Protein Structure Prediction and Protein-Ligand Docking
|
|
|
- Branden Gibbs
- 6 years ago
- Views:
Transcription
1 Protein Structure Prediction and Protein-Ligand Docking Björn Wallner Jan. 24, 2014
2 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of a protein How can we tell which part of structure that needs to be improved. Protein-Ligand docking
3 From sequence to structure

4 Why do we need structure prediction?

5 DNA sequencing is getting cheap

6 From sequence to structure
7 Two main routes EQLTKCEVFQKLKDLKD... folding human baboon mouse cow Physics De Novo prediction Evolution Comparative Modeling Homology Modeling
8 De Novo Prediction Requires only the amino acid sequence Assumption: The native fold is the structure with the lowest free energy Aim: Find the structure with the lowest free energy
9 De Novo Prediction Energy function - measure how good a certain protein structure is. A method to sample different structures. energy energy reaction coordinate reaction coordinate
10 What determines how proteins fold? Are protein folding pathways designed into sequences by natural selection? Make large random libraries, select out those sequences which fold based on function Compare their folding and stability to the starting structure
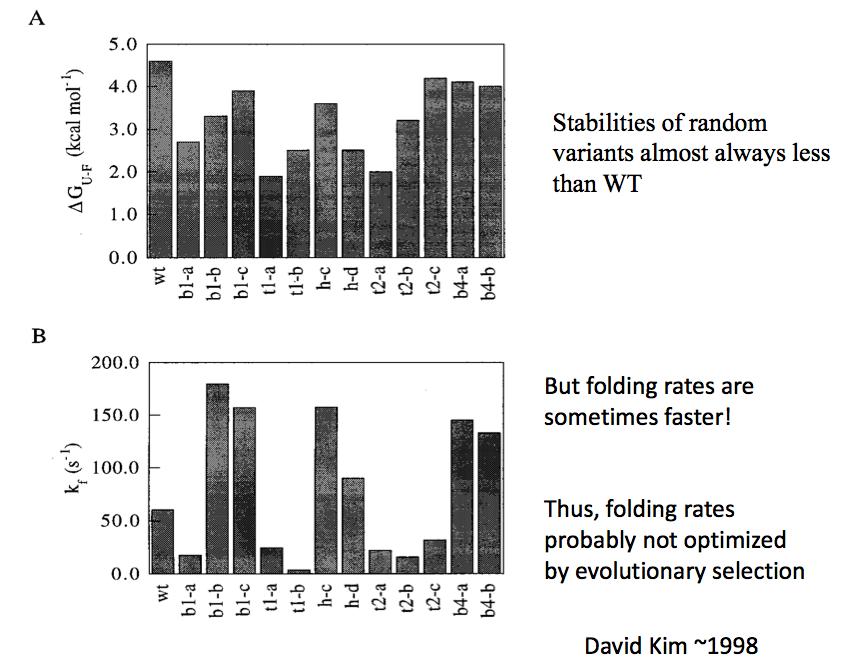
11
12 What determines the rate of folding? If not dependent on details of sequence, perhaps topology of native structure? Contact Order: average sequence separation between residues in contact in native structure
13 Folding rates correlates with contact order
14 What have we learnt from folding studies? 1. Local interactions bias but do not uniquely determine conformations sampled by short segments of the chain. 2. Folding occurs when local structure segments oriented so as to bury hydrophobic residues, pair beta strands, etc. 3. Stability determined by detailed sidechain-sidechain interactions in folded structure. 4. Folding rates are largely determined by contact order of native structure. Short folding times -> low contact order structures. 5. Native interactions on average stronger/ more consistent than non native interactions -> native minima broader than non native minima.

15 Assume distribution of conformations sampled by sequence segment during folding similar to distribution of conformations adopted by segment in protein 3D structures.

16 ,000 short simulations to generate a population of 'decoys' Remove very low contact order structures Select lowest energy structures Select broadest minima using cluster analysis Kim Simons, David Shortle ~1998


17 Lowest energy structures sampled on independent trajectories Energy gap native structures lowest energy structures
18 Native free energy gaps recurrent feature of structure prediction Soluble proteins, multimeric proteins, heterodimers, RNAs, membrane proteins, etc. Reflection of very large free energy gaps required for existence of single unique native state - think about statistical mechanics! exp(-[energy gap]/(kt)) Prediction possible because (magnitude of actual free energy gap) >> (error in free energy calculation) Challenge: how to sample close to native state?

19 Rosetta folding video
20 Two main routes EQLTKCEVFQKLKDLKD... folding human baboon mouse cow Physics De Novo prediction Evolution Comparative Modeling Homology Modeling
21 Protein Evolution - In the Lab


22 Protein Evolution - In the vivo Survival?
23 Two related proteins (homologs)
24 Homology modeling Assumption: Proteins with high sequence similarity have the same overall structure Idea: Search in the PDB _if_ there is a structure with a similar sequence that can be used as starting point (template) for modeling. Example: If the human protein (red) is not known and we want to create a model, using the fly protein (yellow) as a template is really good starting point
25 structural similarity (ångströms) Does high sequence similarity <-> similar structure? sequence similarity (%)

26 Look in the PDB Perform sequence search against all proteins with known 3D- structure (e.g. BLAST search against PDB, or more sensitive methods like PSI-blast, HHblits or jackhmmer) Compare sequences using Sequence Alignment BLAST PDB


27 Alignments against PDB - finding equivalent residues in known structures Sequence Alignment Structure determines function Similar Similar Similar Sequence Structure Function

28 Model Building Steps in Practice Find template Align target sequence with template Generate model: add loops add side chains Refine model
29 Homology Modeling Overview START TARGET SEQUENCE TEMPLATE STRUCTURE Identify related structures (templates)...acghtkildikgidywiahkalcteklftkcelsqnlydidgy... Align target sequence to template structures Build a model for the target sequence using information from template structures ALIGNMENT TARGET...SCDKLLDDELDDDIACAKKILAIKGID... TEMPLATE...SCDKFLDDDITDDIMCAKKILDIKGID... TARGET MODEL Evaluate the model NO model OK? YES QUALITY PROFILE QUALITY protein model X 0.1 END RESIDUE POSITION

30 Model Evaluation How can we tell if a model is good or bad?
31 Sometimes it is easy

32 Sometimes it is _not_ so easy Native 85% correct 70% correct
33 Assessing models in 3D Scoring Function Sequence-Structure alignment must make sense i 3D. Few gaps in secondary structure Good strand pairing
34 Scoring Function
35 Statistical Potentials Construct a database of all 20x20 or 21*20/2 amino acid pairs b Derive a potential using: P(a,b) = exp(-e(a,b)/kt) E(a,b)=-kT ln P(a,b) Predict a given sequence using the pairwise potentials Frequency of X-Y distance X-Y distance a
36 Problem: Many different parameters
37 Neural Network Could Make the Decision
38 Neural Network Example Output X X Lower alignment score can be compensated by better pairwise energy
39 Neural Network provides Confidence Scores Low ( Medium(80% High (99%) Certain (100%)
40 Why does homology modeling work? The structure of a protein is uniquely determined by its amino acid sequence except for: disorder proteins ph, ions, cofactors, chaperones Structure is more conserved than sequence Sequence -> Structure -> Function Possible to infer: Sequence -> Function
41 Modeling summary HM Example EQLTKCEVFQKLKDLKD... folding human baboon mouse cow Physics De Novo prediction Evolution Homology Modeling Comparative Modeling
42 Protein - Ligand Docking Introduction to protein-ligand docking Searching for poses Scoring functions Assessing performance Practical aspects Slides adopted from: Dr. Noel O Boyle University College Cork n.oboyle@ucc.ie
43 Protein-ligand docking Computational method that mimics the binding of a ligand to a protein. Example: Structure-Based Drug Design (SBDD) method structure means using protein structure Given... Predicts... The pose of the molecule in the binding site The binding affinity or a score representing the strength of binding Images from Charaka Goonatilake s web page, Glen Group, Unilever Centre, Cambridge
44 Pose vs. binding site Binding site (or active site ) the part of the protein where the ligand binds generally a cavity on the protein surface can be identified by looking at the crystal structure of the protein bound with a known inhibitor Pose (or binding mode ) The geometry of the ligand in the binding site Geometry = location, orientation and conformation Protein-ligand docking is not about identifying the binding site
45 Uses of docking The main uses of protein-ligand docking are for Virtual screening, to identify potential lead compounds from a large dataset Pose prediction, find out how the ligand binds Pose prediction If we know exactly where and how a known ligand binds... We can see which parts are important for binding We can suggest changes to improve affinity Avoid changes that will clash with the protein
46 Virtual screening Virtual screening is the computational or in silico analogue of biological screening The aim is to score, rank or filter a set of structures using one or more computational procedures It can be used to help decide which compounds to screen (experimentally) which libraries to synthesis which compounds to purchase from an external company to analyse the results of an experiment, such as a HTS run
47 Components of docking software Typically, protein-ligand docking software consist of two main components which work together: 1. Search algorithm Generates a large number of poses of a molecule in the binding site 2. Scoring function Calculates a score or binding affinity for a particular pose To give: The pose of the molecule in the binding site The binding affinity or a score representing the strength of binding
48 Large number of docking programs available AutoDock, DOCK, e-hits, FlexX, FRED, Glide, GOLD, LigandFit, QXP, Surflex-Dock among others Different approaches different scoring functions different search algorithms You will use AutoDock in the practical exercise, next Friday (31/ , two groups). Note: protein-ligand docking is not to be confused with the field of protein-protein docking ( protein docking )
49 Outline Introduction to protein-ligand docking Searching for poses Scoring functions Assessing performance Practical aspects
50 The search space The difficulty with protein ligand docking is in part due to the fact that it involves many degrees of freedom The translation and rotation of one molecule relative to another involves six degrees of freedom There are in addition the conformational degrees of freedom of both the ligand and the protein The solvent may also play a significant role in determining the protein ligand geometry (often ignored though) The search algorithm generates poses, orientations of particular conformations of the molecule in the binding site Tries to cover the search space, if not exhaustively, then as extensively as possible There is a tradeoff between time and search space coverage


51 Ligand conformations Conformations are different three-dimensional structures of molecules that result from rotation about single bonds That is, they have the same bond lengths and angles but different torsion angles For a molecule with N rotatable bonds, if each torsion angle is rotated in increments of θ degrees, number of conformations is (360º/ θ) N If the torsion angles are incremented in steps of 30º, this is 12 N Having too many rotatable bonds results in combinatorial explosion Taxol Wikipedia Taxol ; Lakdawala et al, BMC Chem Biol, 2001, 1, 2.

52 Sample different ligand conformations
53 Search Algorithms We can classify the various search algorithms according to the degrees of freedom that they consider Rigid docking or flexible docking With respect to the ligand structure Rigid docking The ligand is treated as a rigid structure during the docking Only the translational and rotational degrees of freedom are considered To deal with the problem of ligand conformations, a large number of conformations of each ligand are generated in advance and each is docked separately Examples: FRED (Fast Rigid Exhaustive Docking) from OpenEye, and one of the earliest docking programs, DOCK
54 The DOCK algorithm Rigid docking The DOCK algorithm developed by Kuntz and co-workers is generally considered one of the major advances in protein ligand docking [Kuntz et al., JMB, 1982, 161, 269] The earliest version of the DOCK algorithm only considered rigid body docking and was designed to identify molecules with a high degree of shape complementarity to the protein binding site. The first stage of the DOCK method involves the construction of a negative image of the binding site consisting of a series of overlapping spheres of varying radii, derived from the molecular surface of the protein AR Leach, VJ Gillet, An Introduction to Cheminformatics
55 The DOCK algorithm Rigid docking Ligand atoms are then matched to the sphere centres so that the distances between the atoms equal the distances between the corresponding sphere centres, within some tolerance. The ligand conformation is then oriented into the binding site. After checking to ensure that there are no unacceptable steric interactions, it is then scored. New orientations are produced by generating new sets of matching ligand atoms and sphere centres. The procedure continues until all possible matches have been considered. AR Leach, VJ Gillet, An Introduction to Cheminformatics
56 Flexible docking Flexible docking is the most common form of docking today Conformations of each molecule are generated on-thefly by the search algorithm during the docking process The algorithm can avoid considering conformations that do not fit Exhaustive (systematic) searching computationally too expensive as the search space is very large One common approach is to use stochastic search methods These don t guarantee optimum solution, but good solution within reasonable length of time Stochastic means that they incorporate a degree of randomness Such algorithms include genetic algorithms and simulated annealing
57 Flexible docking using a Genetic Algorithm (GA) A genetic algorithm is a stochastic algorithm that can be used to solve global optimisation problems It is based on ideas from Darwinian evolution An initial population of chromosomes is generated randomly Each chromosome represents a pose, so a large number of poses are randomly generated in the binding site Pairs of high-scoring chromosomes ( parents ) are combined to generate children For example, the location of one high-scoring pose may be combined with the torsion angles of another high-scoring pose to generate a new child pose Children are randomly mutated For example, a torsion angle or the orientation of the child pose might be altered randomly Selection of the fittest to produce the next generation The highest scoring of the new poses are combined with the highest scoring of the original poses to make the next generation Repeat for N generations or until no significant improvement is observed We have identified a high scoring pose
 protein side chains in the active site may adopt different conformations Some docking programs allow protein")
58 Handling protein conformations Most docking software treats the protein as rigid Rigid Receptor Approximation This approximation may be invalid for a particular protein-ligand complex as... the protein may deform slightly to accommodate different ligands (ligand-induced fit) protein side chains in the active site may adopt different conformations Some docking programs allow protein side-chain flexibility For example, selected side chains are allowed to undergo torsional rotation around acyclic bonds Increases the search space Larger protein movements can only be handled by separate dockings to different protein conformations Image of Tyrosine rotamers from CMBI, Univeristy of Nijmegen at
59 Outline Introduction to protein-ligand docking Searching for poses Scoring functions Assessing performance Practical aspects
60 Components of docking software Typically, protein-ligand docking software consist of two main components which work together: 1. Search algorithm Generates a large number of poses of a molecule in the binding site 2. Scoring function Calculates a score or binding affinity for a particular pose To give: The pose of the molecule in the binding site The binding affinity or a score representing the strength of binding
61 The perfect scoring function will Accurately calculate the binding affinity Will allow actives to be identified in a virtual screen Be able to rank different ligands in terms of affinity Score the poses of active ligands higher than poses of an inactive Score the correct pose of the active higher than an incorrect pose of the active Will allow the correct pose of the active to be identified
62 Classes of scoring function Broadly speaking, scoring functions can be divided into the following classes: Forcefield-based Based on terms from molecular mechanics forcefields GoldScore, DOCK, AutoDock Empirical Parameterised against experimental binding affinities ChemScore, PLP, Glide SP/XP Knowledge-based potentials Based on statistical analysis of observed pairwise distributions PMF, DrugScore, ASP
63 Statistical Potentials = Knowledge-based potential Construct a database of all 20x20 or 21*20/2 amino acid pairs b Derive a potential using: P(a,b) = exp(-e(a,b)/kt) E(a,b)=-kT ln P(a,b) Predict a given sequence using the pairwise potentials Frequency of X-Y distance X-Y distance a
64 Knowledge-based potentials Based on a comparison between the observed number of contacts between certain atom types and the number of contacts one would expect if there were no interaction between the atoms (the reference state) Derived from an analysis of pairs of non-bonded interactions between proteins and ligands in PDB Observed distributions of geometries of ligands in crystal structures are used to deduce the potential that gave rise to the distribution Hence knowledge-based potential
65 Knowledge-based potentials For example, creating the distributions of ligand carbonyl oxygens to protein hydroxyl groups: Ligand Ligand O O Ligand O HO HO HO Protein Protein Protein (Invented) distribution of a particular pairwise interaction (imagine the minimum at 3.0Ang) 1000 Number of observations Distance (Angstrom)
66 Knowledge-based potentials Some pairwise interactions may occur seldom in the PDB Resulting distribution may be inaccurate Doesn t take into account directionality of interactions, e.g. hydrogen bonds Just based on pairwise distances Resulting score contains contributions from a large number of pairwise interactions Difficult to identify problems and to improve Sensitive to definition of reference state DrugScore has a different reference state than ASP (Astex Statistical Potential)
67 Alternative scoring strategies Consensus docking Carry out two (or more) separate docking experiments and rank based on the mean of the ranks from the two dockings (rank-by-rank) Rationale: a true active will be scored well by both scoring functions Rescoring Use one scoring function during the docking, but evaluate the final poses using another scoring function Rationale: One scoring function is better at pose prediction, the other is better at ranking actives versus inactives Consensus scoring Combine the results from several rescorings
68 Outline Introduction to protein-ligand docking Searching for poses Scoring functions Assessing performance Practical aspects
69 Pose prediction accuracy Given a set of actives with known crystal poses, can they be docked accurately? Accuracy measured by RMSD (root mean squared deviation) compared to known crystal structures RMSD = square root of the average of (the difference between a particular coordinate in the crystal and that coordinate in the pose) 2 Within 2.0Å RMSD considered cut-off for accuracy More sophisticated measures have been proposed, but are not widely adopted In general, the best docking software predicts the correct pose about 70% of the time Note: it s always easier to find the correct pose when docking back into the active s own crystal structure More difficult to cross-dock
70 Outline Introduction to protein-ligand docking Searching for poses Scoring functions Assessing performance Practical aspects
71 Ligand Preparation A reasonable 3D structure is required as starting point Even during flexible docking, bond lengths and angles are held fixed The protonation state and tautomeric form of a particular ligand could influence its hydrogen bonding ability Either protonate as expected for physiological ph and use a single tautomer Or generate and dock all possible protonation states and tautomers, and retain the one with the highest score OH H + O Enol Ketone
72 Preparing the protein structure The Protein Data Bank (PDB) is a repository of protein crystal structures, often in complexes with inhibitors PDB structures often contain water molecules In general, all water molecules are removed except where it is known that they play an important role in coordinating to the ligand PDB structures are missing all hydrogen atoms Many docking programs require the protein to have explicit hydrogens. In general these can be added unambiguously, except in the case of acidic/basic side chains An incorrect assignment of protonation states in the active site will give poor results Glutamate, Aspartate have COO- or COOH OH is hydrogen bond donor, O- is not Histidine is a base and its neutral form has two tautomers N R HN R NH N HN R + NH
73 Preparing the protein structure For particular protein side chains, the PDB structure can be incorrect Crystallography gives electron density, not molecular structure In poorly resolved crystal structures of proteins, isoelectronic groups can give make it difficult to deduce the correct structure R O NH 2 R NH 2 O R N N R N N Affects asparagine, glutamine, histidine Important? Affects hydrogen bonding pattern May need to flip amide or imidazole How to decide? Look at hydrogen bonding pattern in crystal structures containing ligands
74 Final thoughts Protein-ligand docking is an essential tool for computational drug design Widely used in pharmaceutical companies Many success stories (see Kolb et al. Curr. Opin. Biotech., 2009, 20, 429) But it s not a golden bullet The perfect scoring function has yet to be found The performance varies from target to target, and scoring function to scoring function Care needs to be taken when preparing both the protein and the ligands The more information you have (and use!), the better your chances Targeted library, docking constraints, filtering poses, seeding with known actives, comparing with known crystal poses
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
What is Protein-Ligand Docking?
 MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein-Ligand Docking Evaluations
 Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
BUDE. A General Purpose Molecular Docking Program Using OpenCL. Richard B Sessions
 BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Flexibility and Constraints in GOLD
 Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Cheminformatics platform for drug discovery application
 EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Building 3D models of proteins
 Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
High Throughput In-Silico Screening Against Flexible Protein Receptors
 John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Advanced in silico Drug Design KFC/ADD
 Advanced in silico Drug Design S pozdravem KFC/ADD Molecular Karel Berka Docking Intro Karel Berka, Ph.D. Jindřich Fanfrlík, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. UP Olomouc, 30.1.-1.2. 2017
Advanced in silico Drug Design S pozdravem KFC/ADD Molecular Karel Berka Docking Intro Karel Berka, Ph.D. Jindřich Fanfrlík, Ph.D. Martin Lepšík, Ph.D. Pavel Polishchuk, Ph.D. UP Olomouc, 30.1.-1.2. 2017
Molecular Modeling Lecture 7. Homology modeling insertions/deletions manual realignment
 Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Neural Networks for Protein Structure Prediction Brown, JMB CS 466 Saurabh Sinha
 Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Introduction to" Protein Structure
 Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
CHAPTER 29 HW: AMINO ACIDS + PROTEINS
 CAPTER 29 W: AMI ACIDS + PRTEIS For all problems, consult the table of 20 Amino Acids provided in lecture if an amino acid structure is needed; these will be given on exams. Use natural amino acids (L)
CAPTER 29 W: AMI ACIDS + PRTEIS For all problems, consult the table of 20 Amino Acids provided in lecture if an amino acid structure is needed; these will be given on exams. Use natural amino acids (L)
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Computational modeling of G-Protein Coupled Receptors (GPCRs) has recently become
 has recently become") Homology Modeling and Docking of Melatonin Receptors Andrew Kohlway, UMBC Jeffry D. Madura, Duquesne University 6/18/04 INTRODUCTION Computational modeling of G-Protein Coupled Receptors (GPCRs) has recently
Homology Modeling and Docking of Melatonin Receptors Andrew Kohlway, UMBC Jeffry D. Madura, Duquesne University 6/18/04 INTRODUCTION Computational modeling of G-Protein Coupled Receptors (GPCRs) has recently
Protein-Ligand Docking Methods
 Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University
 Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Drug Design. Molecular docking. RNDr. Karel Berka, PhD RNDr. Jindřich Fanfrlík, PhD RNDr. Martin Lepšík, PhD
 Drug Design Molecular docking RNDr. Karel Berka, PhD RNDr. Jindřich Fanfrlík, PhD RNDr. Martin Lepšík, PhD Dpt. Physical Chemistry, RCPTM, Faculty of Science, Palacky University in Olomouc Motto tj. 18
Drug Design Molecular docking RNDr. Karel Berka, PhD RNDr. Jindřich Fanfrlík, PhD RNDr. Martin Lepšík, PhD Dpt. Physical Chemistry, RCPTM, Faculty of Science, Palacky University in Olomouc Motto tj. 18
Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions Van der Waals Interactions
Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions Van der Waals Interactions
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Build_model v User Guide
 Build_model v.2.0.1 User Guide MolTech Build_model User Guide 2008-2011 Molecular Technologies Ltd. www.moltech.ru Please send your comments and suggestions to contact@moltech.ru. Table of Contents Input
Build_model v.2.0.1 User Guide MolTech Build_model User Guide 2008-2011 Molecular Technologies Ltd. www.moltech.ru Please send your comments and suggestions to contact@moltech.ru. Table of Contents Input
Towards Physics-based Models for ADME/Tox. Tyler Day
 Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Protein-Ligand Docking
 Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015,
 Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Identifying Interaction Hot Spots with SuperStar
 Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
FlexPepDock In a nutshell
 FlexPepDock In a nutshell All Tutorial files are located in http://bit.ly/mxtakv FlexPepdock refinement Step 1 Step 3 - Refinement Step 4 - Selection of models Measure of fit FlexPepdock Ab-initio Step
FlexPepDock In a nutshell All Tutorial files are located in http://bit.ly/mxtakv FlexPepdock refinement Step 1 Step 3 - Refinement Step 4 - Selection of models Measure of fit FlexPepdock Ab-initio Step
SAM Teacher s Guide Protein Partnering and Function
 SAM Teacher s Guide Protein Partnering and Function Overview Students explore protein molecules physical and chemical characteristics and learn that these unique characteristics enable other molecules
SAM Teacher s Guide Protein Partnering and Function Overview Students explore protein molecules physical and chemical characteristics and learn that these unique characteristics enable other molecules
Protein Structure Determination
 Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Portal. User Guide Version 1.0. Contributors
 Portal www.dockthor.lncc.br User Guide Version 1.0 Contributors Diogo A. Marinho, Isabella A. Guedes, Eduardo Krempser, Camila S. de Magalhães, Hélio J. C. Barbosa and Laurent E. Dardenne www.gmmsb.lncc.br
Portal www.dockthor.lncc.br User Guide Version 1.0 Contributors Diogo A. Marinho, Isabella A. Guedes, Eduardo Krempser, Camila S. de Magalhães, Hélio J. C. Barbosa and Laurent E. Dardenne www.gmmsb.lncc.br
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today
 est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
NMR, X-ray Diffraction, Protein Structure, and RasMol
 NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
HOMOLOGY MODELING. The sequence alignment and template structure are then used to produce a structural model of the target.
 HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Life Science Webinar Series
 Life Science Webinar Series Elegant protein- protein docking in Discovery Studio Francisco Hernandez-Guzman, Ph.D. November 20, 2007 Sr. Solutions Scientist fhernandez@accelrys.com Agenda In silico protein-protein
Life Science Webinar Series Elegant protein- protein docking in Discovery Studio Francisco Hernandez-Guzman, Ph.D. November 20, 2007 Sr. Solutions Scientist fhernandez@accelrys.com Agenda In silico protein-protein
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009
 114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Viewing and Analyzing Proteins, Ligands and their Complexes 2
 2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Using AutoDock 4 with ADT: A Tutorial
 Using AutoDock 4 with ADT: A Tutorial Ruth Huey Sargis Dallakyan Alex Perryman David S. Goodsell (Garrett Morris) 9/2/08 Using AutoDock 4 with ADT 1 What is Docking? Predicting the best ways two molecules
Using AutoDock 4 with ADT: A Tutorial Ruth Huey Sargis Dallakyan Alex Perryman David S. Goodsell (Garrett Morris) 9/2/08 Using AutoDock 4 with ADT 1 What is Docking? Predicting the best ways two molecules
Interpreting and evaluating biological NMR in the literature. Worksheet 1
 Interpreting and evaluating biological NMR in the literature Worksheet 1 1D NMR spectra Application of RF pulses of specified lengths and frequencies can make certain nuclei detectable We can selectively
Interpreting and evaluating biological NMR in the literature Worksheet 1 1D NMR spectra Application of RF pulses of specified lengths and frequencies can make certain nuclei detectable We can selectively
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
CHAPTER-5 MOLECULAR DOCKING STUDIES
 CHAPTER-5 MOLECULAR DOCKING STUDIES 156 CHAPTER -5 MOLECULAR DOCKING STUDIES 5. Molecular docking studies This chapter discusses about the molecular docking studies of the synthesized compounds with different
CHAPTER-5 MOLECULAR DOCKING STUDIES 156 CHAPTER -5 MOLECULAR DOCKING STUDIES 5. Molecular docking studies This chapter discusses about the molecular docking studies of the synthesized compounds with different
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows
 MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
Conformational Geometry of Peptides and Proteins:
 Conformational Geometry of Peptides and Proteins: Before discussing secondary structure, it is important to appreciate the conformational plasticity of proteins. Each residue in a polypeptide has three
Conformational Geometry of Peptides and Proteins: Before discussing secondary structure, it is important to appreciate the conformational plasticity of proteins. Each residue in a polypeptide has three
Overview & Applications. T. Lezon Hands-on Workshop in Computational Biophysics Pittsburgh Supercomputing Center 04 June, 2015
 Overview & Applications T. Lezon Hands-on Workshop in Computational Biophysics Pittsburgh Supercomputing Center 4 June, 215 Simulations still take time Bakan et al. Bioinformatics 211. Coarse-grained Elastic
Overview & Applications T. Lezon Hands-on Workshop in Computational Biophysics Pittsburgh Supercomputing Center 4 June, 215 Simulations still take time Bakan et al. Bioinformatics 211. Coarse-grained Elastic
STRUCTURAL BIOINFORMATICS II. Spring 2018
 STRUCTURAL BIOINFORMATICS II Spring 2018 Syllabus Course Number - Classification: Chemistry 5412 Class Schedule: Monday 5:30-7:50 PM, SERC Room 456 (4 th floor) Instructors: Ronald Levy, SERC 718 (ronlevy@temple.edu)
STRUCTURAL BIOINFORMATICS II Spring 2018 Syllabus Course Number - Classification: Chemistry 5412 Class Schedule: Monday 5:30-7:50 PM, SERC Room 456 (4 th floor) Instructors: Ronald Levy, SERC 718 (ronlevy@temple.edu)
Programme Last week s quiz results + Summary Fold recognition Break Exercise: Modelling remote homologues
 Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Conformational Searching using MacroModel and ConfGen. John Shelley Schrödinger Fellow
 Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Introduction to Structure Preparation and Visualization
 Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Modeling lecture 2
 Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
BSc and MSc Degree Examinations
 Examination Candidate Number: Desk Number: BSc and MSc Degree Examinations 2018-9 Department : BIOLOGY Title of Exam: Molecular Biology and Biochemistry Part I Time Allowed: 1 hour and 30 minutes Marking
Examination Candidate Number: Desk Number: BSc and MSc Degree Examinations 2018-9 Department : BIOLOGY Title of Exam: Molecular Biology and Biochemistry Part I Time Allowed: 1 hour and 30 minutes Marking
Can protein model accuracy be. identified? NO! CBS, BioCentrum, Morten Nielsen, DTU
 Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Physiochemical Properties of Residues
 Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Bioinformatics. Macromolecular structure
 Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
Copyright Mark Brandt, Ph.D A third method, cryogenic electron microscopy has seen increasing use over the past few years.
 Structure Determination and Sequence Analysis The vast majority of the experimentally determined three-dimensional protein structures have been solved by one of two methods: X-ray diffraction and Nuclear
Structure Determination and Sequence Analysis The vast majority of the experimentally determined three-dimensional protein structures have been solved by one of two methods: X-ray diffraction and Nuclear
GC and CELPP: Workflows and Insights
 GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
Chapter 4. Glutamic Acid in Solution - Correlations
 Chapter 4 Glutamic Acid in Solution - Correlations 4. Introduction Glutamic acid crystallises from aqueous solution, therefore the study of these molecules in an aqueous environment is necessary to understand
Chapter 4 Glutamic Acid in Solution - Correlations 4. Introduction Glutamic acid crystallises from aqueous solution, therefore the study of these molecules in an aqueous environment is necessary to understand
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Analyzing Molecular Conformations Using the Cambridge Structural Database. Jason Cole Cambridge Crystallographic Data Centre
 Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Other Cells. Hormones. Viruses. Toxins. Cell. Bacteria
 Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Visualization of Macromolecular Structures
 Visualization of Macromolecular Structures Present by: Qihang Li orig. author: O Donoghue, et al. Structural biology is rapidly accumulating a wealth of detailed information. Over 60,000 high-resolution
Visualization of Macromolecular Structures Present by: Qihang Li orig. author: O Donoghue, et al. Structural biology is rapidly accumulating a wealth of detailed information. Over 60,000 high-resolution
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors
 MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases