Biologically Relevant Molecular Comparisons. Mark Mackey
|
|
|
- Wilfrid McKinney
- 5 years ago
- Views:
Transcription

1 Biologically Relevant Molecular Comparisons Mark Mackey
2 Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime
3 About Cresset > Specialist modelling software and services company > Started in 2001 > Virtual screening service run on >80 projects with 80% success rate > Software used in big pharma and biotechs > Focus on ligand-based drug design, virtual screening, QSAR
 2D 3D Molecular Electrostatic Potential (MEP) = Positive = Negative = Shape = Hydrophobic Field")
4 Field Points Condensed representation of electrostatic, hydrophobic and shape properties ( protein s view ) > Molecular Field Extrema ( Field Points ) 2D 3D Molecular Electrostatic Potential (MEP) = Positive = Negative = Shape = Hydrophobic Field Points
 = Positive = Negative = Shape =")
5 Explanatory Power of Fields Field points give you new insights into your molecule +ve ionic H H H N O O Aromatic p cloud H acceptor H-bond acceptor N H O Aromatic inplane H donor H-bond donor Hydrophobes Field point size show importance -ve ionic Stickiest surfaces (high vdw) = Positive = Negative = Shape = Hydrophobic

6 XEDs make Fields Work > Field patterns from Cresset s proprietary XED force field reproduce experimental results Experimental Using XEDs Not using XEDs Interaction of Acetone and Any-OH from small molecule crystal structures XED adds p-orbitals to get better representation of atoms
7 Alignment and scoring > Fast and accurate 3D field similarity method > J. Chem. Inf. Model., 2006, 46 (2), pp > J. Chem. Inf. Model., 2008, 48 (11), pp > Uses field points + full analytical molecular fields > Pharmacophore identification > Virtual screening > Bioisosteres > Alignment > SAR > QSAR
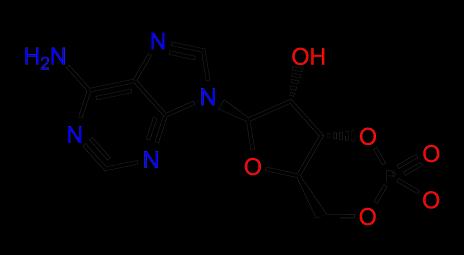
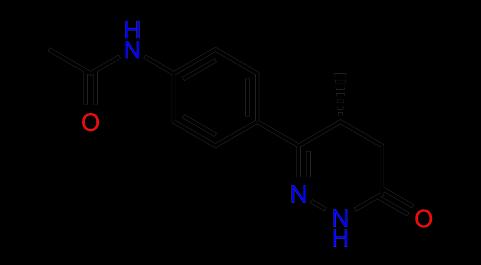
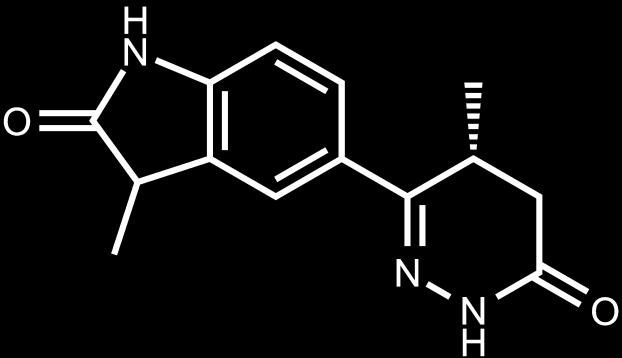
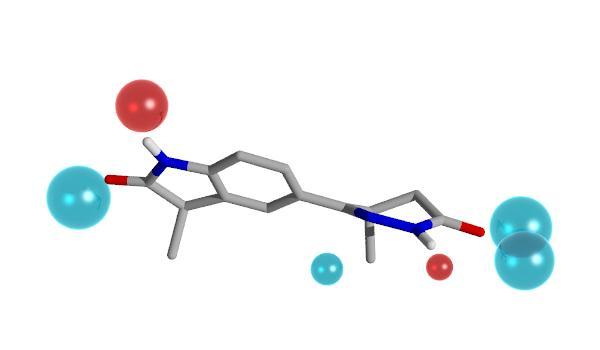

8 Bioisosteres PDE III Biologically relevant method for comparing molecules Bioisosteres Bioisosteric groups
9 Applications of Fields > Hit Identification > Evaluating scope of new targets > Discovering hit compounds with better properties > Acquiring information about the target s active site without need for X-ray crystal structure > Patent busting
10 Applications of Fields > Hit/ Lead Optimization > Efficiently discovering better active compounds > Designing out unwanted ADME and toxicity properties in hits/leads > Gaining valuable insight into Structure Activity Relationships > Back-up Candidates > Identifying new core templates (chemotypes) with good properties > Patent Protection > Patent Exclusion
11 Cresset Products > Diverse lead like hits > Large library design > HTS rescue > New directions > Patent protection > Patent busting > Understand SAR > Improve design > Singles > Libraries > Understand binding > Relate chemical series > Pharmacophores
12 Product Deployment > Linux cluster > Web interface > FieldView > Desktop GUI, Win, Linux > Desktop GUI > Windows, Linux, MacOS > Command line > Workflow Spring 2011 > Desktop GUI > Windows, Linux, MacOS > Command line Spring 2011 > Workflow Spring 2011 > Desktop GUI > Windows, Linux > Command Line Fall 2011 > Workflow Fall 2011
13 KNIME support > Initial set of KNIME nodes in beta > Xedex > Biologically relevant diverse conformations > Xedmin > Minimisation using XED force field > FieldView > Cresset molecule viewer (free!) > Fieldalign > Molecular alignment and 3D similarity > Parallelisable across cluster > Fieldstere > Intelligent bioisosteres > No SDF reader(!)
14 Future > FieldTemplater > Model bioactive conformations from ligand data > FieldScreen > Field-based VS on large databases > 3D QSAR > Integrated > Data matrix
15 Questions welcomed
16 Comparing Molecules > Clique based initial alignment > Uses the distance matrix of Field Points > Detailed score > Single point > Uses Field-Point on Field Scoring > Optional simplex based optimisation of alignment > Rigid body - no bonds twisted > Uses detailed score as optimisation parameter


17 Field Alignment Imidazole N-methyl acetamide


18 Field Scoring Imidazole N-methyl acetamide To score a particular alignment, we use the field points of molecule 1 to sample the actual field of molecule 2 Cheeseright et al, J. Chem Inf. Mod., 2006, 665

19 Field Scoring Imidazole N-methyl acetamide To score a particular alignment, we use the field points of molecule 1 to sample the actual field of molecule 2 and vice-versa Cheeseright et al, J. Chem Inf. Mod., 2006, 665
20 Similarity Scoring function - 1 E A B size( fp A ) F B ( position ( fp A )) fp A A B
21 Similarity Scoring function - 2 E E A B AB E fp A A B size( E 2 fp A B A ) F B ( position ( S AB E 2E AA E A B = Energy, S AB = Similiarity fp A AB E )) BB A B
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Introduction to Spark
 1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Cres s et Bi omol ecul a r Di s covery Ltd, a l l ri ghts res erved.
 10.5 2017 Cres s et Bi omol ecul a r Di s covery Ltd, a l l ri ghts res erved. Table of contents Introduction... 8 What are field points?... 8 Interpretation of field point patterns... 9 About this document...
10.5 2017 Cres s et Bi omol ecul a r Di s covery Ltd, a l l ri ghts res erved. Table of contents Introduction... 8 What are field points?... 8 Interpretation of field point patterns... 9 About this document...
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Design and Synthesis of the Comprehensive Fragment Library
 YOUR INNOVATIVE CHEMISTRY PARTNER IN DRUG DISCOVERY Design and Synthesis of the Comprehensive Fragment Library A 3D Enabled Library for Medicinal Chemistry Discovery Warren S Wade 1, Kuei-Lin Chang 1,
YOUR INNOVATIVE CHEMISTRY PARTNER IN DRUG DISCOVERY Design and Synthesis of the Comprehensive Fragment Library A 3D Enabled Library for Medicinal Chemistry Discovery Warren S Wade 1, Kuei-Lin Chang 1,
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Zurich, February 2014 The Schrödinger extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Zurich, February 2014 The Schrödinger extensions
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Ignasi Belda, PhD CEO. HPC Advisory Council Spain Conference 2015
 Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Practical QSAR and Library Design: Advanced tools for research teams
 DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS
 EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS PETER GUND Pharmacopeia Inc., CN 5350 Princeton, NJ 08543, USA pgund@pharmacop.com Empirical and theoretical approaches to drug discovery have often
EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS PETER GUND Pharmacopeia Inc., CN 5350 Princeton, NJ 08543, USA pgund@pharmacop.com Empirical and theoretical approaches to drug discovery have often
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
Cheminformatics Role in Pharmaceutical Industry. Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS
 Cheminformatics Role in Pharmaceutical Industry Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS Agenda The big picture for pharmaceutical industry Current technological/scientific issues Types
Cheminformatics Role in Pharmaceutical Industry Randal Chen Ph.D. Abbott Laboratories Aug. 23, 2004 ACS Agenda The big picture for pharmaceutical industry Current technological/scientific issues Types
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Bridging the Dimensions:
 Bridging the Dimensions: Seamless Integration of 3D Structure-based Design and 2D Structure-activity Relationships to Guide Medicinal Chemistry ACS Spring National Meeting. COMP, March 13 th 2016 Marcus
Bridging the Dimensions: Seamless Integration of 3D Structure-based Design and 2D Structure-activity Relationships to Guide Medicinal Chemistry ACS Spring National Meeting. COMP, March 13 th 2016 Marcus
Navigation in Chemical Space Towards Biological Activity. Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland
 Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Data Mining in the Chemical Industry. Overview of presentation
 Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Marvin. Sketching, viewing and predicting properties with Marvin - features, tips and tricks. Gyorgy Pirok. Solutions for Cheminformatics
 Marvin Sketching, viewing and predicting properties with Marvin - features, tips and tricks Gyorgy Pirok Solutions for Cheminformatics The Marvin family The Marvin toolkit provides web-enabled components
Marvin Sketching, viewing and predicting properties with Marvin - features, tips and tricks Gyorgy Pirok Solutions for Cheminformatics The Marvin family The Marvin toolkit provides web-enabled components
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Structure-Activity Modeling - QSAR. Uwe Koch
 Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
MSc Drug Design. Module Structure: (15 credits each) Lectures and Tutorials Assessment: 50% coursework, 50% unseen examination.
 Lectures and Tutorials Assessment: 50% coursework, 50% unseen examination.") Module Structure: (15 credits each) Lectures and Assessment: 50% coursework, 50% unseen examination. Module Title Module 1: Bioinformatics and structural biology as applied to drug design MEDC0075 In the
Module Structure: (15 credits each) Lectures and Assessment: 50% coursework, 50% unseen examination. Module Title Module 1: Bioinformatics and structural biology as applied to drug design MEDC0075 In the
Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery
 21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining
![Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining](/thumbs/91/107031261.jpg "Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining") Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Targeting protein-protein interactions: A hot topic in drug discovery
 Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit
 October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
AMRI COMPOUND LIBRARY CONSORTIUM: A NOVEL WAY TO FILL YOUR DRUG PIPELINE
 AMRI COMPOUD LIBRARY COSORTIUM: A OVEL WAY TO FILL YOUR DRUG PIPELIE Muralikrishna Valluri, PhD & Douglas B. Kitchen, PhD Summary The creation of high-quality, innovative small molecule leads is a continual
AMRI COMPOUD LIBRARY COSORTIUM: A OVEL WAY TO FILL YOUR DRUG PIPELIE Muralikrishna Valluri, PhD & Douglas B. Kitchen, PhD Summary The creation of high-quality, innovative small molecule leads is a continual
Analyzing Building Blocks Diversity for DNA Encoded Library Design. Cresset User Group Meeting Nik Stiefl & Finton Sirockin, Novartis
 Analyzing Building Blocks Diversity for DA Encoded Library Design Cresset User Group Meeting ik Stiefl & Finton Sirockin, ovartis 2016.06.16 Outline DA Encoded Libraries (DEL) Building blocks selection
Analyzing Building Blocks Diversity for DA Encoded Library Design Cresset User Group Meeting ik Stiefl & Finton Sirockin, ovartis 2016.06.16 Outline DA Encoded Libraries (DEL) Building blocks selection
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Early Stages of Drug Discovery in the Pharmaceutical Industry
 Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Cross Discipline Analysis made possible with Data Pipelining. J.R. Tozer SciTegic
 Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Medicinal Chemistry/ CHEM 458/658 Chapter 3- SAR and QSAR
 Medicinal Chemistry/ CHEM 458/658 Chapter 3- SAR and QSAR Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Introduction Structure-Activity Relationship (SAR) - similar
Medicinal Chemistry/ CHEM 458/658 Chapter 3- SAR and QSAR Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Introduction Structure-Activity Relationship (SAR) - similar
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Database Speaks. Ling-Kang Liu ( 劉陵崗 ) Institute of Chemistry, Academia Sinica Nangang, Taipei 115, Taiwan
 Institute of Chemistry, Academia Sinica Nangang, Taipei 115, Taiwan") Database Speaks Ling-Kang Liu ( 劉陵崗 ) Institute of Chemistry, Academia Sinica Nangang, Taipei 115, Taiwan Email: liuu@chem.sinica.edu.tw 1 OUTLINES -- Personal experiences Publication types Secondary publication
Database Speaks Ling-Kang Liu ( 劉陵崗 ) Institute of Chemistry, Academia Sinica Nangang, Taipei 115, Taiwan Email: liuu@chem.sinica.edu.tw 1 OUTLINES -- Personal experiences Publication types Secondary publication
Chemoinformatics and information management. Peter Willett, University of Sheffield, UK
 Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Molecular Modelling. Computational Chemistry Demystified. RSC Publishing. Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK
 Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation
 Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation Charles Blundell charles.blundell@c4xdiscovery.com www.c4xdiscovery.com Rigid: single
Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation Charles Blundell charles.blundell@c4xdiscovery.com www.c4xdiscovery.com Rigid: single
Farewell, PipelinePilot Migrating the Exquiron cheminformatics platform to KNIME and the ChemAxon technology
 Farewell, PipelinePilot Migrating the Exquiron cheminformatics platform to KNIME and the ChemAxon technology Serge P. Parel, PhD ChemAxon User Group Meeting, Budapest 21 st May, 2014 Outline Exquiron Who
Farewell, PipelinePilot Migrating the Exquiron cheminformatics platform to KNIME and the ChemAxon technology Serge P. Parel, PhD ChemAxon User Group Meeting, Budapest 21 st May, 2014 Outline Exquiron Who
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
The Changing Requirements for Informatics Systems During the Growth of a Collaborative Drug Discovery Service Company. Sally Rose BioFocus plc
 The Changing Requirements for Informatics Systems During the Growth of a Collaborative Drug Discovery Service Company Sally Rose BioFocus plc Overview History of BioFocus and acquisition of CDD Biological
The Changing Requirements for Informatics Systems During the Growth of a Collaborative Drug Discovery Service Company Sally Rose BioFocus plc Overview History of BioFocus and acquisition of CDD Biological
CSD. Unlock value from crystal structure information in the CSD
 CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
Fast similarity searching making the virtual real. Stephen Pickett, GSK
 Fast similarity searching making the virtual real Stephen Pickett, GSK Introduction Introduction to similarity searching Use cases Why is speed so crucial? Why MadFast? Some performance stats Implementation
Fast similarity searching making the virtual real Stephen Pickett, GSK Introduction Introduction to similarity searching Use cases Why is speed so crucial? Why MadFast? Some performance stats Implementation
Part 6. 3D Pharmacophore Modeling
 279 Part 6 3D Pharmacophore Modeling 281 20 3D Pharmacophore Modeling Techniques in Computer Aided Molecular Design Using LigandScout Thomas Seidel, Sharon D. Bryant, Gökhan Ibis, Giulio Poli, and Thierry
279 Part 6 3D Pharmacophore Modeling 281 20 3D Pharmacophore Modeling Techniques in Computer Aided Molecular Design Using LigandScout Thomas Seidel, Sharon D. Bryant, Gökhan Ibis, Giulio Poli, and Thierry
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
COMBINATORIAL CHEMISTRY IN A HISTORICAL PERSPECTIVE
 NUE FEATURE T R A N S F O R M I N G C H A L L E N G E S I N T O M E D I C I N E Nuevolution Feature no. 1 October 2015 Technical Information COMBINATORIAL CHEMISTRY IN A HISTORICAL PERSPECTIVE A PROMISING
NUE FEATURE T R A N S F O R M I N G C H A L L E N G E S I N T O M E D I C I N E Nuevolution Feature no. 1 October 2015 Technical Information COMBINATORIAL CHEMISTRY IN A HISTORICAL PERSPECTIVE A PROMISING
Reaxys Medicinal Chemistry Fact Sheet
 R&D SOLUTIONS FOR PHARMA & LIFE SCIENCES Reaxys Medicinal Chemistry Fact Sheet Essential data for lead identification and optimization Reaxys Medicinal Chemistry empowers early discovery in drug development
R&D SOLUTIONS FOR PHARMA & LIFE SCIENCES Reaxys Medicinal Chemistry Fact Sheet Essential data for lead identification and optimization Reaxys Medicinal Chemistry empowers early discovery in drug development
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Different conformations of the drugs within the virtual library of FDA approved drugs will be generated.
 Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
KNIME-based scoring functions in Muse 3.0. KNIME User Group Meeting 2013 Fabian Bös
 KIME-based scoring functions in Muse 3.0 KIME User Group Meeting 2013 Fabian Bös Certara Mission: End-to-End Model-Based Drug Development Certara was formed by acquiring and integrating Tripos, Pharsight,
KIME-based scoring functions in Muse 3.0 KIME User Group Meeting 2013 Fabian Bös Certara Mission: End-to-End Model-Based Drug Development Certara was formed by acquiring and integrating Tripos, Pharsight,
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
3D-QSAR STUDIES OF ANTI-BACTERIAL CHALCONES
 International Journal of ChemTech Research CDEN (USA): IJCRGG ISSN : 0974-4290 Vol.1, No.3, pp 717-721, July-Sept 2009 3D-QSAR STUDIES F ANTI-BACTERIAL CHALCNES G.B.Kulkarni*, A siva reddy, A Padmavathi,
International Journal of ChemTech Research CDEN (USA): IJCRGG ISSN : 0974-4290 Vol.1, No.3, pp 717-721, July-Sept 2009 3D-QSAR STUDIES F ANTI-BACTERIAL CHALCNES G.B.Kulkarni*, A siva reddy, A Padmavathi,
Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,..
 3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
Merck Virtual Library (MVL): Deployment, Application, and Future Enhancement
: Deployment, Application, and Future Enhancement") Merck Virtual Library (MVL): Deployment, Application, and Future Enhancement Zhengwei Peng Informatics, Discovery Chemistry, Merck & Co., Inc., Kenilworth, NJ, USA, and ChemAxon UGM, Boston, MA, USA Contents
Merck Virtual Library (MVL): Deployment, Application, and Future Enhancement Zhengwei Peng Informatics, Discovery Chemistry, Merck & Co., Inc., Kenilworth, NJ, USA, and ChemAxon UGM, Boston, MA, USA Contents
SCULPT 3.0. Using SCULPT to Gain Competitive Insights. Brings 3D Visualization to the Lab Bench SPECIAL REPORT. 4 Molecular Connection Fall 1999
 SPECIAL REPORT SCULPT 3.0 Brings 3D Visualization to the Lab Bench ith the acquisition of Interactive Simulations, Inc., last April, MDL added to its arsenal of discovery informatics tools a powerful analysis
SPECIAL REPORT SCULPT 3.0 Brings 3D Visualization to the Lab Bench ith the acquisition of Interactive Simulations, Inc., last April, MDL added to its arsenal of discovery informatics tools a powerful analysis
Hydrogen Bonding & Molecular Design Peter
 Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
DivCalc: A Utility for Diversity Analysis and Compound Sampling
 Molecules 2002, 7, 657-661 molecules ISSN 1420-3049 http://www.mdpi.org DivCalc: A Utility for Diversity Analysis and Compound Sampling Rajeev Gangal* SciNova Informatics, 161 Madhumanjiri Apartments,
Molecules 2002, 7, 657-661 molecules ISSN 1420-3049 http://www.mdpi.org DivCalc: A Utility for Diversity Analysis and Compound Sampling Rajeev Gangal* SciNova Informatics, 161 Madhumanjiri Apartments,
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Chemical Data Retrieval and Management
 Chemical Data Retrieval and Management ChEMBL, ChEBI, and the Chemistry Development Kit Stephan A. Beisken What is EMBL-EBI? Part of the European Molecular Biology Laboratory International, non-profit
Chemical Data Retrieval and Management ChEMBL, ChEBI, and the Chemistry Development Kit Stephan A. Beisken What is EMBL-EBI? Part of the European Molecular Biology Laboratory International, non-profit
Building innovative drug discovery alliances. Just in KNIME: Successful Process Driven Drug Discovery
 Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
An Integrated Approach to in-silico
 An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today
 est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
Identification of the Beer Component Hordenine as Food-Derived Dopamine D2. Receptor Agonist by Virtual Screening a 3D Compound Database
 Supplementary information Identification of the Beer Component Hordenine as Food-Derived Dopamine D2 Receptor Agonist by Virtual Screening a 3D Compound Database Thomas Sommer 1, 2, Harald Hübner 3, Ahmed
Supplementary information Identification of the Beer Component Hordenine as Food-Derived Dopamine D2 Receptor Agonist by Virtual Screening a 3D Compound Database Thomas Sommer 1, 2, Harald Hübner 3, Ahmed
Cheminformatics platform for drug discovery application
 EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
Nonlinear QSAR and 3D QSAR
 onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
Dock Ligands from a 2D Molecule Sketch
 Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
How IJC is Adding Value to a Molecular Design Business
 How IJC is Adding Value to a Molecular Design Business James Mills Sexis LLP ChemAxon TechTalk Stevenage, ov 2012 james.mills@sexis.co.uk Overview Introduction to Sexis Sexis IJC use cases Data visualisation
How IJC is Adding Value to a Molecular Design Business James Mills Sexis LLP ChemAxon TechTalk Stevenage, ov 2012 james.mills@sexis.co.uk Overview Introduction to Sexis Sexis IJC use cases Data visualisation
Toward an Understanding of GPCR-ligand Interactions. Alexander Heifetz
 Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
A reliable computational workflow for the selection of optimal screening libraries
 DOI 10.1186/s13321-015-0108-0 RESEARCH ARTICLE Open Access A reliable computational workflow for the selection of optimal screening libraries Yocheved Gilad 1, Katalin Nadassy 2 and Hanoch Senderowitz
DOI 10.1186/s13321-015-0108-0 RESEARCH ARTICLE Open Access A reliable computational workflow for the selection of optimal screening libraries Yocheved Gilad 1, Katalin Nadassy 2 and Hanoch Senderowitz
GC and CELPP: Workflows and Insights
 GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
CHAPTER 3. Pharmacophore modelling studies:
 CHAPTER 3 Pharmacophore modelling studies: 3.1 Introduction: According to Langer & Wolber (2004), the key goal of computer-aided molecular design methods in medicinal chemistry is to reduce the time and
CHAPTER 3 Pharmacophore modelling studies: 3.1 Introduction: According to Langer & Wolber (2004), the key goal of computer-aided molecular design methods in medicinal chemistry is to reduce the time and
MassHunter Software Overview
 MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
PROVIDING CHEMINFORMATICS SOLUTIONS TO SUPPORT DRUG DISCOVERY DECISIONS
 179 Molecular Informatics: Confronting Complexity, May 13 th - 16 th 2002, Bozen, Italy PROVIDING CHEMINFORMATICS SOLUTIONS TO SUPPORT DRUG DISCOVERY DECISIONS CARLETON R. SAGE, KEVIN R. HOLME, NIANISH
179 Molecular Informatics: Confronting Complexity, May 13 th - 16 th 2002, Bozen, Italy PROVIDING CHEMINFORMATICS SOLUTIONS TO SUPPORT DRUG DISCOVERY DECISIONS CARLETON R. SAGE, KEVIN R. HOLME, NIANISH
Advanced Medicinal Chemistry SLIDES B
 Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification
 Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Ultra High Throughput Screening using THINK on the Internet
 Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures