Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
|
|
|
- Mervin Lucas
- 5 years ago
- Views:
Transcription
1 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller
2 Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics The study of small-molecular-weight drug candidates on gene/protein function.

3 Chemogenomic Chemogenomics defines, in principle, the screening of the chemical universe, i.e., all possible chemical compounds, against the target universe, i.e., all proteins and other potential drug targets. Mission impossible!!
4 Chemogenomic The solution the method defines the screening of congeneric chemical libraries against certain target families, e.g., the G proteincoupled receptors, nuclear receptors, different protease families, kinases, phosphodiesterases, ion channels, transporters, etc.
5 Chemogenomic Requirements A compound library A representative biological system Target library Single cell Organism A reliable readout Gene/protein expression High-throughput screening (binding or functionality assays)
6 Chemogenomic Completion of a two-dimentional matrix, representing the interaction of tragets/genes and compounds by values of binding affinities (Ki) or functional effect (IC50).
7 Ligand and Target Spaces Assumptions for any chemogenomi-based approach (a) Compounds sharing some chemical similarity should also share targets. (b) Targets sharing similar ligands should share similar patterns (binding sites). Question How do we measure the distances between two ligands or two targets?

8 Ligand Space Distance measuring between two compounds is done by solving a similarity matrix The compounds properties are often described using descriptors Descriptor classification One-dimensional Two-dimensional Three-dimensional
9 Ligand Space (Descriptors) 1D descriptors Easy and fast to compute Describe global properties (MW, atom and bond counts) Based on the chemical formulae Prediction of physicochemical properties Polar surface area Solubility Rings Discrimination between compound sets Drugs vs. nondrugs Ligands from targetfamilies 1D linear representations of compounds SMILES (Simplified Molecular Input Entry System)
10 Ligand Space (Descriptors) 2D descriptors Most common ligand descriptors Describe topological properties (maximum common substructure, structural keys) Encode both atomic and bond properties 2D sketch figure Scanning libraries for similar substructures or fragments Graph-based method Molecular graph (subfamily clustering) Computational slow Fingerprint-based method Bit strings (0 and 1 = atoms, fragments, rings..) Fingerprints easy for comparison Also used in receptor-ligand recognition
11 Ligand Space (Descriptors) 3D descriptors Describe conformational properties (atomic coordinates, potentials, fields, shapes) Necessities for proper alignment Comparison in same 3D Cartesian space Conformational space accessible to each ligand Bit strings vs. structure comparison Structure comparison can produce false positives 3D information is stored in bit strings Binary representation of 2D or 3D properties Tanimoto coefficient (simple similarity indicies)
12 Ligand Space (Descriptors)


13 Target Space Chemoproteomics Traget = proteins Dimension Classification scheme Databases 1D 2D 3D By sequence By patterns By secondary structure, fold By atomic coordinates By binding site UniProt, Pfam PRINTS, PROSITE SCOP, CATH PDB, MODBASE BindingMOAD, sc-pdb
14 Target Space The amino acid sequence (1D) Clustering of targets into target-families Large variation in sequence length even among family members e.g., human GPCRs range from 290 to 6200 residues Structural motifs (2D) Mapping of a-helices, b-sheets, coils and random structures 3D Structure Atomic coordinates derived by X-ray diffraction or NMR Structural fold Ligand-binding site, higher similarity among related targets Pharmacological profile Binding affinity for a panel of ligands Modifying pharmacological profiles of druges are widely used in drug design

15 Protein-Ligand Space Full matrices (affinity or structural information) Experimental data are stored in the matrices Affinity of a new compound to a known target Measuring structure-activity relationships Prediction of a global pharmacological profile Advantages Based on experimental data Superior to computed descriptors Disadvantages Enormous amount of data is necessary Highly cost consuming (not realistic in academic environments) Interaction fingerprints (IFPs) Replacement of affinity with molecular interaction descriptors Conversion of atomic coordinates of protein-ligand complexes into bit strings.
16 Ligand-based Chemogenomic Annotating ligand libraries Molecules sharing enough similarity to existing ligands for which a target profile is known have enhanced probability of sharing the same biological profile. Ligand libraries Targets In vitro affinity data ADME properties Biological annotated compound libraries AurSCOPE ( GPCR ligands and kinase inhibitors) MedChem database (Biological and pharmacological information of compounds) ChemBank ( compounds in 441 high-troughput screening assays) Natural product-oriented chemical libraries Evolutionary pressure Highly specific binding mechnisms
 A privileged structure is defined as a substructure or scaffold exhibiting strong preference for a particular area of the target space.")
17 Ligand-based: Privileged Structures Coined by Evans et al. (1,4-benzodiazepine scaffold) A privileged structure is defined as a substructure or scaffold exhibiting strong preference for a particular area of the target space. Suitable to orient design of trageted compound libraries Biphenyl: protein-binding motif No particular preference for target family 2-tetrazolo-biphenyl GPCRs Only few are really selective
18 Ligand-based In silico Screening Target fishing Reference compounds set (known 2D or 3D descriptors) Screening procedure (QSAR, Bayesian analysis or pharmacophore) Screening collection for identification of new compounds
19 Ligand-based In silico Screening Mestres et al. Library of molecules targeting nuclear hormone receptors NHR 2000 ligands 25 receptors Easily distinction between selective and promiscuous scaffolds SHannon Entropy Descriptors SHED Novartis Prediction of target profiles from extended connectivity fingerprints Machine learning algorithm based on Bayesian statistics Wombat database (1230 unique SMILES) Bayesian models was produced (trained) for each activity class Prediction is done by calculating the probability of each test compound to become a ligand for each of the tragets Improvement by concatenate all target-associated probabilities Bayes affinity fingerprint 2D descriptors was more predictive than 3D (not for singletons)
20 Ligand-based In silico Screening Drawback Categorization of training set compounds according to their molecular target, without checking: Does it really bind? Where it binds? How it binds? Training a machine learning algorithm with incorrect data Alternatives 3D pharmacophores from protein-ligand complexes Experimentally determined atomic coordinates Experimentally determined pharmacological activities Limited chemical diversity observed among PDB ligands
21 Target-based Chemogenomic Selectivity control Selectivity of ligands among family related targets Proteome-wide comparative modeling Structural data (X-ray or NMR) Sequence-based comparison Structure-based comparison Comparing Molecular fields Comparing 3D structures
22 Sequence-based Comparison Multiple alignment of all targets Comparison of any kind of target families Lack of high-resolution structural data GPCRs are ideal candidates for sequence-based comparison Only bovine rhodopsin has been crystallised Important target family for drug design Key residues are extracted and concatenated Ungapped sequence (30 residues) Distance matrix based on: Sequence identity Sequence similarity Physicochemical properties Cavity-based clustering of 372 human GPCRs Reproduced a perfect full sequence based tree Target comparison across a family is possible using only few residues Applications Simple analysis of binding site regions by residue conservation Target hopping used to discover receptor ligands to a particular receptor
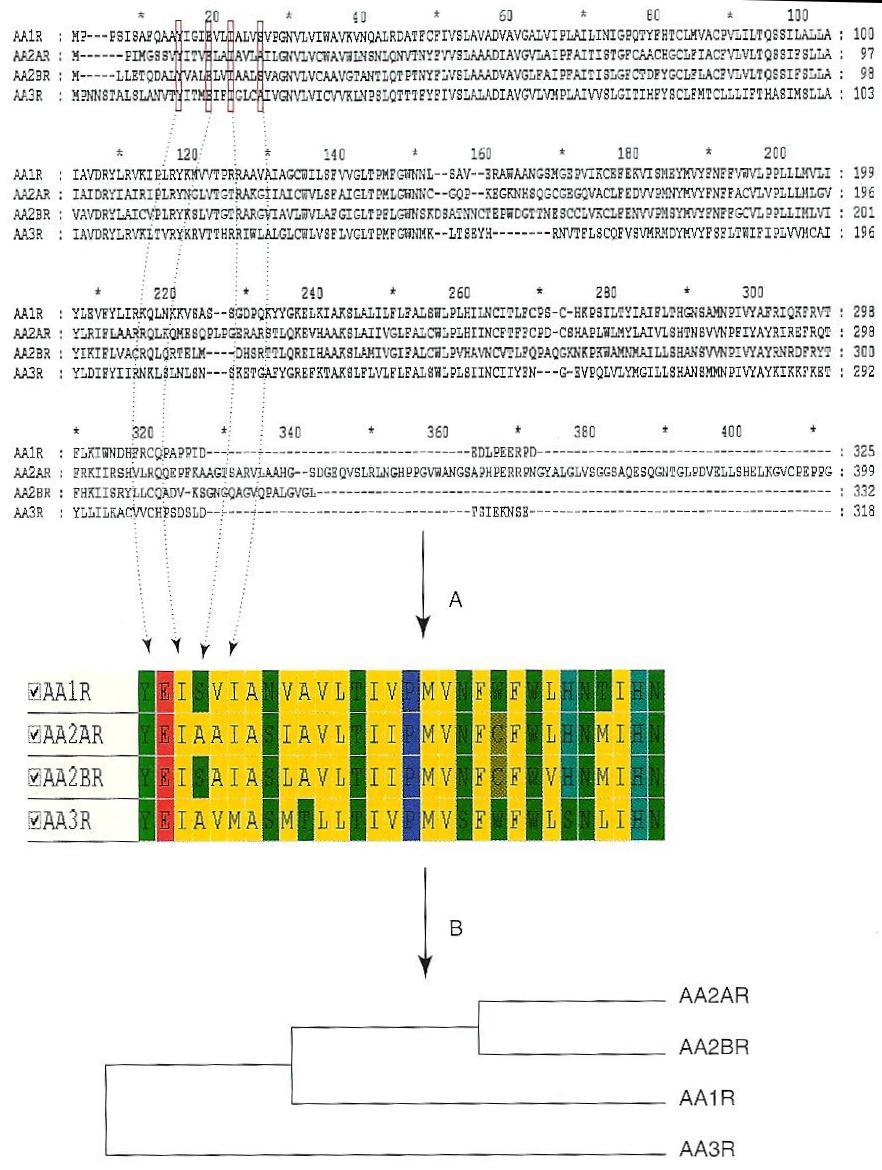
23 Sequence-based Comparison
24 Structure-based Comparison High-resolution structural data is crucial for homology modeling, however only the ligand-binding site are compared Comparing Molecular Fields Molecular interaction fields (MIFs) Structural alignment of targets Interaction energies Probe atoms at each point of a 3D grid (binding site) MIFs placed in a global matrix Rows: Targets Columns: Interaction energies Analysis either by: Principal component analysis Hierarchical clustering Highly dependent on: Structural alignment, grid resolution and probe atoms
25 Structure-based Comparison Comparing 3D Structures Global structural alignment methods GASH DaliLite CE Alignment of predefined structural motifs Matching templates to a reference protein Not all proteins sharing binding sites for a particular ligand share any structural template similarities Structural alignment by physicochemical property description Surface-based comparison Relatively slow and thus incompatible with proteome-wide comparison SuMO, Cavbase, SiteEngine, SitesBase and CPASS Emerged in the last years Represent active site by pseudocenters encoding physicochemical properties (H-bonding, capacity, aromaticity, hydrophobicity and charge) Pseudocentres are linked by edges providing a molecular graph Detection of maximal common subgraphs (clique detection) Detection of local similarities at ligand-binding subpockets for proteins with totally different fold and catalytic activities

26 Structure-based Comparison Comparing 3D Structures Interpretation of computated similarity scores often difficult Active sites of different dimensions Larger sites tend to present more matches even if the smallest is more similar Surgand et al. projected an active site on a dimensionless 80- triangled sphere of cavity descriptors Measuring normalized distance in descriptor space
27 Target-Ligand-based Chemogenomic Chemical annotation of target binding sites Various chemical compound libraries exist Binding information is crucial Protein/binding site must annotated by ligand chemotype SMID (Small Molecule Interaction Database) annotate protein sequence by domain-specific ligands Browse likely ligands to a protein of unknown 3D structure Ligand-annotated binding sites from PDB BindingMOAD and sc-pdb Pharmacological point of view Prioritize ligands for designing targeted compound libraries
28 Target-Ligand-based Chemogenomic To browse and predict protein-ligand complexes, one needs to set up simple descriptors for both ligands and proteins from knowledge databases and concatenate them into a single protein-ligands description. Two dimensional searches Use experimental binding affinity matrices and define appropriate QSAR models to predict affinity of new compounds Three-dimensional searches Dock each ligand of compound library into each active site of target library Molecular inverse docking approach Scoring functions cannot quantify very heterogeneous proteinligand complexes Computation of IFP strings Converts 3D information about protein-ligand interaction to 1D
29 Target-Ligand-based Chemogenomic Three-dimensional searches 3D-based docking-independent methods Retrieving ligand from protein and vice versa Encode protein and ligand properties with similar descriptors CoLiBRI (complementary ligands based on receptor information) Ligand and protein described using same molecular descriptors (TAE-RECON) Shape and electronic properties of isolated atoms Mapping patterns of active sites onto patterns of their complementary ligands and vice versa High test results when similar training set
30 Final remarks High-troughput data (structure, binding affinity, etc.) Ligand Target Linking data either by ligand or target focusing Target-based Chemogenomics Ligand-based Chemogenomics Target-Ligand-bases Chemogenomics Selectivity profiles for therapeutic usage Not more selective ligands In silico approach
31 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Navigation in Chemical Space Towards Biological Activity. Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland
 Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Machine learning for ligand-based virtual screening and chemogenomics!
 Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
CSCE555 Bioinformatics. Protein Function Annotation
 CSCE555 Bioinformatics Protein Function Annotation Why we need to do function annotation? Fig from: Network-based prediction of protein function. Molecular Systems Biology 3:88. 2007 What s function? The
CSCE555 Bioinformatics Protein Function Annotation Why we need to do function annotation? Fig from: Network-based prediction of protein function. Molecular Systems Biology 3:88. 2007 What s function? The
Machine Learning Concepts in Chemoinformatics
 Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
An Integrated Approach to in-silico
 An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Structure to Function. Molecular Bioinformatics, X3, 2006
 Structure to Function Molecular Bioinformatics, X3, 2006 Structural GeNOMICS Structural Genomics project aims at determination of 3D structures of all proteins: - organize known proteins into families
Structure to Function Molecular Bioinformatics, X3, 2006 Structural GeNOMICS Structural Genomics project aims at determination of 3D structures of all proteins: - organize known proteins into families
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Notes of Dr. Anil Mishra at 1
 Introduction Quantitative Structure-Activity Relationships QSPR Quantitative Structure-Property Relationships What is? is a mathematical relationship between a biological activity of a molecular system
Introduction Quantitative Structure-Activity Relationships QSPR Quantitative Structure-Property Relationships What is? is a mathematical relationship between a biological activity of a molecular system
Structure-Activity Modeling - QSAR. Uwe Koch
 Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Statistical concepts in QSAR.
 Statistical concepts in QSAR. Computational chemistry represents molecular structures as a numerical models and simulates their behavior with the equations of quantum and classical physics. Available programs
Statistical concepts in QSAR. Computational chemistry represents molecular structures as a numerical models and simulates their behavior with the equations of quantum and classical physics. Available programs
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Plan. Day 2: Exercise on MHC molecules.
 Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Plan. Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics.
 and its used in Chemoinformatics.") Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Chemoinformatics and information management. Peter Willett, University of Sheffield, UK
 Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors
 A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
Development of a Structure Generator to Explore Target Areas on Chemical Space
 Development of a Structure Generator to Explore Target Areas on Chemical Space Kimito Funatsu Department of Chemical System Engineering, This materials will be published on Molecular Informatics Drug Development
Development of a Structure Generator to Explore Target Areas on Chemical Space Kimito Funatsu Department of Chemical System Engineering, This materials will be published on Molecular Informatics Drug Development
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
György M. Keserű H2020 FRAGNET Network Hungarian Academy of Sciences
 Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structure-Based Drug Discovery An Overview
 Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Bioinformatics. Macromolecular structure
 Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Data Mining in the Chemical Industry. Overview of presentation
 Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression
 APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
proteins Comparison of structure-based and threading-based approaches to protein functional annotation Michal Brylinski, and Jeffrey Skolnick*
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS Comparison of structure-based and threading-based approaches to protein functional annotation Michal Brylinski, and Jeffrey Skolnick* Center for the Study
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Comparison of structure-based and threading-based approaches to protein functional annotation Michal Brylinski, and Jeffrey Skolnick* Center for the Study
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery
 21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
Virtual affinity fingerprints in drug discovery: The Drug Profile Matching method
 Ágnes Peragovics Virtual affinity fingerprints in drug discovery: The Drug Profile Matching method PhD Theses Supervisor: András Málnási-Csizmadia DSc. Associate Professor Structural Biochemistry Doctoral
Ágnes Peragovics Virtual affinity fingerprints in drug discovery: The Drug Profile Matching method PhD Theses Supervisor: András Málnási-Csizmadia DSc. Associate Professor Structural Biochemistry Doctoral
CS612 - Algorithms in Bioinformatics
 Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
Fall 2017 Databases and Protein Structure Representation October 2, 2017 Molecular Biology as Information Science > 12, 000 genomes sequenced, mostly bacterial (2013) > 5x10 6 unique sequences available
A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery
 AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
Bioinformatics. Proteins II. - Pattern, Profile, & Structure Database Searching. Robert Latek, Ph.D. Bioinformatics, Biocomputing
 Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Xia Ning,*, Huzefa Rangwala, and George Karypis
 J. Chem. Inf. Model. XXXX, xxx, 000 A Multi-Assay-Based Structure-Activity Relationship Models: Improving Structure-Activity Relationship Models by Incorporating Activity Information from Related Targets
J. Chem. Inf. Model. XXXX, xxx, 000 A Multi-Assay-Based Structure-Activity Relationship Models: Improving Structure-Activity Relationship Models by Incorporating Activity Information from Related Targets
Author Index Volume
 Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Computational Methods and Drug-Likeness. Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004
 Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Introduction to FBDD Fragment screening methods and library design
 Introduction to FBDD Fragment screening methods and library design Samantha Hughes, PhD Fragments 2013 RSC BMCS Workshop 3 rd March 2013 Copyright 2013 Galapagos NV Why fragment screening methods? Guess
Introduction to FBDD Fragment screening methods and library design Samantha Hughes, PhD Fragments 2013 RSC BMCS Workshop 3 rd March 2013 Copyright 2013 Galapagos NV Why fragment screening methods? Guess
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
In Silico Investigation of Off-Target Effects
 PHARMA & LIFE SCIENCES WHITEPAPER In Silico Investigation of Off-Target Effects STREAMLINING IN SILICO PROFILING In silico techniques require exhaustive data and sophisticated, well-structured informatics
PHARMA & LIFE SCIENCES WHITEPAPER In Silico Investigation of Off-Target Effects STREAMLINING IN SILICO PROFILING In silico techniques require exhaustive data and sophisticated, well-structured informatics
STRUCTURAL BIOINFORMATICS II. Spring 2018
 STRUCTURAL BIOINFORMATICS II Spring 2018 Syllabus Course Number - Classification: Chemistry 5412 Class Schedule: Monday 5:30-7:50 PM, SERC Room 456 (4 th floor) Instructors: Ronald Levy, SERC 718 (ronlevy@temple.edu)
STRUCTURAL BIOINFORMATICS II Spring 2018 Syllabus Course Number - Classification: Chemistry 5412 Class Schedule: Monday 5:30-7:50 PM, SERC Room 456 (4 th floor) Instructors: Ronald Levy, SERC 718 (ronlevy@temple.edu)
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Similarity methods for ligandbased virtual screening
 Similarity methods for ligandbased virtual screening Peter Willett, University of Sheffield Computers in Scientific Discovery 5, 22 nd July 2010 Overview Molecular similarity and its use in virtual screening
Similarity methods for ligandbased virtual screening Peter Willett, University of Sheffield Computers in Scientific Discovery 5, 22 nd July 2010 Overview Molecular similarity and its use in virtual screening
Molecular Similarity Searching Using Inference Network
 Molecular Similarity Searching Using Inference Network Ammar Abdo, Naomie Salim* Faculty of Computer Science & Information Systems Universiti Teknologi Malaysia Molecular Similarity Searching Search for
Molecular Similarity Searching Using Inference Network Ammar Abdo, Naomie Salim* Faculty of Computer Science & Information Systems Universiti Teknologi Malaysia Molecular Similarity Searching Search for
Early Stages of Drug Discovery in the Pharmaceutical Industry
 Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Amino Acid Structures from Klug & Cummings. 10/7/2003 CAP/CGS 5991: Lecture 7 1
 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 1 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 2 Amino Acid Structures from Klug & Cummings
Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 1 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 2 Amino Acid Structures from Klug & Cummings
Basics of protein structure
 Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Cheminformatics analysis and learning in a data pipelining environment
 Molecular Diversity (2006) 10: 283 299 DOI: 10.1007/s11030-006-9041-5 c Springer 2006 Review Cheminformatics analysis and learning in a data pipelining environment Moises Hassan 1,, Robert D. Brown 1,
Molecular Diversity (2006) 10: 283 299 DOI: 10.1007/s11030-006-9041-5 c Springer 2006 Review Cheminformatics analysis and learning in a data pipelining environment Moises Hassan 1,, Robert D. Brown 1,
Introduction to Computational Structural Biology
 Introduction to Computational Structural Biology Part I 1. Introduction The disciplinary character of Computational Structural Biology The mathematical background required and the topics covered Bibliography
Introduction to Computational Structural Biology Part I 1. Introduction The disciplinary character of Computational Structural Biology The mathematical background required and the topics covered Bibliography
Design and Synthesis of the Comprehensive Fragment Library
 YOUR INNOVATIVE CHEMISTRY PARTNER IN DRUG DISCOVERY Design and Synthesis of the Comprehensive Fragment Library A 3D Enabled Library for Medicinal Chemistry Discovery Warren S Wade 1, Kuei-Lin Chang 1,
YOUR INNOVATIVE CHEMISTRY PARTNER IN DRUG DISCOVERY Design and Synthesis of the Comprehensive Fragment Library A 3D Enabled Library for Medicinal Chemistry Discovery Warren S Wade 1, Kuei-Lin Chang 1,
How Diverse Are Diversity Assessment Methods? A Comparative Analysis and Benchmarking of Molecular Descriptor Space
 pubs.acs.org/jcim How Diverse Are Diversity Assessment Methods? A Comparative Analysis and Benchmarking of Molecular Descriptor Space Alexios Koutsoukas,, Shardul Paricharak,,, Warren R. J. D. Galloway,
pubs.acs.org/jcim How Diverse Are Diversity Assessment Methods? A Comparative Analysis and Benchmarking of Molecular Descriptor Space Alexios Koutsoukas,, Shardul Paricharak,,, Warren R. J. D. Galloway,
Protein Structure Analysis and Verification. Course S Basics for Biosystems of the Cell exercise work. Maija Nevala, BIO, 67485U 16.1.
 Protein Structure Analysis and Verification Course S-114.2500 Basics for Biosystems of the Cell exercise work Maija Nevala, BIO, 67485U 16.1.2008 1. Preface When faced with an unknown protein, scientists
Protein Structure Analysis and Verification Course S-114.2500 Basics for Biosystems of the Cell exercise work Maija Nevala, BIO, 67485U 16.1.2008 1. Preface When faced with an unknown protein, scientists
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Detection of Protein Binding Sites II
 Detection of Protein Binding Sites II Goal: Given a protein structure, predict where a ligand might bind Thomas Funkhouser Princeton University CS597A, Fall 2007 1hld Geometric, chemical, evolutionary
Detection of Protein Binding Sites II Goal: Given a protein structure, predict where a ligand might bind Thomas Funkhouser Princeton University CS597A, Fall 2007 1hld Geometric, chemical, evolutionary
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining
![Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining](/thumbs/91/107031261.jpg "Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining") Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
QSAR in Green Chemistry
 QSAR in Green Chemistry Activity Relationship QSAR is the acronym for Quantitative Structure-Activity Relationship Chemistry is based on the premise that similar chemicals will behave similarly The behavior/activity
QSAR in Green Chemistry Activity Relationship QSAR is the acronym for Quantitative Structure-Activity Relationship Chemistry is based on the premise that similar chemicals will behave similarly The behavior/activity
Bioinformatics. Dept. of Computational Biology & Bioinformatics
 Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
COMPARISON OF SIMILARITY METHOD TO IMPROVE RETRIEVAL PERFORMANCE FOR CHEMICAL DATA
 http://www.ftsm.ukm.my/apjitm Asia-Pacific Journal of Information Technology and Multimedia Jurnal Teknologi Maklumat dan Multimedia Asia-Pasifik Vol. 7 No. 1, June 2018: 91-98 e-issn: 2289-2192 COMPARISON
http://www.ftsm.ukm.my/apjitm Asia-Pacific Journal of Information Technology and Multimedia Jurnal Teknologi Maklumat dan Multimedia Asia-Pasifik Vol. 7 No. 1, June 2018: 91-98 e-issn: 2289-2192 COMPARISON
Drug Informatics for Chemical Genomics...
 Drug Informatics for Chemical Genomics... An Overview First Annual ChemGen IGERT Retreat Sept 2005 Drug Informatics for Chemical Genomics... p. Topics ChemGen Informatics The ChemMine Project Library Comparison
Drug Informatics for Chemical Genomics... An Overview First Annual ChemGen IGERT Retreat Sept 2005 Drug Informatics for Chemical Genomics... p. Topics ChemGen Informatics The ChemMine Project Library Comparison
Ignasi Belda, PhD CEO. HPC Advisory Council Spain Conference 2015
 Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Benchmarking of protein descriptor sets in proteochemometric modeling (part 2): modeling performance of 13 amino acid descriptor sets
: modeling performance of 13 amino acid descriptor sets") van Westen et al. Journal of Cheminformatics 2013, 5:42 RESEARCH ARTICLE Open Access Benchmarking of protein descriptor sets in proteochemometric modeling (part 2): modeling performance of 13 amino acid
van Westen et al. Journal of Cheminformatics 2013, 5:42 RESEARCH ARTICLE Open Access Benchmarking of protein descriptor sets in proteochemometric modeling (part 2): modeling performance of 13 amino acid
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Computational Molecular Biology (
 Computational Molecular Biology (http://cmgm cmgm.stanford.edu/biochem218/) Biochemistry 218/Medical Information Sciences 231 Douglas L. Brutlag, Lee Kozar Jimmy Huang, Josh Silverman Lecture Syllabus
Computational Molecular Biology (http://cmgm cmgm.stanford.edu/biochem218/) Biochemistry 218/Medical Information Sciences 231 Douglas L. Brutlag, Lee Kozar Jimmy Huang, Josh Silverman Lecture Syllabus
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Overview. Descriptors. Definition. Descriptors. Overview 2D-QSAR. Number Vector Function. Physicochemical property (log P) Atom
 Atom") verview D-QSAR Definition Examples Features counts Topological indices D fingerprints and fragment counts R-group descriptors ow good are D descriptors in practice? Summary Peter Gedeck ovartis Institutes
verview D-QSAR Definition Examples Features counts Topological indices D fingerprints and fragment counts R-group descriptors ow good are D descriptors in practice? Summary Peter Gedeck ovartis Institutes
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Functional Group Fingerprints CNS Chemistry Wilmington, USA
 Functional Group Fingerprints CS Chemistry Wilmington, USA James R. Arnold Charles L. Lerman William F. Michne James R. Damewood American Chemical Society ational Meeting August, 2004 Philadelphia, PA
Functional Group Fingerprints CS Chemistry Wilmington, USA James R. Arnold Charles L. Lerman William F. Michne James R. Damewood American Chemical Society ational Meeting August, 2004 Philadelphia, PA
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence
 Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
COMP 598 Advanced Computational Biology Methods & Research. Introduction. Jérôme Waldispühl School of Computer Science McGill University
 COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Quantitative Structure-Activity Relationship (QSAR) computational-drug-design.html
 computational-drug-design.html") Quantitative Structure-Activity Relationship (QSAR) http://www.biophys.mpg.de/en/theoretical-biophysics/ computational-drug-design.html 07.11.2017 Ahmad Reza Mehdipour 07.11.2017 Course Outline 1. 1.Ligand-
Quantitative Structure-Activity Relationship (QSAR) http://www.biophys.mpg.de/en/theoretical-biophysics/ computational-drug-design.html 07.11.2017 Ahmad Reza Mehdipour 07.11.2017 Course Outline 1. 1.Ligand-
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface