Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
|
|
|
- Claude Derek Young
- 5 years ago
- Views:
Transcription

1 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
2 The road to new drugs. How to find new hits? High Throughput Screening (HTS) Virtual Screening (VS) Integration HTS and VS Molecular docking. Considering protein flexibility. Structure-based drug design in practice: Influenza case study.
. is very expensive (~ 1 billion $).")
3 WHAT DO WE WANT? The goal of a drug is to modulate the function of its target receptor which result in a pharmacological effect in the human body. HOW DO WE GET THERE? Developing a new drug: is extremely difficult. takes a lot of time (> 10 years). is very expensive (~ 1 billion $). target compound pharmacological effect

4 CN F O N H S Target Discovery Lead Discovery Lead Optimization Pre-Clinical Development Clinical Development Registration Marketing & Sales RESEARCH DEVELOPMENT


5 CN F O N H S Bioinformatics Computer Aided Drug Design Target Discovery Lead Discovery Lead Optimization Target Validation In Vitro In Vivo Target Identification Assay Development Hit / Lead Candidate Development Candidate
6 Computer-Aided Drug Design (CADD) refers to the application of informatics methods within rational drug design, to discover, design and optimize biologically active compounds.
7 Target Discovery Target gene Ligands unknown TWO SCENARIOS Ligands unknown Target protein structure unknown high-throughput screening Target protein structure known




8


9 HTS is the most important source of new hits. Compound collection Pharmaceutical companies have screening libraries up to a few million compounds. Chemical space of drug-like molecules is > Building a good screening collection is crucial! Active compounds

10 In-house collection Available to buy Screening set? Everything Diverse selection Focused selection
11 DIVERSE SELECTION FOCUSED SELECTION Diverse selection: identify dissimilar compounds Focused selection: Identify similar compounds
12 Chemical Descriptor 1 DIVERSE SELECTION Chemical Descriptor 2 HOW CAN WE MEASURE SIMILARITY?

13 In the selection of screening compounds the Rule of Five is often used. It summarizes typical properties of known drugs. These rules are often used as a first filtering step. In 1997 Chris Lipinski observed for many drugs: molecular weight < 500 lipophilicity (LogP) < 5 H-bond donors < 5 H-bond acceptors < 10 rotatable bonds < 10
14 Chemical Descriptor 1 Chemical Descriptor 1 DIVERSE SELECTION FOCUSED SELECTION Chemical Descriptor 2 Chemical Descriptor 2

15 Sampling around known active sub-structures or structural fragments can improve the quality of the library. dopamine derivative
16 Chemical Descriptor 1 The use of chemical (or molecular) descriptors is based on the similar property principle. Molecules with similar structures and similar properties should also exhibit similar activity. Chemical Descriptor 2


17 Fingerprints consist of various descriptors encoded into bit strings. These descriptors can be fragments or the presence or absence of other properties. dopamine derivative
18 HOW CAN WE MEASURE SIMILARITY?


19 Tanimoto coefficient S AB x A x x AB B x AB x A = 8 x B = 6 x AB = 5 Note: this is just one of many different similarity measures! S AB


20 BEB HIV Protease inhibitor Tanimoto Similarity 0.60 PZQ NOT HIV Prot. inhibitor Tanimoto Similarity 0.49 VAC HIV Protease inhibitor MK1 HIV Protease inhibitor Tanimoto Similarity 0.63 A high Tanimoto Similarity can be useful for prioritization. However, no guarantees! TI3 NOT HIV. inhibitor TS Tanimoto Similarity 0.48 XN1 HIV Protease inhibitor Tanimoto Similarity 0.63 BEH HIV Protease inhibitor Tanimoto Similarity IN HIV Protease inhibitor Tanimoto Similarity 0.47
21 O 2 N CF 3 H N O OH Br O 2 N CF 3 O N H OH Br flutamide retro-flutamide flutamide retro-flutamide progesterone receptor 4 nm 6 nm glucocorticoid receptor 25 nm 38 nm androgen receptor 0.5 nm 55 nm
22 Despite being the major source of new hits, HTS has its drawbacks: It s expensive. In practice only accessible to industry. Logistical errors. e.g. frequent hitters Measurement errors. e.g. suboptimal readout Strategic errors. e.g. assay variability Compound collection Active compounds
23 Target Discovery Target gene Ligand(s) unknown TWO SCENARIOS Ligand(s) unknown Target protein structure unknown Target protein structure known high-throughput screening virtual screening

24 In Virtual Screening (VS) compounds are selected using computer programs to predict receptor binding. Compound database VS is much cheaper and is able to process much more compounds in less time. Experimental validation is however always required! Active compounds
25 A few success stories from virtual screening
26 STRUCTURE-BASED VS Predict the orientation (and affinity) of a small molecule binding to a protein target. Requires the availability of a 3D target structure! Structure-based virtual screening Compound database Compounds to purchase. Compounds from in-house library. Virtual compounds. Active compounds



27 Target protein Compound database Docking program Docking program Target-Compound complexes Active compounds
28 Despite its advantages VS also has its drawbacks: Experimental validation is always required. Protein structure errors. e.g. induced fit Sampling errors. e.g. faulty poses due to solvent Scoring errors. e.g. false positives / negatives Compound database Active compounds
29 Screening library Focused and sequential screening Virtual Screening High Throughput Screening Focused library Hits Hypothesis generation

30 Screening library Parallel and independent screening Virtual Screening VS hits Analysis High Throughput Screening Hits HTS hits
 Virtual Screening (VS)")

31 The road to new drugs. How to find new hits? High Throughput Screening (HTS) Virtual Screening (VS) Integration HTS and VS Molecular docking. Considering protein flexibility. Structure-based drug design in practice: Influenza case study.
32 The docking problem involves many degrees freedom: Translational. Rotational. Configurational (Ligand + Receptor!) Since the early eighties several docking algorithms have been devised. Target protein Docking program Compound These can be characterized by the number of degrees freedom that they ignore. Target-Compound complex
33 Ligand rotations Ligand translations Ligand flexibility Receptor flexibility Rigid body docking Flexible ligand docking Induced fit docking Fully flexible docking
34 A number of flexible ligand docking programs: Dock [Kuntz et al, J Mol Biol, 161: , 1982] Autodock [Morris et al, J Comput Chem, 19: , 1994] FlexX [Rarey et al, J Mol Biol, 261: , 1996] Gold [Jones et al, J Mol Biol, 267: , 1997] Glide [Friesner et al, J Med Chem, 47: , 2004]
35 Molecular docking typically consists of two separate stages: Target protein Compound 1. Exploration of conformational and configurational space. Sampling Docking program 2. Evaluation of the strength of the receptor-ligand interaction. Scoring Target-Compound complex
36 Prior to ligand placement, most docking programs will create a simplified description of the target binding site. Receptor This is typically done using simple geometry descriptors, like spheres or triangles. These geometrical descriptors are usually combined with chemical and electrostatic descriptors to guide ligand placement. Ligand
37 Example 1 Example 2
38 Docking programs generate a large number of different docking poses. In general one can distinguish two different scenarios: 1. Many different poses of the same ligand need to be ranked for accuracy. 2. Different poses of different ligands need to be ranked based on their receptor affinity The ideal scoring function works well in both cases
39 First principles scoring functions generally use a Molecular Mechanics force field. Such force fields typically contain intra-molecular terms: Bond lengths Bond angles Dihedral terms And inter-molecular terms: Van der Waals contacts (non-polar) Electrostatic interactions (polar) E bind = E intra + E nonpolar + E polar
 + ΔG non-polar Σ f(complex) + ΔG rot N rotatable-bonds ΔG 0, ΔG polar, ΔG non-polar, and ΔG rot empirically parameterized weights.")
40 Empirical scoring functions have been developed to score ligands very rapidly. ΔG bind = ΔG 0 + ΔG polar Σ f(complex) + ΔG non-polar Σ f(complex) + ΔG rot N rotatable-bonds ΔG 0, ΔG polar, ΔG non-polar, and ΔG rot empirically parameterized weights. are f(complex) is a penalty function aimed at penalizing any unfavorable interaction geometries.
41 In practice molecular docking is generally used to answer two different types of questions: + docking 1. Which compounds in my compound collection could be active on receptor A? Receptor A Compound collection Actives? 2. How does the complex look that is formed by receptor A and compound B? + docking Receptor A Compound B Complex?
42 The road to new drugs. How to find new hits? High Throughput Screening (HTS) Virtual Screening (VS) Integration HTS and VS Molecular docking. Considering protein flexibility. Structure-based drug design in practice: Influenza case study.
 and optimizing lead compounds.")
43 Drug targets are flexible biomolecules and their dynamics play an important role in ligand binding. Insight in receptor flexibility can be valuable when interpreting structure activity relationships (SAR) and optimizing lead compounds. 1 ligand 1 receptor conformation 10 ligands 10 receptor conformations Predicting ligand binding in flexible binding sites is however problematic!
44 LOCK AND KEY INDUCED FIT Receptor A + X Receptor A + Y Complex A-X Complex A -Y

45 Introduce flexibility Generate complexes Optimize complexes Compound Target Induced fit complex





46 flexible residue selection binding site rotamer sampling binding site His/Gln/Asn sampling Single receptor structure Selection is based on structural analyses of: apo structures other holo structures temperature factors Gln Asn His flexible ensemble

47 The Fleksy approach docks into an ensemble of receptor structures. The approach is based on a united protein description generated from an ensemble of protein structures. In our case the ensemble contains the generated set of side chain rotamers and sampled Asn/Gln/His side chains.



48 crystal structure apo form? crystal structure holo form identify flexible residues sample Asn/His 2 Asn states 8 His states 15 side chain rotamers

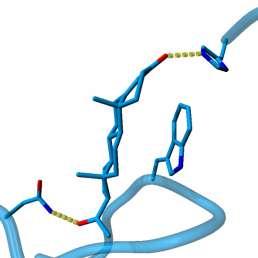
49 RMSD 0.6 Å
50 Target Discovery Target gene Ligand(s) unknown known! TWO SCENARIOS Ligand(s) unknown Target protein structure unknown Target protein structure known high-throughput screening virtual screening by docking
51 SBDD Structure based drug design relies on structural knowledge of the target protein to design and optimize lead compounds. This knowledge is obtained from either experimental structures or computational predictions. A requirement is the availability of receptor structures: NMR spectroscopy X-ray crystallography In practice protein X-ray crystallography is the major source of structural information.

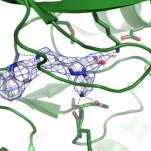

52 Protein Expression and Purification Crystallisation Data Collection Structure Building Refinement Analysis / Design

53 A receptor structure can often explain: Binding Specificity Inhibition Flexibility Reaction mechanism And it allows predictions to be made!


54 1918 Influenza Epidemic Influenza Virus
55 NEURAMIDASE POCKET SIALIC ACID
56 RELENZA SIALIC ACID
57 RELENZA doses in NL TAMIFLU

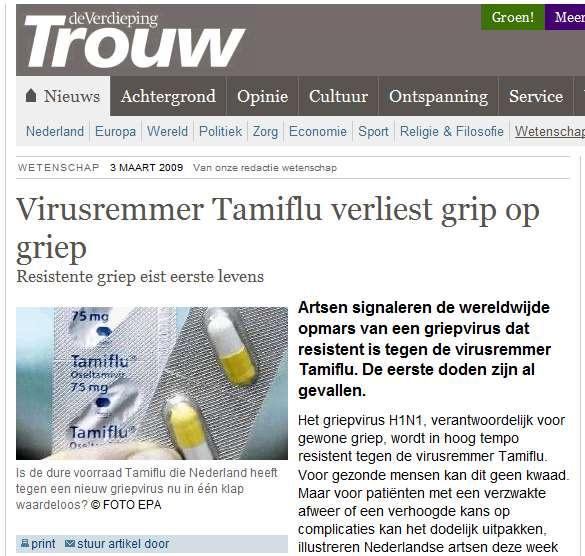
58 Trouw 3 maart 2009
59 RELENZA SIALIC ACID TAMIFLU WT K i = 1.0 H274Y K i = 1.9 WT K i = 1.0 H274Y K i =265 H274Y H274Y
60 UMC St. Radboud Jacob de Vlieg Gijs Schaftenaar Software BiosolveIT Accelrys Molecular Networks Schering-Plough Scott Lusher Markus Wagener Ross McGuire Hans Raaijmakers Funding Dutch Organization for Scientific Research (NWO)
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
György M. Keserű H2020 FRAGNET Network Hungarian Academy of Sciences
 Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Protein-Ligand Docking Evaluations
 Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Early Stages of Drug Discovery in the Pharmaceutical Industry
 Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Protein-Ligand Docking Methods
 Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors
 MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery
 21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Advanced Medicinal Chemistry SLIDES B
 Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Protein-Ligand Docking
 Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
BUDE. A General Purpose Molecular Docking Program Using OpenCL. Richard B Sessions
 BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
Cheminformatics platform for drug discovery application
 EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
Ignasi Belda, PhD CEO. HPC Advisory Council Spain Conference 2015
 Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Protein-Ligand Docking Methods
 Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Structure-based maximal affinity model predicts small-molecule druggability
 Structure-based maximal affinity model predicts small-molecule druggability Alan Cheng alan.cheng@amgen.com IMA Workshop (Jan 17, 2008) Druggability prediction Introduction Affinity model Some results
Structure-based maximal affinity model predicts small-molecule druggability Alan Cheng alan.cheng@amgen.com IMA Workshop (Jan 17, 2008) Druggability prediction Introduction Affinity model Some results
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
High Throughput In-Silico Screening Against Flexible Protein Receptors
 John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
FRAUNHOFER IME SCREENINGPORT
 FRAUNHOFER IME SCREENINGPORT Design of screening projects General remarks Introduction Screening is done to identify new chemical substances against molecular mechanisms of a disease It is a question of
FRAUNHOFER IME SCREENINGPORT Design of screening projects General remarks Introduction Screening is done to identify new chemical substances against molecular mechanisms of a disease It is a question of
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Structure-Based Drug Discovery An Overview
 Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
Author Index Volume
 Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
What is Protein-Ligand Docking?
 MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
Virtual screening for drug discovery. Markus Lill Purdue University
 Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Targeting protein-protein interactions: A hot topic in drug discovery
 Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Cross Discipline Analysis made possible with Data Pipelining. J.R. Tozer SciTegic
 Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Protein Structure Prediction and Protein-Ligand Docking
 Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Plan. Day 2: Exercise on MHC molecules.
 Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors
 A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
Computer Graphics Applications on Molecular Biology and Drug Design
 Computer Graphics Applications on Molecular Biology and Drug Design Katerina PERDIKURI 1, Athanasios TSAKALIDIS 1 1 Department of Computer Engineering and Informatics University of Patras,26500 Patras,
Computer Graphics Applications on Molecular Biology and Drug Design Katerina PERDIKURI 1, Athanasios TSAKALIDIS 1 1 Department of Computer Engineering and Informatics University of Patras,26500 Patras,
Binding Response: A Descriptor for Selecting Ligand Binding Site on Protein Surfaces
 J. Chem. Inf. Model. 2007, 47, 2303-2315 2303 Binding Response: A Descriptor for Selecting Ligand Binding Site on Protein Surfaces Shijun Zhong and Alexander D. MacKerell, Jr.* Computer-Aided Drug Design
J. Chem. Inf. Model. 2007, 47, 2303-2315 2303 Binding Response: A Descriptor for Selecting Ligand Binding Site on Protein Surfaces Shijun Zhong and Alexander D. MacKerell, Jr.* Computer-Aided Drug Design
Introduction to FBDD Fragment screening methods and library design
 Introduction to FBDD Fragment screening methods and library design Samantha Hughes, PhD Fragments 2013 RSC BMCS Workshop 3 rd March 2013 Copyright 2013 Galapagos NV Why fragment screening methods? Guess
Introduction to FBDD Fragment screening methods and library design Samantha Hughes, PhD Fragments 2013 RSC BMCS Workshop 3 rd March 2013 Copyright 2013 Galapagos NV Why fragment screening methods? Guess
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification
 Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Combining Docking and Molecular Dynamic Simulations in Drug Design
 Combining Docking and Molecular Dynamic Simulations in Drug Design Hernán Alonso, 1 Andrey A. Bliznyuk, 2 Jill E. Gready 1 1 Computational Proteomics Group, John Curtin School of Medical Research, The
Combining Docking and Molecular Dynamic Simulations in Drug Design Hernán Alonso, 1 Andrey A. Bliznyuk, 2 Jill E. Gready 1 1 Computational Proteomics Group, John Curtin School of Medical Research, The
FRAGMENT SCREENING IN LEAD DISCOVERY BY WEAK AFFINITY CHROMATOGRAPHY (WAC )
") FRAGMENT SCREENING IN LEAD DISCOVERY BY WEAK AFFINITY CHROMATOGRAPHY (WAC ) SARomics Biostructures AB & Red Glead Discovery AB Medicon Village, Lund, Sweden Fragment-based lead discovery The basic idea:
FRAGMENT SCREENING IN LEAD DISCOVERY BY WEAK AFFINITY CHROMATOGRAPHY (WAC ) SARomics Biostructures AB & Red Glead Discovery AB Medicon Village, Lund, Sweden Fragment-based lead discovery The basic idea:
Virtual affinity fingerprints in drug discovery: The Drug Profile Matching method
 Ágnes Peragovics Virtual affinity fingerprints in drug discovery: The Drug Profile Matching method PhD Theses Supervisor: András Málnási-Csizmadia DSc. Associate Professor Structural Biochemistry Doctoral
Ágnes Peragovics Virtual affinity fingerprints in drug discovery: The Drug Profile Matching method PhD Theses Supervisor: András Málnási-Csizmadia DSc. Associate Professor Structural Biochemistry Doctoral
Mechanistic insight into inhibition of two-component system signaling
 Supporting Information Mechanistic insight into inhibition of two-component system signaling Samson Francis, a Kaelyn E. Wilke, a Douglas E. Brown a and Erin E. Carlson a,b* a Department of Chemistry,
Supporting Information Mechanistic insight into inhibition of two-component system signaling Samson Francis, a Kaelyn E. Wilke, a Douglas E. Brown a and Erin E. Carlson a,b* a Department of Chemistry,
In Silico Investigation of Off-Target Effects
 PHARMA & LIFE SCIENCES WHITEPAPER In Silico Investigation of Off-Target Effects STREAMLINING IN SILICO PROFILING In silico techniques require exhaustive data and sophisticated, well-structured informatics
PHARMA & LIFE SCIENCES WHITEPAPER In Silico Investigation of Off-Target Effects STREAMLINING IN SILICO PROFILING In silico techniques require exhaustive data and sophisticated, well-structured informatics
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining
![Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining](/thumbs/91/107031261.jpg "Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining") Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit
 October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
COMPARISON OF SIMILARITY METHOD TO IMPROVE RETRIEVAL PERFORMANCE FOR CHEMICAL DATA
 http://www.ftsm.ukm.my/apjitm Asia-Pacific Journal of Information Technology and Multimedia Jurnal Teknologi Maklumat dan Multimedia Asia-Pasifik Vol. 7 No. 1, June 2018: 91-98 e-issn: 2289-2192 COMPARISON
http://www.ftsm.ukm.my/apjitm Asia-Pacific Journal of Information Technology and Multimedia Jurnal Teknologi Maklumat dan Multimedia Asia-Pasifik Vol. 7 No. 1, June 2018: 91-98 e-issn: 2289-2192 COMPARISON
ENERGY MINIMIZATION AND CONFORMATION SEARCH ANALYSIS OF TYPE-2 ANTI-DIABETES DRUGS
 Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Supplementary Material. Table of Contents. Evaluation of the test compounds using Lipinski s rule 2
 Supplementary Material Table of Contents Pages Evaluation of the test compounds using Lipinski s rule 2 Characterization of compounds PB3, PB6, PB10, PB11, PB12, PB14, PB15, PB17, PB19 2-6 Docking Methodology
Supplementary Material Table of Contents Pages Evaluation of the test compounds using Lipinski s rule 2 Characterization of compounds PB3, PB6, PB10, PB11, PB12, PB14, PB15, PB17, PB19 2-6 Docking Methodology
Implementation of novel tools to facilitate fragment-based drug discovery by NMR:
 Implementation of novel tools to facilitate fragment-based drug discovery by NMR: Automated analysis of large sets of ligand-observed NMR binding data and 19 F methods Andreas Lingel Global Discovery Chemistry
Implementation of novel tools to facilitate fragment-based drug discovery by NMR: Automated analysis of large sets of ligand-observed NMR binding data and 19 F methods Andreas Lingel Global Discovery Chemistry
BioSolveIT. A Combinatorial Approach for Handling of Protonation and Tautomer Ambiguities in Docking Experiments
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
Supporting Information
 S-1 Supporting Information Flaviviral protease inhibitors identied by fragment-based library docking into a structure generated by molecular dynamics Dariusz Ekonomiuk a, Xun-Cheng Su b, Kiyoshi Ozawa
S-1 Supporting Information Flaviviral protease inhibitors identied by fragment-based library docking into a structure generated by molecular dynamics Dariusz Ekonomiuk a, Xun-Cheng Su b, Kiyoshi Ozawa
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites. J. Andrew Surface
 Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses
 Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today
 est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
Docking: Modeling the structure of the ligand protein complex
 1/41 Docking: Modeling the structure of the ligand protein complex G. Marcou 1, E. Kellenberger 2 (author) 1 Faculté de Chimie, UMR7140 2 Faculté de Pharmacie, UMR 7200, Illkirch introduction 3D Protein
1/41 Docking: Modeling the structure of the ligand protein complex G. Marcou 1, E. Kellenberger 2 (author) 1 Faculté de Chimie, UMR7140 2 Faculté de Pharmacie, UMR 7200, Illkirch introduction 3D Protein
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows
 MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
Similarity methods for ligandbased virtual screening
 Similarity methods for ligandbased virtual screening Peter Willett, University of Sheffield Computers in Scientific Discovery 5, 22 nd July 2010 Overview Molecular similarity and its use in virtual screening
Similarity methods for ligandbased virtual screening Peter Willett, University of Sheffield Computers in Scientific Discovery 5, 22 nd July 2010 Overview Molecular similarity and its use in virtual screening
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
Fragment based drug discovery in teams of medicinal and computational chemists. Carsten Detering
 Fragment based drug discovery in teams of medicinal and computational chemists Carsten Detering BioSolveIT Quick Facts Founded in 2001 by the developers of FlexX ~20 people Core expertise: docking, screening,
Fragment based drug discovery in teams of medicinal and computational chemists Carsten Detering BioSolveIT Quick Facts Founded in 2001 by the developers of FlexX ~20 people Core expertise: docking, screening,
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS
 EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS PETER GUND Pharmacopeia Inc., CN 5350 Princeton, NJ 08543, USA pgund@pharmacop.com Empirical and theoretical approaches to drug discovery have often
EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS PETER GUND Pharmacopeia Inc., CN 5350 Princeton, NJ 08543, USA pgund@pharmacop.com Empirical and theoretical approaches to drug discovery have often
ESPRESSO (Extremely Speedy PRE-Screening method with Segmented compounds) 1
 1") Vol.2016-MPS-108 o.18 Vol.2016-BI-46 o.18 ESPRESS 1,4,a) 2,4 2,4 1,3 1,3,4 1,3,4 - ESPRESS (Extremely Speedy PRE-Screening method with Segmented cmpounds) 1 Glide HTVS ESPRESS 2,900 200 ESPRESS: An ultrafast
Vol.2016-MPS-108 o.18 Vol.2016-BI-46 o.18 ESPRESS 1,4,a) 2,4 2,4 1,3 1,3,4 1,3,4 - ESPRESS (Extremely Speedy PRE-Screening method with Segmented cmpounds) 1 Glide HTVS ESPRESS 2,900 200 ESPRESS: An ultrafast
Cheminformatics Role in Pharmaceutical Industry. Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS
 Cheminformatics Role in Pharmaceutical Industry Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS Agenda The big picture for pharmaceutical industry Current technological/scientific issues Types
Cheminformatics Role in Pharmaceutical Industry Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS Agenda The big picture for pharmaceutical industry Current technological/scientific issues Types
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
Development of a Structure Generator to Explore Target Areas on Chemical Space
 Development of a Structure Generator to Explore Target Areas on Chemical Space Kimito Funatsu Department of Chemical System Engineering, This materials will be published on Molecular Informatics Drug Development
Development of a Structure Generator to Explore Target Areas on Chemical Space Kimito Funatsu Department of Chemical System Engineering, This materials will be published on Molecular Informatics Drug Development
Protein Structures. 11/19/2002 Lecture 24 1
 Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Robust Search Methods for Rational Drug Design Applications
 Robust Search Methods for Rational Drug Design Applications by Bashir S. Sadjad A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Doctor of Philosophy
Robust Search Methods for Rational Drug Design Applications by Bashir S. Sadjad A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Doctor of Philosophy
DivCalc: A Utility for Diversity Analysis and Compound Sampling
 Molecules 2002, 7, 657-661 molecules ISSN 1420-3049 http://www.mdpi.org DivCalc: A Utility for Diversity Analysis and Compound Sampling Rajeev Gangal* SciNova Informatics, 161 Madhumanjiri Apartments,
Molecules 2002, 7, 657-661 molecules ISSN 1420-3049 http://www.mdpi.org DivCalc: A Utility for Diversity Analysis and Compound Sampling Rajeev Gangal* SciNova Informatics, 161 Madhumanjiri Apartments,
Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation
 Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation Charles Blundell charles.blundell@c4xdiscovery.com www.c4xdiscovery.com Rigid: single
Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation Charles Blundell charles.blundell@c4xdiscovery.com www.c4xdiscovery.com Rigid: single
MOLECULAR RECOGNITION DOCKING ALGORITHMS. Natasja Brooijmans 1 and Irwin D. Kuntz 2
 Annu. Rev. Biophys. Biomol. Struct. 2003. 32:335 73 doi: 10.1146/annurev.biophys.32.110601.142532 Copyright c 2003 by Annual Reviews. All rights reserved First published online as a Review in Advance on
Annu. Rev. Biophys. Biomol. Struct. 2003. 32:335 73 doi: 10.1146/annurev.biophys.32.110601.142532 Copyright c 2003 by Annual Reviews. All rights reserved First published online as a Review in Advance on
- Introduction of x-ray crystallography: what it s used for, how it works, applications in science - Different methods used to generate data - Case
 - Introduction of x-ray crystallography: what it s used for, how it works, applications in science - Different methods used to generate data - Case studies emphasizing the importance of the technique -
- Introduction of x-ray crystallography: what it s used for, how it works, applications in science - Different methods used to generate data - Case studies emphasizing the importance of the technique -
Supplementary Methods
 Supplementary Methods MMPBSA Free energy calculation Molecular Mechanics/Poisson Boltzmann Surface Area (MM/PBSA) has been widely used to calculate binding free energy for protein-ligand systems (1-7).
Supplementary Methods MMPBSA Free energy calculation Molecular Mechanics/Poisson Boltzmann Surface Area (MM/PBSA) has been widely used to calculate binding free energy for protein-ligand systems (1-7).
COMBINATORIAL CHEMISTRY: CURRENT APPROACH
 COMBINATORIAL CHEMISTRY: CURRENT APPROACH Dwivedi A. 1, Sitoke A. 2, Joshi V. 3, Akhtar A.K. 4* and Chaturvedi M. 1, NRI Institute of Pharmaceutical Sciences, Bhopal, M.P.-India 2, SRM College of Pharmacy,
COMBINATORIAL CHEMISTRY: CURRENT APPROACH Dwivedi A. 1, Sitoke A. 2, Joshi V. 3, Akhtar A.K. 4* and Chaturvedi M. 1, NRI Institute of Pharmaceutical Sciences, Bhopal, M.P.-India 2, SRM College of Pharmacy,
Data Mining in the Chemical Industry. Overview of presentation
 Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Ultrafast Shape Recognition to Search Compound Databases for Similar Molecular Shapes
 Ultrafast Shape Recognition to Search Compound Databases for Similar Molecular Shapes PEDRO J. BALLESTER, W. GRAHAM RICHARDS Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks
Ultrafast Shape Recognition to Search Compound Databases for Similar Molecular Shapes PEDRO J. BALLESTER, W. GRAHAM RICHARDS Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application