Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
|
|
|
- Sheena Carroll
- 6 years ago
- Views:
Transcription
1 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007
2 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug) Enzyme-substrate Protein-DNA (or RNA) Protein-protein
3 Brief History of Docking Crick (1953) suggested that complementarity in helical coils could be modelled as knobs fitting into holes DOCK (Kuntz, 1982) pioneered the field of molecular docking GRID (Goodford, 1985) too became a part of many subsequent softwares
4 General considerations Molecular representations Abstract or atoms Fixed or flexible Juxtaposition of molecules Interactive or automated Search algorithm to create conformations Evaluating complementarity (ranking) Scoring function Force field energy functions
5 Search Algorithms Potentially several ways of putting two molecules together; possibilities increase exponentially with size of molecules involved Attempt to locate the most stable state in the energy landscape Broadly two types: 1) full solution space search; 2) guided search through solution space
6 Search Algorithms Random Genetic algorithms Monte Carlo methods Tabu search Systematic Fragment-based methods Point complementary methods Distance geometry methods Database Simulation Molecular dynamics Energy minimisation Multiple methods Algorithms
7 Docking Softwares Virtual screening AutoDock DOCK FlexX/E SLIDE Surflex ICM GOLD De novo design LUDI GRID MCSS SMoG GrowMol SPROUT
8 Random methods Sample the conformation space by making single change to a ligand or a population of ligands Alteration performed at each step and accepted or rejected based on a predetermined probability function Include Monte Carlo (MC) methods; Genetic Algorithm (GA) methods; Tabu search methods
9 Monte Carlo methods Use a simple energy function Makes random moves and accepting or rejecting based on Boltzmann probability function More efficient in stepping over energy barriers, allowing more complete searches of conformation space PRODOCK, MC-DOCK, ICM, DockVision, QXP, GLIDE; too slow for extensive flexible docking
10 Energy global minimum conformers generated by Monte Carlo method
11 Genetic Algorithm methods Apply ideas of genetics and evolution in docking Start with an initial population of random ligand conformers wrt protein, each defined by a set of variables called genes Genetic operators (mutations, crossovers) applied to sample conformation space till optimal population is derived AUTODOCK, GOLD, DIVALI, DARWIN; too slow for extensive flexible docking
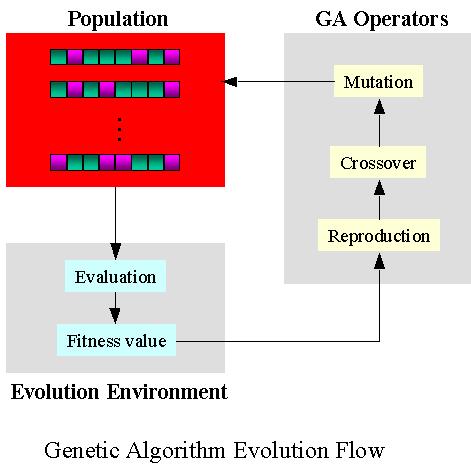
12
13 Autodock Suite of automated docking tools Designed to predict how small molecules (ligands drug candidates) bind to a receptor; AMBER force field Three constituent programs -Autotors- define torsions in the ligand -Autogrid- calculate grids -Autodock- docking tool -AutoDockTools (ADT)- GUI to facilitate above and other modules accompanying AutoDock
14 Autodock Lamarckian GA LGA encompasses a genotypic and phenotypic phase i.e. genetic operations and energy function to be optimised Energy minimisation performed after genotypic changes and these phenotypic changes mapped back onto genes (by changing ligand coordinates. Most efficient and reliable of random methods
 Consists of 3D lattice of regularly spaced points, surrounding and centered on region of interest in the macromolecule Typical")
15 Autodock Grid maps Pre-calculated Grid for each atom type (e.g. C, H, O, N) Consists of 3D lattice of regularly spaced points, surrounding and centered on region of interest in the macromolecule Typical spacing is Å Probe atom placed at each grid point and energy calculated
16 GOLD Genetic Optimisation and Ligand Docking, uses multiple subpopulations of ligand Force-field based scoring function, includes three terms: H-bonding term, intermolecular dispersion potential, intramolecular potential 71% success in identifying experimental binding mode in 100 protein complexes
17 Tabu Search methods Impose restrictions preventing searches from repeating already explored conformations New conformation is compared to the previous ones based on RMSD values which determine acceptance PRO-LEADS
18 Systematic Search methods Attempt to explore all degrees of freedom in a molecule Can be divided into three types: conformational search methods, fragmentation methods, and database methods
19 Conformational Search methods Brute force or shotgun methods of docking All rotatable bonds in ligand rotated through 360 till in fixed increments till all possible combinations generated and evaluated Number of structures generated increases exponentially with number of rotatable bondscombinatorial explosion
20 Fragmentation Search methods Incrementally grow ligand into the active site, by docking several fragments into the active site followed by covalent-linking to recreate the initial ligand Rigid core-fragment of the ligand is docked first followed by addition of flexible regions DOCK, FlexX, LUDI, ADAM, Hammerhead
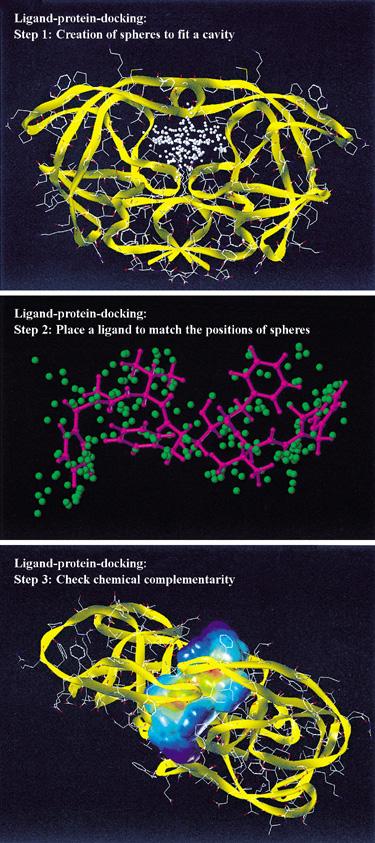
21 DOCK Methodology
22 FlexX Base fragment is picked up and docked using pose-clustering algorithm Clustering algorithm is implemented to merge similar ligand transformations into active site Flexible fragments are added incrementally using MIMUMBA and evaluated using overlap function, followed by energy calculations till the ligand is completely built Final evaluation through Böhm s scoring function that includes H-bonds, ionic, aromatic and lipophilic terms
23 Database methods Tackle combinatorial explosion by using libraries of pregenerated conformations to deal with ligand flexibility FLOG generates and docks conformational libraries called Flexibases using distance geometry EUDOC uses conformational searches of ligands to generate different structures, which are placed into receptor active-site followed by energy evaluation
24 Scoring Essential to rank the ligand conformations determined by the search algorithms Scoring function must be able to distinguish between true binding modes and others Speed and accuracy are most desirable Three major classes: force-field based; empirical; knowledge-based
25 GoldScore, G-SCORE, D-SCORE, AMBER, CHARRM, GROMOS Force-field based Scoring Quantify sum of two energies-interaction energy between receptor-ligand; internal energy of the ligand Consist of van der Waals (Lennard-Jones potential) + electrostatic energy terms (Coulombic function) Do not include solvation and entropic terms
26 Empirical Scoring Designed to reproduce experimental data; binding energy can be approximated by sum of individual uncorrelated terms Experimentally determined binding energies used to quantify individual terms Easy computation, but non-versatile due to dependence on experimental datasets ChemScore, Böhm s scoring function, F-Score, X-Score
27 Knowledge-based Scoring Statistically derived principles that aim to replicate experimentally determined structures Employ simple interactions to screen large databases Dependent on information available in preexisting datasets DrugScore, SMoG score, Potential of Mean force (PMF)
28 Consensus Scoring Combines information from different scoring schemes to compensate for individual limitations Correlation of individual scoring systems may be a problem X-SCORE combines functions from PMF, ChemScore, PMF with FlexX
29 Protein-protein Docking Prediction of protein complex structure given individual components structures Huge number of degrees of freedom; docking largely performed as rigid body docking Z-DOCK, a Fast Fourier Transform-based rigid body docking program, is one of the most accurate programs as rated in Critical Assessment of Predicted Interactions (CAPRI)
30 Docking- strengths and limitations Most available softwares are able to predict known protein-bound conformations with an accuracy of Å; 70-80% success rate Scoring function- major limitation factor due to simplifications and assumptions Solvation effects, quality of crystallographic data
31 Comparing Docking softwares in difficult Several studies compare docking programs but conclusions of general applicability are not evident Minor differences in methodology can have significant impact on success rates of various docking programs Cole et al., 2005 PROTEINS 60, provide a list of recommendations in assessing docking programs

32 Docking softwares representations in citations

33 Docking Softwares- Citations per year
34
35 Challenges Predicting structures of multi-domain, multisubunit protein complexes Prediction and specificity in protein-nucleic acid interactions Protein-docking with backbone flexibility
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
What is Protein-Ligand Docking?
 MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
MOLECULAR RECOGNITION DOCKING ALGORITHMS. Natasja Brooijmans 1 and Irwin D. Kuntz 2
 Annu. Rev. Biophys. Biomol. Struct. 2003. 32:335 73 doi: 10.1146/annurev.biophys.32.110601.142532 Copyright c 2003 by Annual Reviews. All rights reserved First published online as a Review in Advance on
Annu. Rev. Biophys. Biomol. Struct. 2003. 32:335 73 doi: 10.1146/annurev.biophys.32.110601.142532 Copyright c 2003 by Annual Reviews. All rights reserved First published online as a Review in Advance on
Protein-Ligand Docking Evaluations
 Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Protein-Ligand Docking Methods
 Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Protein-Ligand Docking
 Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Using AutoDock 4 with ADT: A Tutorial
 Using AutoDock 4 with ADT: A Tutorial Ruth Huey Sargis Dallakyan Alex Perryman David S. Goodsell (Garrett Morris) 9/2/08 Using AutoDock 4 with ADT 1 What is Docking? Predicting the best ways two molecules
Using AutoDock 4 with ADT: A Tutorial Ruth Huey Sargis Dallakyan Alex Perryman David S. Goodsell (Garrett Morris) 9/2/08 Using AutoDock 4 with ADT 1 What is Docking? Predicting the best ways two molecules
Protein-Ligand Docking Methods
 Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Protein Structure Prediction and Protein-Ligand Docking
 Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
BCB410 Protein-Ligand Docking Exercise Set Shirin Shahsavand December 11, 2011
 BCB410 Protein-Ligand Docking Exercise Set Shirin Shahsavand December 11, 2011 1. Describe the search algorithm(s) AutoDock uses for solving protein-ligand docking problems. AutoDock uses 3 different approaches
BCB410 Protein-Ligand Docking Exercise Set Shirin Shahsavand December 11, 2011 1. Describe the search algorithm(s) AutoDock uses for solving protein-ligand docking problems. AutoDock uses 3 different approaches
Cheminformatics platform for drug discovery application
 EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
High Throughput In-Silico Screening Against Flexible Protein Receptors
 John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Using AutoDock With AutoDockTools: A Tutorial
 Using AutoDock With AutoDockTools: A Tutorial Dr. Ruth Huey & Dr. Garrett M. Morris 6/6/06 AutoDock & ADT Tutorial 1 What is Docking? Best ways to put two molecules together. Three steps: (1) Obtain the
Using AutoDock With AutoDockTools: A Tutorial Dr. Ruth Huey & Dr. Garrett M. Morris 6/6/06 AutoDock & ADT Tutorial 1 What is Docking? Best ways to put two molecules together. Three steps: (1) Obtain the
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Ranking of HIV-protease inhibitors using AutoDock
 Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Assessing Scoring Functions for Protein-Ligand Interactions
 3032 J. Med. Chem. 2004, 47, 3032-3047 Assessing Scoring Functions for Protein-Ligand Interactions Philippe Ferrara,, Holger Gohlke,, Daniel J. Price, Gerhard Klebe, and Charles L. Brooks III*, Department
3032 J. Med. Chem. 2004, 47, 3032-3047 Assessing Scoring Functions for Protein-Ligand Interactions Philippe Ferrara,, Holger Gohlke,, Daniel J. Price, Gerhard Klebe, and Charles L. Brooks III*, Department
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
BUDE. A General Purpose Molecular Docking Program Using OpenCL. Richard B Sessions
 BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
Computational modeling of G-Protein Coupled Receptors (GPCRs) has recently become
 has recently become") Homology Modeling and Docking of Melatonin Receptors Andrew Kohlway, UMBC Jeffry D. Madura, Duquesne University 6/18/04 INTRODUCTION Computational modeling of G-Protein Coupled Receptors (GPCRs) has recently
Homology Modeling and Docking of Melatonin Receptors Andrew Kohlway, UMBC Jeffry D. Madura, Duquesne University 6/18/04 INTRODUCTION Computational modeling of G-Protein Coupled Receptors (GPCRs) has recently
Small Molecule & Protein Docking CHEM 430
 Small Molecule & Protein Docking CHEM 430 Molecular Docking Models Over the years biochemists have developed numerous models to capture the key elements of the molecular recognition process. Although very
Small Molecule & Protein Docking CHEM 430 Molecular Docking Models Over the years biochemists have developed numerous models to capture the key elements of the molecular recognition process. Although very
Pedro Alexandrino Fernandes, Dep. Chemistry & Biochemistry, University of Porto, Portugal
 Pedro Alexandrino Fernandes, Dep. Chemistry & Biochemistry, University of Porto, Portugal pedro.fernandes@fc.up.pt 1. Introduc3on Intermolecular Associa3ons 1. Introduc3on What type of forces govern these
Pedro Alexandrino Fernandes, Dep. Chemistry & Biochemistry, University of Porto, Portugal pedro.fernandes@fc.up.pt 1. Introduc3on Intermolecular Associa3ons 1. Introduc3on What type of forces govern these
From Small Molecules to Biological Molecules: Modelling Interactions. Dr. Antonio Chana Milano, 22 nd April 2008
 From Small Molecules to Biological Molecules: Modelling Interactions Dr. Antonio Chana Milano, 22 nd April 2008 Computational Methods & Applicability 2 *Diagram taken from S. C. Glotzer The University
From Small Molecules to Biological Molecules: Modelling Interactions Dr. Antonio Chana Milano, 22 nd April 2008 Computational Methods & Applicability 2 *Diagram taken from S. C. Glotzer The University
PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS
 TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
New techniques of molecular modelling and structural chemistry for the development of bioactive compounds
 UNIVERSITY OF SALERNO DEPARTMENT OF PHARMACY PhD course in Scienza e Tecnologie per l Industria Chimica, Farmaceutica e Alimentare XI course NS (XXV) 2009-2012 New techniques of molecular modelling and
UNIVERSITY OF SALERNO DEPARTMENT OF PHARMACY PhD course in Scienza e Tecnologie per l Industria Chimica, Farmaceutica e Alimentare XI course NS (XXV) 2009-2012 New techniques of molecular modelling and
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Introduction to AutoDock and AutoDock Tools
 Introduction to AutoDock and AutoDock Tools Alexander B. Pacheco User Services Consultant LSU HPC & LONI sys-help@loni.org HPC Training Series Louisiana State University Baton Rouge Mar. 28, 2012 HPC@LSU
Introduction to AutoDock and AutoDock Tools Alexander B. Pacheco User Services Consultant LSU HPC & LONI sys-help@loni.org HPC Training Series Louisiana State University Baton Rouge Mar. 28, 2012 HPC@LSU
Using AutoDock 4 with ADT: A Tutorial
 Using AutoDock 4 with ADT: A Tutorial Dr. Garrett M. Morris 1 What is Docking? A computational procedure for predicting the best way(s) two molecules will interact in 3D. (1) Obtain the 3D structures of
Using AutoDock 4 with ADT: A Tutorial Dr. Garrett M. Morris 1 What is Docking? A computational procedure for predicting the best way(s) two molecules will interact in 3D. (1) Obtain the 3D structures of
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Portal. User Guide Version 1.0. Contributors
 Portal www.dockthor.lncc.br User Guide Version 1.0 Contributors Diogo A. Marinho, Isabella A. Guedes, Eduardo Krempser, Camila S. de Magalhães, Hélio J. C. Barbosa and Laurent E. Dardenne www.gmmsb.lncc.br
Portal www.dockthor.lncc.br User Guide Version 1.0 Contributors Diogo A. Marinho, Isabella A. Guedes, Eduardo Krempser, Camila S. de Magalhães, Hélio J. C. Barbosa and Laurent E. Dardenne www.gmmsb.lncc.br
Principles of Drug Design
 Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Using Molecular Dynamics and Enhanced Sampling to Predict Binding Poses Beyond The Rigid Docking Approximation
 Using Molecular Dynamics and Enhanced Sampling to Predict Binding Poses Beyond The Rigid Docking Approximation Emil Åberg Supervisor: Pär Söderhjelm Center for Molecular Protein Science, CMPS Faculty of
Using Molecular Dynamics and Enhanced Sampling to Predict Binding Poses Beyond The Rigid Docking Approximation Emil Åberg Supervisor: Pär Söderhjelm Center for Molecular Protein Science, CMPS Faculty of
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation
 Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
Crystal Structure Prediction using CRYSTALG program
 Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation
 OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation Gareth Price Computational MiniProject OpenDiscovery Aims + Achievements Produce a high-throughput protocol to screen a
OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation Gareth Price Computational MiniProject OpenDiscovery Aims + Achievements Produce a high-throughput protocol to screen a
Cross Discipline Analysis made possible with Data Pipelining. J.R. Tozer SciTegic
 Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
A genetic algorithm for the ligand-protein docking problem
 Research Article Genetics and Molecular Biology, 27, 4, 605-610 (2004) Copyright by the Brazilian Society of Genetics. Printed in Brazil www.sbg.org.br A genetic algorithm for the ligand-protein docking
Research Article Genetics and Molecular Biology, 27, 4, 605-610 (2004) Copyright by the Brazilian Society of Genetics. Printed in Brazil www.sbg.org.br A genetic algorithm for the ligand-protein docking
Challenges, Applications, and Recent Advances of Protein-Ligand Docking in Structure-Based Drug Design
 Molecules 2014, 19, 10150-10176; doi:10.3390/molecules190710150 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Challenges, Applications, and Recent Advances of Protein-Ligand
Molecules 2014, 19, 10150-10176; doi:10.3390/molecules190710150 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Challenges, Applications, and Recent Advances of Protein-Ligand
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Towards Physics-based Models for ADME/Tox. Tyler Day
 Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors
 MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Computational protein design
 Computational protein design There are astronomically large number of amino acid sequences that needs to be considered for a protein of moderate size e.g. if mutating 10 residues, 20^10 = 10 trillion sequences
Computational protein design There are astronomically large number of amino acid sequences that needs to be considered for a protein of moderate size e.g. if mutating 10 residues, 20^10 = 10 trillion sequences
Computational Analyses of Protein-Ligand Interactions
 Computational Analyses of Protein-Ligand Interactions Muhammad Kamran Haider Submitted for the Degree of Doctor of Philosophy University of York Department of Chemistry September 2010 Abstract Protein-ligand
Computational Analyses of Protein-Ligand Interactions Muhammad Kamran Haider Submitted for the Degree of Doctor of Philosophy University of York Department of Chemistry September 2010 Abstract Protein-ligand
BioSolveIT. A Combinatorial Approach for Handling of Protonation and Tautomer Ambiguities in Docking Experiments
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
Author Index Volume
 Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Studying complex macromolecules
 Studying complex macromolecules M2 SERP-Chem Fabien Cailliez LCP Bât 349 fabien.cailliez@u-psud.fr Outline I. Classical forcefields II. III. IV. Molecular simulations and biomolecules One typical example:
Studying complex macromolecules M2 SERP-Chem Fabien Cailliez LCP Bât 349 fabien.cailliez@u-psud.fr Outline I. Classical forcefields II. III. IV. Molecular simulations and biomolecules One typical example:
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
Computational Identification of Inhibitors of Protein-Protein Interactions
 Current Topics in Medicinal Chemistry, 2007, 7, 63-82 63 Computational Identification of Inhibitors of Protein-Protein Interactions Shun Zhong, Alba T. Macias and Alexander D. MacKerell Jr.* Department
Current Topics in Medicinal Chemistry, 2007, 7, 63-82 63 Computational Identification of Inhibitors of Protein-Protein Interactions Shun Zhong, Alba T. Macias and Alexander D. MacKerell Jr.* Department
Molecular Dynamics. A very brief introduction
 Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Molecular Dynamics A very brief introduction Sander Pronk Dept. of Theoretical Physics KTH Royal Institute of Technology & Science For Life Laboratory Stockholm, Sweden Why computer simulations? Two primary
Presenter: She Zhang
 Presenter: She Zhang Introduction Dr. David Baker Introduction Why design proteins de novo? It is not clear how non-covalent interactions favor one specific native structure over many other non-native
Presenter: She Zhang Introduction Dr. David Baker Introduction Why design proteins de novo? It is not clear how non-covalent interactions favor one specific native structure over many other non-native
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
Molecular Simulation III
 Molecular Simulation III Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Molecular Dynamics Jeffry D. Madura Department of Chemistry & Biochemistry Center
Molecular Simulation III Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Molecular Dynamics Jeffry D. Madura Department of Chemistry & Biochemistry Center
Structure Investigation of Fam20C, a Golgi Casein Kinase
 Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015,
 Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Distance Constraint Model; Donald J. Jacobs, University of North Carolina at Charlotte Page 1 of 11
 Distance Constraint Model; Donald J. Jacobs, University of North Carolina at Charlotte Page 1 of 11 Taking the advice of Lord Kelvin, the Father of Thermodynamics, I describe the protein molecule and other
Distance Constraint Model; Donald J. Jacobs, University of North Carolina at Charlotte Page 1 of 11 Taking the advice of Lord Kelvin, the Father of Thermodynamics, I describe the protein molecule and other
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Investigation of physiochemical interactions in
 Investigation of physiochemical interactions in Bulk and interfacial water Aqueous salt solutions (new endeavor) Polypeptides exhibiting a helix-coil transition Aqueous globular proteins Protein-solvent
Investigation of physiochemical interactions in Bulk and interfacial water Aqueous salt solutions (new endeavor) Polypeptides exhibiting a helix-coil transition Aqueous globular proteins Protein-solvent
Chimica Farmaceutica
 Chimica Farmaceutica Drug Targets Why should chemicals, some of which have remarkably simple structures, have such an important effect «in such a complicated and large structure as a human being? The answer
Chimica Farmaceutica Drug Targets Why should chemicals, some of which have remarkably simple structures, have such an important effect «in such a complicated and large structure as a human being? The answer
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Advanced in silico Drug Design KFC/ADD
 Advanced in silico Drug Design S pozdravem KFC/ADD Molecular Karel Berka Docking Intro Karel Berka, Ph.D. Jindřich Fanfrlík, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. UP Olomouc, 30.1.-1.2. 2017
Advanced in silico Drug Design S pozdravem KFC/ADD Molecular Karel Berka Docking Intro Karel Berka, Ph.D. Jindřich Fanfrlík, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. UP Olomouc, 30.1.-1.2. 2017
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Pose and affinity prediction by ICM in D3R GC3. Max Totrov Molsoft
 Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
FDS: Flexible Ligand and Receptor Docking with a Continuum Solvent Model and Soft-Core Energy Function
 FDS: Flexible Ligand and Receptor Docking with a Continuum Solvent Model and Soft-Core Energy Function RICHARD D. TAYLOR, 1, * PHILIP J. JEWSBURY, 2 JONATHAN W. ESSEX 1 1 Department of Chemistry, University
FDS: Flexible Ligand and Receptor Docking with a Continuum Solvent Model and Soft-Core Energy Function RICHARD D. TAYLOR, 1, * PHILIP J. JEWSBURY, 2 JONATHAN W. ESSEX 1 1 Department of Chemistry, University
Introduction to" Protein Structure
 Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Scoring functions. Talk Overview. Eran Eyal. Scoring functions what and why
 Scoring unctions Talk Overview Scoring unctions what and why Force ields based on approximation o molecular orces as we understand them Knowledge-based potentials let the data speak May 2011 Eran Eyal
Scoring unctions Talk Overview Scoring unctions what and why Force ields based on approximation o molecular orces as we understand them Knowledge-based potentials let the data speak May 2011 Eran Eyal
BME Engineering Molecular Cell Biology. Structure and Dynamics of Cellular Molecules. Basics of Cell Biology Literature Reading
 BME 42-620 Engineering Molecular Cell Biology Lecture 05: Structure and Dynamics of Cellular Molecules Basics of Cell Biology Literature Reading BME42-620 Lecture 05, September 13, 2011 1 Outline Review:
BME 42-620 Engineering Molecular Cell Biology Lecture 05: Structure and Dynamics of Cellular Molecules Basics of Cell Biology Literature Reading BME42-620 Lecture 05, September 13, 2011 1 Outline Review:
Automatic Epitope Recognition in Proteins Oriented to the System for Macromolecular Interaction Assessment MIAX
 Genome Informatics 12: 113 122 (2001) 113 Automatic Epitope Recognition in Proteins Oriented to the System for Macromolecular Interaction Assessment MIAX Atsushi Yoshimori Carlos A. Del Carpio yosimori@translell.eco.tut.ac.jp
Genome Informatics 12: 113 122 (2001) 113 Automatic Epitope Recognition in Proteins Oriented to the System for Macromolecular Interaction Assessment MIAX Atsushi Yoshimori Carlos A. Del Carpio yosimori@translell.eco.tut.ac.jp
SCREENED CHARGE ELECTROSTATIC MODEL IN PROTEIN-PROTEIN DOCKING SIMULATIONS
 SCREENED CHARGE ELECTROSTATIC MODEL IN PROTEIN-PROTEIN DOCKING SIMULATIONS JUAN FERNANDEZ-RECIO 1, MAXIM TOTROV 2, RUBEN ABAGYAN 1 1 Department of Molecular Biology, The Scripps Research Institute, 10550
SCREENED CHARGE ELECTROSTATIC MODEL IN PROTEIN-PROTEIN DOCKING SIMULATIONS JUAN FERNANDEZ-RECIO 1, MAXIM TOTROV 2, RUBEN ABAGYAN 1 1 Department of Molecular Biology, The Scripps Research Institute, 10550
NIH Public Access Author Manuscript J Comput Chem. Author manuscript; available in PMC 2011 February 18.
 NIH Public Access Author Manuscript Published in final edited form as: J Comput Chem. 2010 January 30; 31(2): 455 461. doi:10.1002/jcc.21334. AutoDock Vina: improving the speed and accuracy of docking
NIH Public Access Author Manuscript Published in final edited form as: J Comput Chem. 2010 January 30; 31(2): 455 461. doi:10.1002/jcc.21334. AutoDock Vina: improving the speed and accuracy of docking
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
SAM Teacher s Guide Protein Partnering and Function
 SAM Teacher s Guide Protein Partnering and Function Overview Students explore protein molecules physical and chemical characteristics and learn that these unique characteristics enable other molecules
SAM Teacher s Guide Protein Partnering and Function Overview Students explore protein molecules physical and chemical characteristics and learn that these unique characteristics enable other molecules
Advanced in silico drug design
 Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today
 est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
Docking with Water in the Binding Site using GOLD
 Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy