Michael W. Mahoney Department of Physics, Yale University, New Haven, Connecticut 06520
 JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 23 15 DECEMBER 2001 Quantum, intramolecular flexibility, and polarizability effects on the reproduction of the density anomaly of liquid water by simple potential
JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 23 15 DECEMBER 2001 Quantum, intramolecular flexibility, and polarizability effects on the reproduction of the density anomaly of liquid water by simple potential
 Title Theory of solutions in the energy r of the molecular flexibility Author(s) Matubayasi, N; Nakahara, M Citation JOURNAL OF CHEMICAL PHYSICS (2003), 9702 Issue Date 2003-11-08 URL http://hdl.handle.net/2433/50354
Title Theory of solutions in the energy r of the molecular flexibility Author(s) Matubayasi, N; Nakahara, M Citation JOURNAL OF CHEMICAL PHYSICS (2003), 9702 Issue Date 2003-11-08 URL http://hdl.handle.net/2433/50354
1. Hydrogen atom in a box
 1. Hydrogen atom in a box Recall H atom problem, V(r) = -1/r e r exact answer solved by expanding in Gaussian basis set, had to solve secular matrix involving matrix elements of basis functions place atom
1. Hydrogen atom in a box Recall H atom problem, V(r) = -1/r e r exact answer solved by expanding in Gaussian basis set, had to solve secular matrix involving matrix elements of basis functions place atom
Biomolecules are dynamic no single structure is a perfect model
 Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
MARTINI simulation details
 S1 Appendix MARTINI simulation details MARTINI simulation initialization and equilibration In this section, we describe the initialization of simulations from Main Text section Residue-based coarsegrained
S1 Appendix MARTINI simulation details MARTINI simulation initialization and equilibration In this section, we describe the initialization of simulations from Main Text section Residue-based coarsegrained
Force Fields for MD simulations
 Force Fields for MD simulations Topology/parameter files Where do the numbers an MD code uses come from? ow to make topology files for ligands, cofactors, special amino acids, ow to obtain/develop missing
Force Fields for MD simulations Topology/parameter files Where do the numbers an MD code uses come from? ow to make topology files for ligands, cofactors, special amino acids, ow to obtain/develop missing
Molecular Mechanics. C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology. January 2001
 Molecular Mechanics C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology January 2001 Introduction Molecular Mechanics uses classical type models to predict the energy
Molecular Mechanics C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology January 2001 Introduction Molecular Mechanics uses classical type models to predict the energy
Supporting Material for. Microscopic origin of gating current fluctuations in a potassium channel voltage sensor
 Supporting Material for Microscopic origin of gating current fluctuations in a potassium channel voltage sensor J. Alfredo Freites, * Eric V. Schow, * Stephen H. White, and Douglas J. Tobias * * Department
Supporting Material for Microscopic origin of gating current fluctuations in a potassium channel voltage sensor J. Alfredo Freites, * Eric V. Schow, * Stephen H. White, and Douglas J. Tobias * * Department
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
This semester. Books
 Models mostly proteins from detailed to more abstract models Some simulation methods This semester Books None necessary for my group and Prof Rarey Molecular Modelling: Principles and Applications Leach,
Models mostly proteins from detailed to more abstract models Some simulation methods This semester Books None necessary for my group and Prof Rarey Molecular Modelling: Principles and Applications Leach,
Chapter 11 Molecular Mechanics
 Chapter 11 Molecular Mechanics Molecular Mechanics uses an analytical, differentiable, and relatively simple potential energy function, (R), for describing the interactions between a set of atoms specified
Chapter 11 Molecular Mechanics Molecular Mechanics uses an analytical, differentiable, and relatively simple potential energy function, (R), for describing the interactions between a set of atoms specified
Tutorial on the smooth particle-mesh Ewald algorithm
 Tutorial on the smooth particle-mesh Ewald algorithm These notes explain the smooth particle-mesh Ewald algorithm (SPME) in detail. The accompanying library libpme6 is a fully-featured implementation of
Tutorial on the smooth particle-mesh Ewald algorithm These notes explain the smooth particle-mesh Ewald algorithm (SPME) in detail. The accompanying library libpme6 is a fully-featured implementation of
Molecular Modeling -- Lecture 15 Surfaces and electrostatics
 Molecular Modeling -- Lecture 15 Surfaces and electrostatics Molecular surfaces The Hydrophobic Effect Electrostatics Poisson-Boltzmann Equation Electrostatic maps Electrostatic surfaces in MOE 15.1 The
Molecular Modeling -- Lecture 15 Surfaces and electrostatics Molecular surfaces The Hydrophobic Effect Electrostatics Poisson-Boltzmann Equation Electrostatic maps Electrostatic surfaces in MOE 15.1 The
The Molecular Dynamics Simulation Process
 The Molecular Dynamics Simulation Process For textbooks see: M.P. Allen and D.J. Tildesley. Computer Simulation of Liquids.Oxford University Press, New York, 1987. D. Frenkel and B. Smit. Understanding
The Molecular Dynamics Simulation Process For textbooks see: M.P. Allen and D.J. Tildesley. Computer Simulation of Liquids.Oxford University Press, New York, 1987. D. Frenkel and B. Smit. Understanding
Computational Studies of the Photoreceptor Rhodopsin. Scott E. Feller Wabash College
 Computational Studies of the Photoreceptor Rhodopsin Scott E. Feller Wabash College Rhodopsin Photocycle Dark-adapted Rhodopsin hn Isomerize retinal Photorhodopsin ~200 fs Bathorhodopsin Meta-II ms timescale
Computational Studies of the Photoreceptor Rhodopsin Scott E. Feller Wabash College Rhodopsin Photocycle Dark-adapted Rhodopsin hn Isomerize retinal Photorhodopsin ~200 fs Bathorhodopsin Meta-II ms timescale
Molecular dynamics simulations of anti-aggregation effect of ibuprofen. Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov
 Biophysical Journal, Volume 98 Supporting Material Molecular dynamics simulations of anti-aggregation effect of ibuprofen Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov Supplemental
Biophysical Journal, Volume 98 Supporting Material Molecular dynamics simulations of anti-aggregation effect of ibuprofen Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov Supplemental
3. Solutions W = N!/(N A!N B!) (3.1) Using Stirling s approximation ln(n!) = NlnN N: ΔS mix = k (N A lnn + N B lnn N A lnn A N B lnn B ) (3.
 (3.1) Using Stirling s approximation ln(n!) = NlnN N: ΔS mix = k (N A lnn + N B lnn N A lnn A N B lnn B ) (3.") 3. Solutions Many biological processes occur between molecules in aqueous solution. In addition, many protein and nucleic acid molecules adopt three-dimensional structure ( fold ) in aqueous solution.
3. Solutions Many biological processes occur between molecules in aqueous solution. In addition, many protein and nucleic acid molecules adopt three-dimensional structure ( fold ) in aqueous solution.
Advanced Molecular Dynamics
 Advanced Molecular Dynamics Introduction May 2, 2017 Who am I? I am an associate professor at Theoretical Physics Topics I work on: Algorithms for (parallel) molecular simulations including GPU acceleration
Advanced Molecular Dynamics Introduction May 2, 2017 Who am I? I am an associate professor at Theoretical Physics Topics I work on: Algorithms for (parallel) molecular simulations including GPU acceleration
Solutions and Non-Covalent Binding Forces
 Chapter 3 Solutions and Non-Covalent Binding Forces 3.1 Solvent and solution properties Molecules stick together using the following forces: dipole-dipole, dipole-induced dipole, hydrogen bond, van der
Chapter 3 Solutions and Non-Covalent Binding Forces 3.1 Solvent and solution properties Molecules stick together using the following forces: dipole-dipole, dipole-induced dipole, hydrogen bond, van der
Reactive Empirical Force Fields
 Reactive Empirical Force Fields Jason Quenneville jasonq@lanl.gov X-1: Solid Mechanics, EOS and Materials Properties Applied Physics Division Los Alamos National Laboratory Timothy C. Germann, Los Alamos
Reactive Empirical Force Fields Jason Quenneville jasonq@lanl.gov X-1: Solid Mechanics, EOS and Materials Properties Applied Physics Division Los Alamos National Laboratory Timothy C. Germann, Los Alamos
Atomic and molecular interaction forces in biology
 Atomic and molecular interaction forces in biology 1 Outline Types of interactions relevant to biology Van der Waals interactions H-bond interactions Some properties of water Hydrophobic effect 2 Types
Atomic and molecular interaction forces in biology 1 Outline Types of interactions relevant to biology Van der Waals interactions H-bond interactions Some properties of water Hydrophobic effect 2 Types
Molecular Dynamics Simulation of a Nanoconfined Water Film
 Molecular Dynamics Simulation of a Nanoconfined Water Film Kyle Lindquist, Shu-Han Chao May 7, 2013 1 Introduction The behavior of water confined in nano-scale environment is of interest in many applications.
Molecular Dynamics Simulation of a Nanoconfined Water Film Kyle Lindquist, Shu-Han Chao May 7, 2013 1 Introduction The behavior of water confined in nano-scale environment is of interest in many applications.
Solutions to Assignment #4 Getting Started with HyperChem
 Solutions to Assignment #4 Getting Started with HyperChem 1. This first exercise is meant to familiarize you with the different methods for visualizing molecules available in HyperChem. (a) Create a molecule
Solutions to Assignment #4 Getting Started with HyperChem 1. This first exercise is meant to familiarize you with the different methods for visualizing molecules available in HyperChem. (a) Create a molecule
An FPGA Implementation of Reciprocal Sums for SPME
 An FPGA Implementation of Reciprocal Sums for SPME Sam Lee and Paul Chow Edward S. Rogers Sr. Department of Electrical and Computer Engineering University of Toronto Objectives Accelerate part of Molecular
An FPGA Implementation of Reciprocal Sums for SPME Sam Lee and Paul Chow Edward S. Rogers Sr. Department of Electrical and Computer Engineering University of Toronto Objectives Accelerate part of Molecular
Crystal Structure Prediction using CRYSTALG program
 Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Coupling the Level-Set Method with Variational Implicit Solvent Modeling of Molecular Solvation
 Coupling the Level-Set Method with Variational Implicit Solvent Modeling of Molecular Solvation Bo Li Math Dept & CTBP, UCSD Li-Tien Cheng (Math, UCSD) Zhongming Wang (Math & Biochem, UCSD) Yang Xie (MAE,
Coupling the Level-Set Method with Variational Implicit Solvent Modeling of Molecular Solvation Bo Li Math Dept & CTBP, UCSD Li-Tien Cheng (Math, UCSD) Zhongming Wang (Math & Biochem, UCSD) Yang Xie (MAE,
Chem 1140; Molecular Modeling
 P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
The liquid-vapour interface of QDO water. Flaviu Cipcigan Andrew Jones Jason Crain Vlad Sokhan Glenn Martyna
 The liquid-vapour interface of QDO water Flaviu Cipcigan Andrew Jones Jason Crain Vlad Sokhan Glenn Martyna The liquid-vapour interface of QDO water 1. Molecular models 2. The Quantum Drude Oscillator
The liquid-vapour interface of QDO water Flaviu Cipcigan Andrew Jones Jason Crain Vlad Sokhan Glenn Martyna The liquid-vapour interface of QDO water 1. Molecular models 2. The Quantum Drude Oscillator