What is Protein-Ligand Docking?
|
|
|
- Jemima Shana Berry
- 5 years ago
- Views:
Transcription

1 MOLECULAR DOCKING
2 Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes the interaction reveals the most accurate structure of the complex. Importance of complexes - structure -> function 2
3 What is Docking? Given two molecules find their correct association: T + = 3

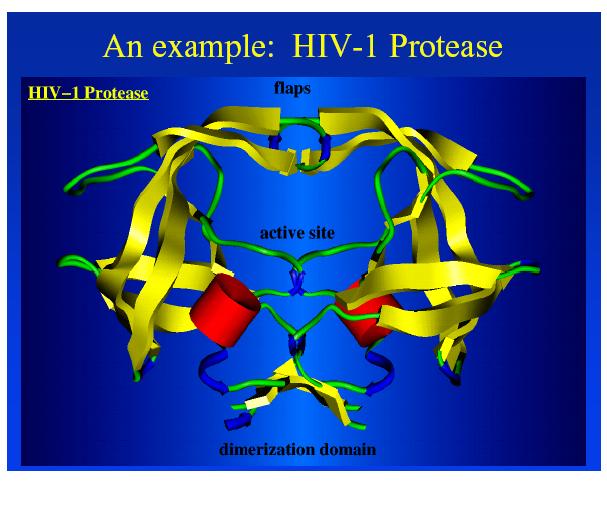
4 3-D Representation of a Protein Binding Site Distances between binding groups in Angstroms and the type of interaction 4 is searchable


5 General Protein Ligand Binding Ligand - Molecule that binds with a protein - DNA, drug lead compounds, etc. Protein active site(s) - Allosteric binding - Competitive binding Function of binding interaction - Natural and artificial 5
6 Issues Involved in Docking Protein Structure and Active Site - Assumed knowledge (PDBs, comparative modeling etc.) - PROCAT database: 3d enzyme active site templates Ligand Structure - Pharmacophore (base fragment) in potential drug compound - well known groups Rigid vs. Flexible - In solution or in vacum - Structure fixed, partly fixed, modeling of flexibility 6
7 Algorithmic Approaches to Docking Qualitative Geometric Shape complementarity and fitting Quantitative Energy calculations Determine global minimum energy Free energy measure Hybrid Geometric and energy complementarity 2 phase process: soft and hard docking 7
8
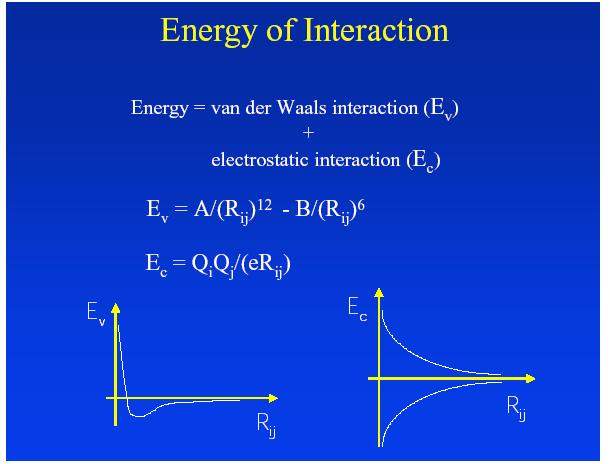
9 It involves: Docking uses a search and score method Finding useful ways of representing the molecules and molecular properties. Exploration of the configuration spaces available for interaction between ligand and receptor. Evaluate and rank configurations using a scoring system, in this case the binding energy However, since it is difficult to evaluate the binding energy because the binding sites may not be easily accessible, the binding energy is modeled as follows: G bind= Gvdw + Ghbond + Gelect + G conform+ G tor + G sol


10 Docking Strategy PDB files Surface Representation Patch Detection Matching Patches Scoring & Filtering Candidate complexes 10
11
12 Adding angles in Cartesian space x = r cos y = r sin converting internal motion to Cartesian motion y (x,y ) (x,y) x' = r cos ( = r (cos cos sin sin = ( r cos cos r sin sin = x cos y sin r x y' = r sin ( = r (sin cos sin cos = ( r sin cos r cos sin = y cos x sin in matrix notation... x' y' cos sin sin x cos y rotation matrix
13 A 3D rotation matrix Is the product of 2D rotation matrices. cos sin 0 cos 0 sin cos cos sin cos sin cos sin cos cos sin sin sin 0 cos sin 0 cos
14 Kinds of Kinds of search:systematic : search Anchor and grow (or) incremental construction algorithm It tries to explore all the degrees of freedom in a molecule, but ultimately face the problem of combinatorial explosion So ligands are often incrementally grown into active sites Exhaustive Deterministic Dependent on granularity of sampling Feasible only for low-dimensional problems DOF, 6D search DOCK (incremental) FlexX (incremental) Glide (incremental)
15 Kinds of search:stochastic Kinds of:::: search Monte Carlo (MC) methods and Evolutionary algorithm It works by making random changes to either a single ligand or a population of ligands Novel ligand is evaluated by pre defined probability function In Tabu search, to accept the novel molecule, it calculates RMSD between current moecular coordinates and every molecule s previously recorded conformation Random Outcome varies Repeat to improve chances of success Feasible for higher-dimensional problems Simulated Annealing (SA) AutoDock (MC/SA,GA/LGA) GOLD (GA) Evolutionary Algorithms (EA) Genetic Algorithm (GA)/Tabu Search (TS) Hybrid Global-Local Search/Lamarckian GA (LGA)
16 Kinds of Search:DETERMINISTIC Energy Minimization methods and Molecular Dynamics simulations Molecular Dynamics simulations are often unable to cross highenergy barriers within feasible simulation time periods, they might accommodate ligands in local minima of the energy surface so, an attempt is made to simulate different parts of a proteinligand system at different temperatures Energy minimization is rarely used as stand alone search techniques, as only local energy minima can be reached DOCK Glide AutoDock
17 Random/stochastic AutoDock (MC) MOE-Dock (MC,TS) GOLD (GA) PRO_LEADS (TS) Systematic DOCK (incremental) FlexX (incremental) Glide (incremental) Hammerhead (incremental) Simulation/Deterministic DOCK Glide MOE-Dock AutoDock Hammerhead
18 SCORING FUNCTIONS:FORCE FIELD BASED SCORING Quantifying the sum of two energies Receptor ligand interaction energy and internal ligand energy Most scoring functions consider a single protein conformation to omit the internal protein energy, which simplifies the scoring Force field scoring functions varies based on different force field parameter sets For E.g.: G Score Tripos force field AutoDock AMBER force field
19 SCORING FUNCTIONS:EMPIRICAL SCORING Based on binding energies and/or conformations It is designed based on idea that binding energies can be approximated by a sum of individual uncorrelated terms The coefficients are obtained from regression analysis using experimentally determined binding energies and X ray structural information Disadvantage it depends on the molecular data sets used to perform regression analysis
20 KNOWLEDGE BASED SCORING It reproduce experimental structures rather than binding energies Protein ligand complex is modelled using relatively simple atomic interaction pair potentials A number of atom type interactions are defined depending on their molecular environment The main attraction is computational simplicity which permits efficient screening of large compound databases Disadvantage derivation is essentially based on information implicitly encoded in limited sets of protein ligand complex
21
22

23 Scoring in Ligand-Protein Docking Potential Energy Description: 23

24

25
26 Type of Scoring Functions FORCE FIELD BASED D-Score G-Score Gold Score Autodock Dock EMPIRICAL Ludi F-Score Chem Score KNOWLEDGE BASED PMF Drug Score CONSENSUS CSCORE X-Score
27 AutoDock
28 Cerius2/LigScore vdw 6-9 C+pol buried polar surface in attractive protein ligand complex Totpol2 square of buried polar surface in attractive repulsive protein ligand complex Cerius2/PLP Cerius2/PMF
29 Cerius2/ LUDI
30 SYBYL/F-Score
31 SYBYL/G-Score:
32 SYBYL/D-Score: SYBYL/ChemScore:
33 DrugScore: X-Score:
34 DOCKING SCORE It based on the bonded and non bonded interactions of ligand binding site Bonded Interactions: Non Bonded Interactions:
35 BINDING AFFINITY The interaction of most ligands with their binding sites can be characterized in terms of a binding affinity The free energy of binding (ΔG) is related to binding affinity by The equilibrium equation is : Where ΔG is Gibb s free energy, R is gas constant, T is temperatures and K is equilibrium constant E is enzyme and I is inhibitor
36
37 172 Protein ligand complexes chosen based on resolution (better than 2.5 Å) 172 Conformational sampling procedure 100 passes 100 Protein Ligand complex has: 43 different proteins Molecular weight KDa Rotatable single bond (ligand) 0 20

38 The selection of suitable sample for study It is done by AutoDock (Genetic Algorithm) Parameter: For best fitting, Translation, rotation, and torsions are set to 0.5 Å, 15, and15, respectively The size of the docking box is 30 Å X 30 Å X 30 Å

39 Screening parameter: RMSD 0-15 Å Distinctive conformational clusters Docked conformation should close to experimental conformation (RMSD 2.0 Å) ga-num-generations determines the quality of the sample runs per complex

40 Force Field based scoring: AutoDock, G-Score, D-Score Empirical scoring: LigScore, PLP,LUDI, F-Score, ChemScore, X-Score Knowledge Based scoring: PMF, DrugScore
41 Success rates of 11 scoring functions under different rmsd criteria
42
43
44
45

46
47
48
49
50
51
52
53 AUTODOCK Simulated Annealing Based on temperature effects Start with high temperature and global search Lower temperature local search Genetic Algorithm Charles Darwin s Theory of Evolution Genotype Phenotype Lamarckian Algorithm ( Jean Baptiste de Lamarck) Phenotype Genotype
54 Search parameters Population size Crossover rate Mutation rate Local search energy evals Termination criteria energy evals generations
55 Genetic function algorithm Start with a random population (50-200) Perform Crossover (Sex, two parents -> 2 children) and Mutation (Cosmic rays, one individual gives 1 mutant child) Compute fitness of each individual Proportional Selection & Elitism New Generation begins if total energy evals or maximum generations reached
56
57
58
59 Dimensionality of molecular docking Degrees of Freedom (DOF) Position or Translation (x,y,z) = 3 Orientation or Quaternion (qx, qy, qz, qw) = 4 Rotatable Bonds or Torsions (tor 1, tor 2, tor n ) = n Total DOF, or Dimensionality, D = n
60 Docking Preparation Grid AutoDock uses gridbased docking Ligand-protein interaction energies are pre-calculated and then used as a look-up table during simulation Grid maps are constructed based on atoms of interest in ligand (here CANOSH)
61

62 Initial X Ray crystallographic positions of protein and ligand (SYBYL)
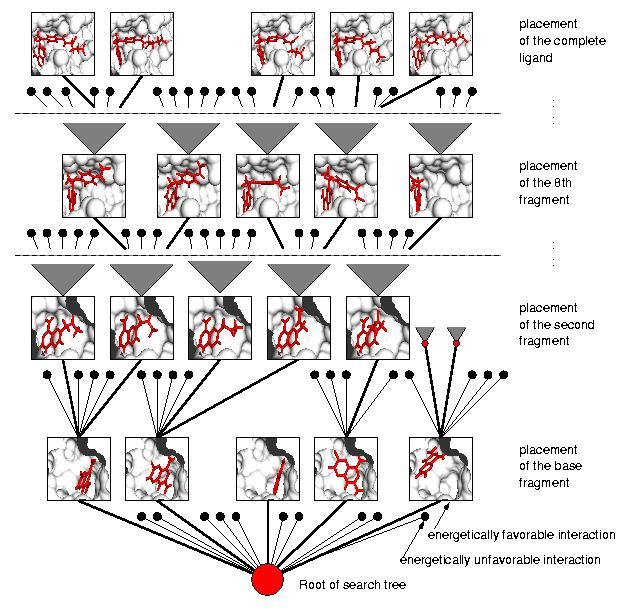
63 Simulated annealing One copy of the ligand (Population = 1) Starts from a random or specific postion/orientation/conformation (=state) Constant temperature annealing cycle (Accepted & Rejected Moves) Temperature reduced before next cycle Stops at maximum cycles
64 Docking Simulated Annealing Runs = 100 Cycles = 50 Initial Temp (RT) = 1,000 Temp reduction factor =.95 Linear temperature reduction Translation reduction factor = 1 Quaternion reduction factor = 1 Torsional reduction factor = 1 # rotatable bonds = 12 Initial coordinates = Random Initial quaternion = Random Initial dihedrals = Random Translation step = 2.0 Å Quaternion step = 50 deg Torsion step = 50 deg Results: 100 different clusters Energy range: to +64,000 Conformation #81: Conformation #67: Conformation #68: Lowest energy conf not close to position but similar to original Conf #67 closest to position and conformation of original ligand; higher energy Conf #68 close to position but not conformation of original ligand; not as high energy

65 Original ligand conf SA conformation #67 Close up of previous (SYBYL)

66 Original ligand conf Best GA conf Best LGA conf Best SA conf Best LS conf (SYBYL)
67 GOLD (CCDC, Cambridge, UK) Flexible docking: match protein and ligand hydrogen bond fitting points optimizes the poses using a Genetic algorithm Flexible rings by flipping ring corners Locally flexible protein: polar hydrogens allowed to move Water switched on and off to maximize interactions SF: GOLDfitness score
68 FRED (OpenEye, Santa Fe, CA, USA) Rigid body docking using a shape-based approach: random generation of poses within active site use of Gaussian functions to represent atoms Use of Gaussian docking functions (combines overlap between ligand with protein atoms and area intersection) and a Quasi-Newton rigid body optimization algorithm to place ligand and select poses Uses Rigid protein SF : ChemScore; Emperical FF
69 FlexX/FlexE (BioSolveIt, Sankt Augustin, Germany) Flexible docking: incremental construction for the ligand combined with a matching of ligand groups to protein interaction types multiple conformations for rings Rigid of flexible protein: all atom representation composite structures assessed (FlexE) water considered using the particle concept (waters placed before docking and only kept during the docking run if favourable interactions are created) SF : Flex X SF;Emperical FF
70 FLEX-X The general schema Ligand conformational flexibility Modeling Receptor-ligand interactions Scoring function Algorithm Base selection Base placement Incremental construction
71 Scoring function Estimates the free binding energy in the complex match score contact score The function is additive in the ligand atoms.
72 Ligand fragmentation Good results are produced if the added fragments are small Every fragment, except for the base fragment consist of only one component.
73
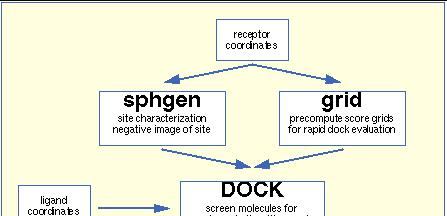
74 DOCK 6.0( (UCSF,CA,USA) Rigid body docking using a clique matching algorithm Flexible ligand using an incremental construction algorithm combined with a simplex minimizer Flexible protein: -negative image of the active site using spheres -use of precomputed grids based on AMBER intermolecular energy and GB/SA(Generalised Born/surface Area) solvation energy -protein flexibility considered using combined grids SF: Dockscore; Amber Force Field
75 DOCK as an Example DOCK works in 5 steps: Step 1 Start with 3D coordinates of target receptor Step 2 Generate molecular surface for receptor Step 3 Generate spheres to fill the active site of the receptor: The spheres become potential locations for ligand atoms Step 4 Matching: Sphere centers are then matched to the ligand atoms, to determine possible orientations for the ligand Step 5 Scoring: Find the top scoring orientation 75


76 DOCK as an Example 4 5 Three scoring schemes: Shape scoring, Electrostatic scoring and Force-field scoring Image 5 is a comparison of the top scoring orientation of the molecule thioketal with the orientation found in the crystal structure
77 The DOCK Algorithm Two steps in rigid ligand mode: Orienting the putative ligand in the site Guided by matching distances, between predefined site points on the target to interatomic distances of the ligand. The RT matrix is used for the transform of the ligand. Scoring the resulting orientation Each orientation is scored for each quality fit. The process is repeated a user-defined number of orientations or maximum orientations 77


78 Site Points Generation in DOCK Program SPHGEN identifies the active site, and other sites of interest. Each invagination is characterized by a set of overlapping spheres. For receptors, a negative image of the surface invaginations is created; For a ligand, the program creates a positive image of the entire molecule. 78
79 The Matching Can be directed by 2 additional features: Chemical matching - labeling the site points such that only particular atom types are allowed to be matched to them. Critical cluster - subsets of interest can be defined as critical clusters, so that at least one member of them will be part of any accepted ligand match. Increase in efficiency and speed due to elimination of potentially less promising orientations! 79
80 Define the target binding site points. O O S S N H N N N H N N N F F Match the distances. 3. Calculate the transformation matrix for the orientation. F H N N O S 4. Dock the molecule. O S H N N N F 5. Score the fit. 80
81 DOCK
82
83 Pharmacophore-Based Docking 83
84 Pharmacophore-based Docking Basic idea: Appropriate spatial disposition of a small number of functional groups in a molecule is sufficient for achieving a desired biological effect. The ensemble formation will be guided by these functional groups. 84

85 Pharmacophore Fingerprint Pharmacophore fingerprint - a set of pharmacophore features and their relative position. Typical pharmacophore features: Hydrogen-bond donors and acceptors Positive and negative ionizable atoms/groups Hydrophobes and ring centroids Implemented in DOCK Hydrogen-bond donors Hydrogen-bond acceptors Dual hydrogen-bond donor and acceptor 5 or 6 membered ring centroids 85
86 Pharmacophore DOCK Prepare target structure Save the best scoring conformer for each molecule Generate a set of chemically labeled site points Score all conformers Orientations tries >MAX Yes No Read a 3D pharmacophore from the database Compare distances between pharmacophore points and site points to determine an orientation matrix Yes No Orientations tries >MAX Use the transformation matrix to dock all conformers associated with the pharmacophore Yes Match? No 86
87 Advantages of Pharmacophore-based Docking Rapid elimination of ligands containing functional groups which would interfere with binding. Speed increase over docking of individual molecules. More information pertaining to the entire molecule is retained (no rigid portions). Chemical matching and critical clusters are encouraged. 87
88 Limitations of Pharmacophore-based Searching A limited subset of key interactions (typically 4-6) which must be extracted from the target site with dozens of potential interactions. Complex queries are extremely slow. The majority of the information contained in the target structure is not considered during the search. There is no scoring function beyond the binary (match/no match). Any steric or electronic constraints imposed by the target, but not defined by the target are ignored. 88
89 Conformational Ensembles Docking Observations: 1. Generating an orientation of a ligand in a binding site may be separated from calculating a conformation of the ligand in that particular orientation. 2. Multiple conformations of a given ligand usually have some portion in common (internally rigid atoms such as ring systems), and therefore, contain redundancies. 89
90 Conformational Ensembles Docking Observations: 1. Generating an orientation of a ligand in a binding site may be separated from calculating a conformation of the ligand in that particular orientation. 2. Multiple conformations of a given ligand usually have some portion in common (internally rigid atoms such as ring systems), and therefore, contain redundancies. 90
91 Overview of the Ligand Ensemble Method 91
92 Disadvantages of Conformational Ensemble Docking Loss of information when the orientations are guided only by a subset of the atoms in molecule. Orientations may be missed because potential distance matches from non-rigid portions of the molecule are not considered. The ensemble method will fail for ligands that lack internally rigid atoms. The use of chemical matching and critical clusters is limited. 92
93 ACTIVE SITE IDENTIFICATION PROGRAMS CASTP: XSITE: Voidoo: APROPOS: apropos/apropos_descrip.html CANGAROO: Surfnet: PASS: Active site templates for Enzymes
94 Protein Ligand Docking Programs AutoDock GOLD FLEXX GLIDE ICM Dock
95 Protein protein Docking Programs ZDOCK : HEX : GRAMM : ICM : CLUSPRO : KORDO : MOLFIT : ort//molfit/
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Using AutoDock 4 with ADT: A Tutorial
 Using AutoDock 4 with ADT: A Tutorial Ruth Huey Sargis Dallakyan Alex Perryman David S. Goodsell (Garrett Morris) 9/2/08 Using AutoDock 4 with ADT 1 What is Docking? Predicting the best ways two molecules
Using AutoDock 4 with ADT: A Tutorial Ruth Huey Sargis Dallakyan Alex Perryman David S. Goodsell (Garrett Morris) 9/2/08 Using AutoDock 4 with ADT 1 What is Docking? Predicting the best ways two molecules
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Protein-Ligand Docking Methods
 Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Protein Structure Prediction and Protein-Ligand Docking
 Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Protein-Ligand Docking
 Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Protein-Ligand Docking Methods
 Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
BCB410 Protein-Ligand Docking Exercise Set Shirin Shahsavand December 11, 2011
 BCB410 Protein-Ligand Docking Exercise Set Shirin Shahsavand December 11, 2011 1. Describe the search algorithm(s) AutoDock uses for solving protein-ligand docking problems. AutoDock uses 3 different approaches
BCB410 Protein-Ligand Docking Exercise Set Shirin Shahsavand December 11, 2011 1. Describe the search algorithm(s) AutoDock uses for solving protein-ligand docking problems. AutoDock uses 3 different approaches
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
5.1. Hardwares, Softwares and Web server used in Molecular modeling
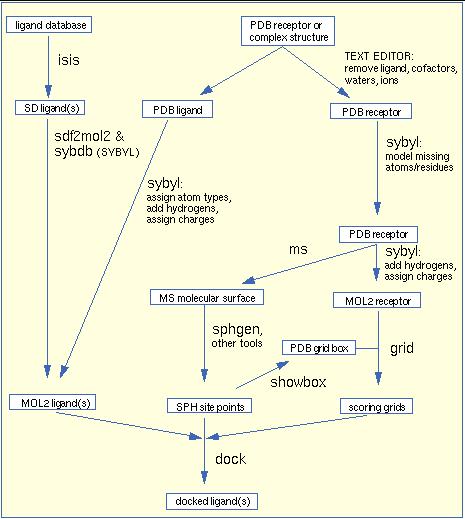
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
MOLECULAR RECOGNITION DOCKING ALGORITHMS. Natasja Brooijmans 1 and Irwin D. Kuntz 2
 Annu. Rev. Biophys. Biomol. Struct. 2003. 32:335 73 doi: 10.1146/annurev.biophys.32.110601.142532 Copyright c 2003 by Annual Reviews. All rights reserved First published online as a Review in Advance on
Annu. Rev. Biophys. Biomol. Struct. 2003. 32:335 73 doi: 10.1146/annurev.biophys.32.110601.142532 Copyright c 2003 by Annual Reviews. All rights reserved First published online as a Review in Advance on
Protein-Ligand Docking Evaluations
 Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Conformational Searching using MacroModel and ConfGen. John Shelley Schrödinger Fellow
 Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Computational modeling of G-Protein Coupled Receptors (GPCRs) has recently become
 has recently become") Homology Modeling and Docking of Melatonin Receptors Andrew Kohlway, UMBC Jeffry D. Madura, Duquesne University 6/18/04 INTRODUCTION Computational modeling of G-Protein Coupled Receptors (GPCRs) has recently
Homology Modeling and Docking of Melatonin Receptors Andrew Kohlway, UMBC Jeffry D. Madura, Duquesne University 6/18/04 INTRODUCTION Computational modeling of G-Protein Coupled Receptors (GPCRs) has recently
Using AutoDock With AutoDockTools: A Tutorial
 Using AutoDock With AutoDockTools: A Tutorial Dr. Ruth Huey & Dr. Garrett M. Morris 6/6/06 AutoDock & ADT Tutorial 1 What is Docking? Best ways to put two molecules together. Three steps: (1) Obtain the
Using AutoDock With AutoDockTools: A Tutorial Dr. Ruth Huey & Dr. Garrett M. Morris 6/6/06 AutoDock & ADT Tutorial 1 What is Docking? Best ways to put two molecules together. Three steps: (1) Obtain the
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Cheminformatics platform for drug discovery application
 EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Receptor Based Drug Design (1)
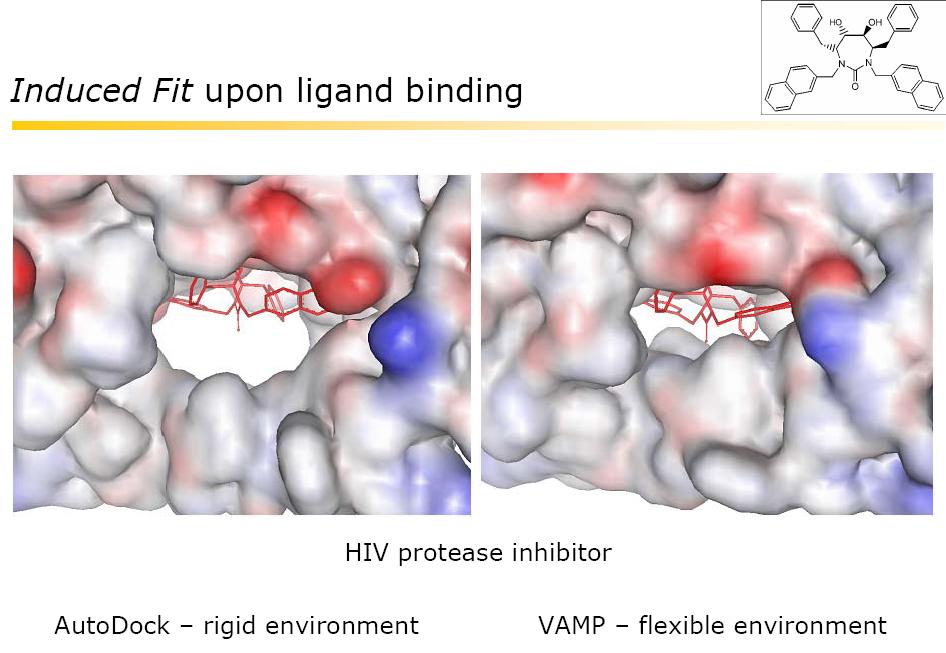
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Pose and affinity prediction by ICM in D3R GC3. Max Totrov Molsoft
 Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS
 TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
High Throughput In-Silico Screening Against Flexible Protein Receptors
 John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
Ranking of HIV-protease inhibitors using AutoDock
 Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
BioSolveIT. A Combinatorial Approach for Handling of Protonation and Tautomer Ambiguities in Docking Experiments
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
Author Index Volume
 Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Computational Identification of Inhibitors of Protein-Protein Interactions
 Current Topics in Medicinal Chemistry, 2007, 7, 63-82 63 Computational Identification of Inhibitors of Protein-Protein Interactions Shun Zhong, Alba T. Macias and Alexander D. MacKerell Jr.* Department
Current Topics in Medicinal Chemistry, 2007, 7, 63-82 63 Computational Identification of Inhibitors of Protein-Protein Interactions Shun Zhong, Alba T. Macias and Alexander D. MacKerell Jr.* Department
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Binding Response: A Descriptor for Selecting Ligand Binding Site on Protein Surfaces
 J. Chem. Inf. Model. 2007, 47, 2303-2315 2303 Binding Response: A Descriptor for Selecting Ligand Binding Site on Protein Surfaces Shijun Zhong and Alexander D. MacKerell, Jr.* Computer-Aided Drug Design
J. Chem. Inf. Model. 2007, 47, 2303-2315 2303 Binding Response: A Descriptor for Selecting Ligand Binding Site on Protein Surfaces Shijun Zhong and Alexander D. MacKerell, Jr.* Computer-Aided Drug Design
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Crystal Structure Prediction using CRYSTALG program
 Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
BUDE. A General Purpose Molecular Docking Program Using OpenCL. Richard B Sessions
 BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites. J. Andrew Surface
 Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Towards Physics-based Models for ADME/Tox. Tyler Day
 Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Chapter 8: Introduction to Evolutionary Computation
 Computational Intelligence: Second Edition Contents Some Theories about Evolution Evolution is an optimization process: the aim is to improve the ability of an organism to survive in dynamically changing
Computational Intelligence: Second Edition Contents Some Theories about Evolution Evolution is an optimization process: the aim is to improve the ability of an organism to survive in dynamically changing
Computational protein design
 Computational protein design There are astronomically large number of amino acid sequences that needs to be considered for a protein of moderate size e.g. if mutating 10 residues, 20^10 = 10 trillion sequences
Computational protein design There are astronomically large number of amino acid sequences that needs to be considered for a protein of moderate size e.g. if mutating 10 residues, 20^10 = 10 trillion sequences
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
GC and CELPP: Workflows and Insights
 GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
Assessing Scoring Functions for Protein-Ligand Interactions
 3032 J. Med. Chem. 2004, 47, 3032-3047 Assessing Scoring Functions for Protein-Ligand Interactions Philippe Ferrara,, Holger Gohlke,, Daniel J. Price, Gerhard Klebe, and Charles L. Brooks III*, Department
3032 J. Med. Chem. 2004, 47, 3032-3047 Assessing Scoring Functions for Protein-Ligand Interactions Philippe Ferrara,, Holger Gohlke,, Daniel J. Price, Gerhard Klebe, and Charles L. Brooks III*, Department
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors
 MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Docking with Water in the Binding Site using GOLD
 Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Flexibility and Constraints in GOLD
 Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
MM-PBSA Validation Study. Trent E. Balius Department of Applied Mathematics and Statistics AMS
 MM-PBSA Validation Study Trent. Balius Department of Applied Mathematics and Statistics AMS 535 11-26-2008 Overview MM-PBSA Introduction MD ensembles one snap-shots relaxed structures nrichment Computational
MM-PBSA Validation Study Trent. Balius Department of Applied Mathematics and Statistics AMS 535 11-26-2008 Overview MM-PBSA Introduction MD ensembles one snap-shots relaxed structures nrichment Computational
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Other Cells. Hormones. Viruses. Toxins. Cell. Bacteria
 Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Analyzing Molecular Conformations Using the Cambridge Structural Database. Jason Cole Cambridge Crystallographic Data Centre
 Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows
 MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
From Small Molecules to Biological Molecules: Modelling Interactions. Dr. Antonio Chana Milano, 22 nd April 2008
 From Small Molecules to Biological Molecules: Modelling Interactions Dr. Antonio Chana Milano, 22 nd April 2008 Computational Methods & Applicability 2 *Diagram taken from S. C. Glotzer The University
From Small Molecules to Biological Molecules: Modelling Interactions Dr. Antonio Chana Milano, 22 nd April 2008 Computational Methods & Applicability 2 *Diagram taken from S. C. Glotzer The University
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Focusing Conformational Ensembles on Bioactive-Like Conformations
 Focusing Conformational Ensembles on Bioactive-Like Conformations Hannah H. Avgy, Boaz Musafia, Hanoch Senderowitz Department of Chemistry Bar-Ilan University 3rd Strasbourg Summer School on Chemoinformatics,
Focusing Conformational Ensembles on Bioactive-Like Conformations Hannah H. Avgy, Boaz Musafia, Hanoch Senderowitz Department of Chemistry Bar-Ilan University 3rd Strasbourg Summer School on Chemoinformatics,
Build_model v User Guide
 Build_model v.2.0.1 User Guide MolTech Build_model User Guide 2008-2011 Molecular Technologies Ltd. www.moltech.ru Please send your comments and suggestions to contact@moltech.ru. Table of Contents Input
Build_model v.2.0.1 User Guide MolTech Build_model User Guide 2008-2011 Molecular Technologies Ltd. www.moltech.ru Please send your comments and suggestions to contact@moltech.ru. Table of Contents Input
Identifying Interaction Hot Spots with SuperStar
 Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
GOLD Configuration File User Guide
 GOLD Configuration File User Guide A Component of the GOLD Suite 5.6.1 GOLDSuite Release Copyright 2018 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The GOLD suite
GOLD Configuration File User Guide A Component of the GOLD Suite 5.6.1 GOLDSuite Release Copyright 2018 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The GOLD suite
Advanced in silico drug design
 Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Advanced in silico drug design RNDr. Martin Lepšík, Ph.D. Lecture: Advanced scoring Palacky University, Olomouc 2016 1 Outline 1. Scoring Definition, Types 2. Physics-based Scoring: Master Equation Terms
Pharmacophore-Based Molecular Docking to Account for Ligand Flexibility
 PROTEINS: Structure, Function, and Genetics 51:172 188 (2003) Pharmacophore-Based Molecular Docking to Account for Ligand Flexibility Diane Joseph-McCarthy,* Bert E. Thomas IV, Michael Belmarsh, Demetri
PROTEINS: Structure, Function, and Genetics 51:172 188 (2003) Pharmacophore-Based Molecular Docking to Account for Ligand Flexibility Diane Joseph-McCarthy,* Bert E. Thomas IV, Michael Belmarsh, Demetri
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
ENERGY MINIMIZATION AND CONFORMATION SEARCH ANALYSIS OF TYPE-2 ANTI-DIABETES DRUGS
 Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Computer Graphics Applications on Molecular Biology and Drug Design
 Computer Graphics Applications on Molecular Biology and Drug Design Katerina PERDIKURI 1, Athanasios TSAKALIDIS 1 1 Department of Computer Engineering and Informatics University of Patras,26500 Patras,
Computer Graphics Applications on Molecular Biology and Drug Design Katerina PERDIKURI 1, Athanasios TSAKALIDIS 1 1 Department of Computer Engineering and Informatics University of Patras,26500 Patras,
Ultra High Throughput Screening using THINK on the Internet
 Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
Portal. User Guide Version 1.0. Contributors
 Portal www.dockthor.lncc.br User Guide Version 1.0 Contributors Diogo A. Marinho, Isabella A. Guedes, Eduardo Krempser, Camila S. de Magalhães, Hélio J. C. Barbosa and Laurent E. Dardenne www.gmmsb.lncc.br
Portal www.dockthor.lncc.br User Guide Version 1.0 Contributors Diogo A. Marinho, Isabella A. Guedes, Eduardo Krempser, Camila S. de Magalhães, Hélio J. C. Barbosa and Laurent E. Dardenne www.gmmsb.lncc.br
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
BioSolveIT. A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
FDS: Flexible Ligand and Receptor Docking with a Continuum Solvent Model and Soft-Core Energy Function
 FDS: Flexible Ligand and Receptor Docking with a Continuum Solvent Model and Soft-Core Energy Function RICHARD D. TAYLOR, 1, * PHILIP J. JEWSBURY, 2 JONATHAN W. ESSEX 1 1 Department of Chemistry, University
FDS: Flexible Ligand and Receptor Docking with a Continuum Solvent Model and Soft-Core Energy Function RICHARD D. TAYLOR, 1, * PHILIP J. JEWSBURY, 2 JONATHAN W. ESSEX 1 1 Department of Chemistry, University
Introduction to AutoDock and AutoDock Tools
 Introduction to AutoDock and AutoDock Tools Alexander B. Pacheco User Services Consultant LSU HPC & LONI sys-help@loni.org HPC Training Series Louisiana State University Baton Rouge Mar. 28, 2012 HPC@LSU
Introduction to AutoDock and AutoDock Tools Alexander B. Pacheco User Services Consultant LSU HPC & LONI sys-help@loni.org HPC Training Series Louisiana State University Baton Rouge Mar. 28, 2012 HPC@LSU
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the