LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
|
|
|
- Nigel Day
- 6 years ago
- Views:
Transcription
1 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * wolber@inteligand.com
2 Pharmacophores from LigandScout Pharmacophores & the Protein Data Bank o 3D pharmacophore methodology o Primary data source: The Protein Data Bank o Why LigandScout? LigandScout o Ligand perception o 3D pharmacophore generation o Shared feature pharmacophores Applications & Future Perspectives

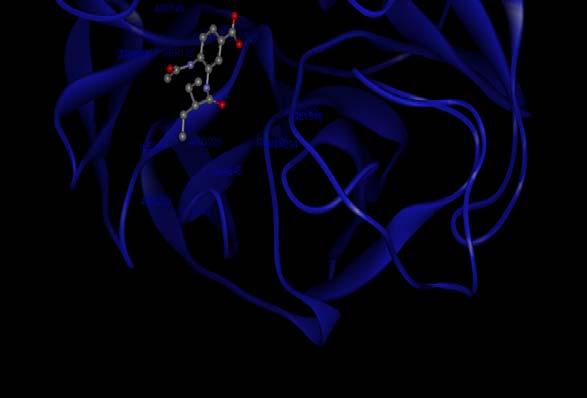
3 Ligand-Protein Interaction Influenza virus neuraminidase inhibition by ligand FDI (4-(N-acetylamino)-3-[N-(2-ethylbutanoylamino]benzoic acid)

4 Pharmacophore Models Definition: Ensemble of universal chemical features that represent a specific mode of action Chemical Features: Hydrogen bonds, charge interactions, hydrophobic areas
5 Why Use Structure-Based Pharmacophores Instead of Docking? Universal Pharmacophores represent chemical functions, valid not only for the currently bound, but also unknown molecules Comprehensive selectivity-tuning by adding or omitting feature constraints Computationally efficient due to simplicity (suitable for virtual screening)
6 PDB Age and Content X Overall number of Structures Stuctures deposited per year
7 Why LigandScout? Structure-based pharmacophore creation from all PDB complexes: 1. Identification & extraction of ligands 2. Interpret ligands (hybridization states, bond types) 3. Create pharmacophores 4. Visualize, allow user interaction and export for virtual screening


8 Hybridization State Determination Quantitative Geometry Templates for all geometry types: sp 3 : sp 2 : sp: tetrahedral trigonal planar linear Align along the first two points, numerically turn to match the third point

9 Geometry Templates: Better Than Bond Angles? 120 degrees
 a i i i= 0 2 d r = n i= 0 ( Ii")
10 Hybridization State: Error Determination Absolute and Relative Geometric Deviation n d = ( I O) a i i i= 0 2 d r = n i= 0 ( Ii Oi) n 2



11 Hybridization State: Planar Rings Planar rings show different bond angles than non-ring sp 2 atoms: all planar ring atoms are to be sp 2 hybridized

![smallest rings (SSSR) [Figueras 2000] (4) Empirical discrimination](/docs-images/80/80562441/images/12-1.jpg "between planar and non-planar rings (Porphyrine pyrroles vs.")
12 Hybridization State: Ring Perception Ring recognition must match graph-theoretical and chemical view 3 out of 6 Smallest set of smallest rings (SSSR) [Figueras 2000] (4) Empirical discrimination between planar and non-planar rings (Porphyrine pyrroles vs. pyrrolidines) (5) (6)
13 Double Bond Distribution Among sp 2 Atoms No exact solution in many cases (e.g. Keto-enol tautomers) Use of patterns explicitely covering all known cases from the view of a central atom Greedy (recursive) scoring algorithm covering the rest of the cases Patterns by Roger Sayle: Bioinformatics Group, Metaphorics LLC, Santa Fe, New Mexico, see
14 Distributed Batch Extraction and Interpretation Extraction and Interpretation is computing-intense o Distance comparison of macromolecular atoms to each ligand atom o Ring detection o Bond distribution Requirements o Client can join or leave any time o Scalable Solution o Central HTTP server distributes PDB files o Central application server collects ligands o Computational clients can arbitrarily join or leave at any time


15 Chemical Feature Constraints Distance Constraints Relation between two points, one located on ligand side, one on macromolecular side. Direction Constraints Relation between two atom groups, one located on ligand side, one on macromolecular side. Feature Type H-Bond Charge Transfer Hydrophobic Distance A A A Groups form a rigid reference geometry, which are the basis for a directed vector. Result: one tolerance sphere on ligand side

16 Chemical Feature Constraints: Rigid H-Bonds

17 Chemical Feature Constraints: Flexible H-Bonds invsin b.sin( χ ) α = δ c
18 Chemical Features Universality Layers Layer 4 Layer 3 Layer 2 Layer 1 Chemical Function Subgraph Without geometry constraint Including geometry constraint Without geometry constraint Including geometry constraint Lipophilic area, positive ionizable area Hydrogen bond donor/acceptor Hydroxylic group, phenol group Phenol group facing a parallel benzene Selectivity Universality
19 Why Universality? Semantic enhancement: Allows the comparison of chemical features Categorization: Prerequisite for the creation of ontologies (classification trees of chemical features) Indexing capabilities: Only categorized features permit indexing: Necessary for efficient pharmcophore search techniques LigandScout creates pharmacophores using the universal Layer 3 and Layer 4 features
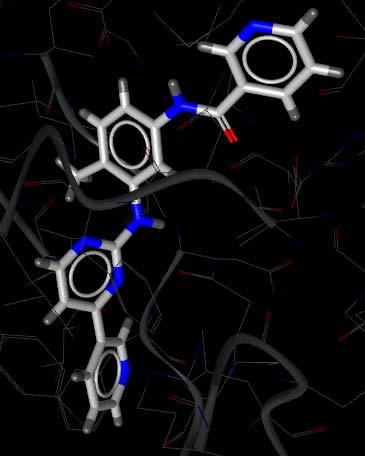
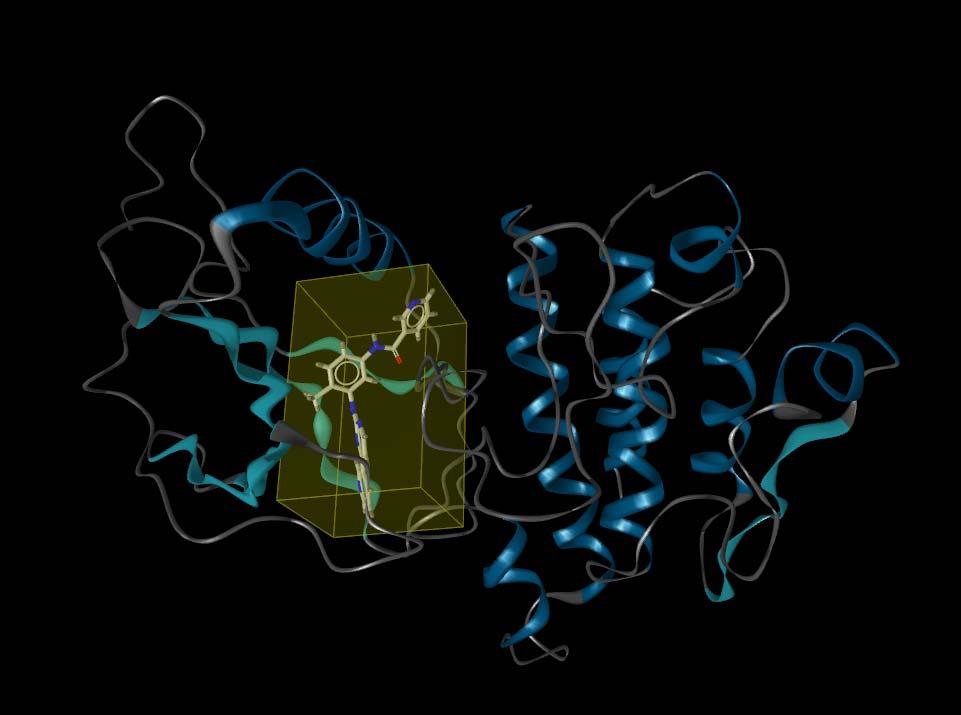
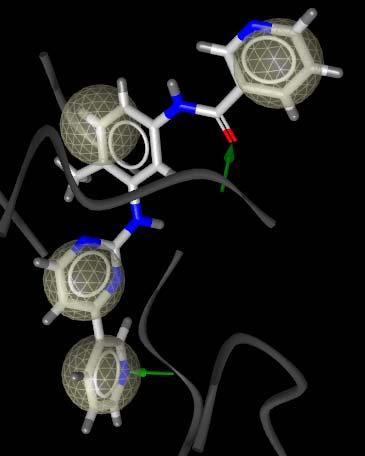
20 Application Example: Gleevec Gleevec modification (PRC) from 1FPU
21 Pharmacophore Overlaying Pharmacophore model derived from one single bound ligand may not be able to retrieve other related compounds Starting set: Several ligandprotein complex pharmacophores Creation of compatibility graphs Maximum clique detection Feature alignment Calculation of combined features newcommon feature pharmacophore

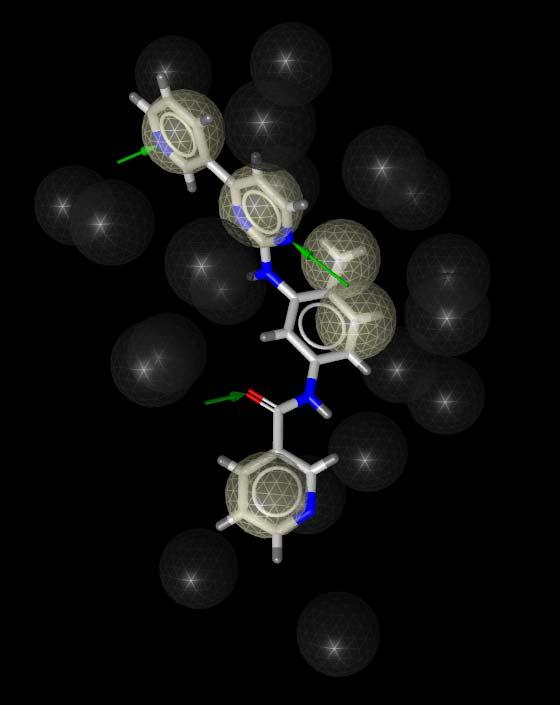
22 Common Feature Pharmacophore 1iep 1fpu 1opj

23 Common Feature Pharmacophore 3 lipophilic aromatic areas 2 hydrogen bond acceptors
24 Virtual Screening Results Database Hits Drug-like hits PDB singleconf (~67k) 7 7 PDB multiconf (~7k) Maybridge (~55k)
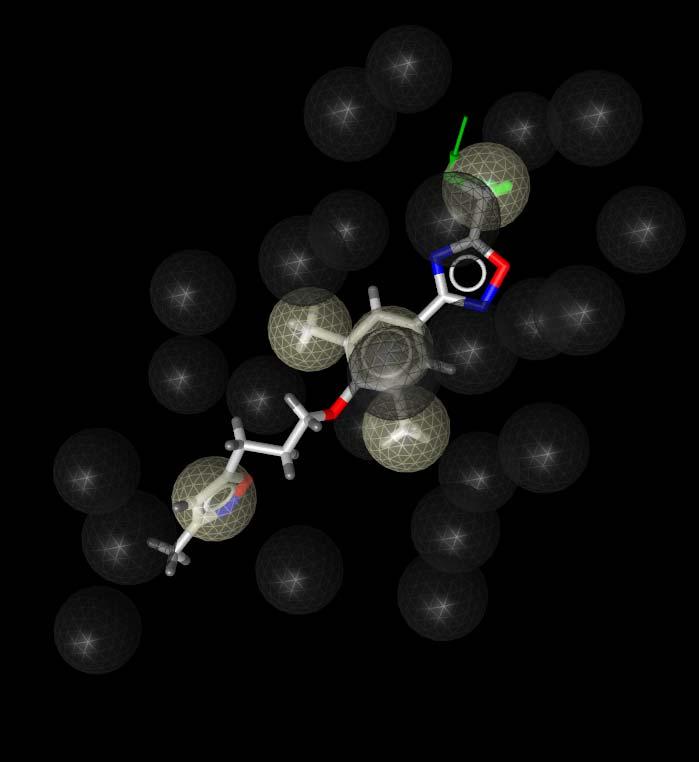
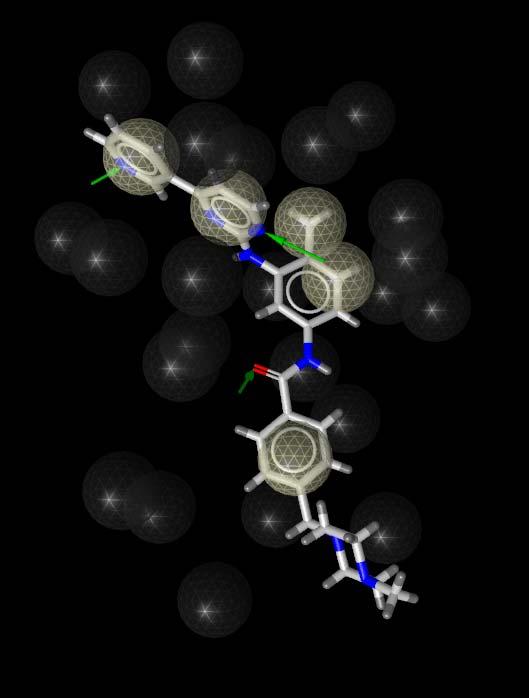
25 Application Example: HRV Coat Protein 1ncr 1nd3 1c8m

26 Common Feature Pharmacophore 2 lipophilic aromatic areas 3 lipophilic areas 1 hydrogen bond acceptor
27 Virtual Screening Results Database Hits Drug-like hits PDB singleconf (~67k) 8 8 PDB multiconf (~7k) Maybridge (~55k)
28 Summary LigandScout Extracts ligands and their protein environment from PDB files Assigns bond characteristics to small molecule ligands in a fully automated and distributed way Creates and visualizes pharmacophore models that represent the interaction between protein and ligand in a universal way
29 Perspectives The collection of all pharmacophores from the PDB can be used to develop 3D pharmacophore cascades in order to create computational models for biological pathways search all pharmacophores for the purpose of screening one compound against all its known biological effects ( activity profiling )
30 Literature G. Wolber and T. Langer. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters J. Chem. Inf. Model; 2005; 45; E. M. Krovat, K. H. Frühwirth, and T. Langer. Pharmacophore Identification, in Silico Screening, and Virtual Library Design for Inhibitors of the Human Factor Xa. J. Chem. Inf. Model.; 2005; 45; T. Steindl and T. Langer. Docking Versus Pharmacophore Model Generation: A Comparison of High-Throughput Virtual Screening Strategies for the Search of Human Rhinovirus Coat Protein Inhibitors. QSAR and Combinatorial Science; 2005; in press

31 Acknowledgements inte:ligand Martin Biely Alois Dornhofer Robert Kosara Judith Rollinger Thierry Langer Oliver Funk Johannes Kirchmair Eva Krovat Christian Laggner Daniela Schuster Theodora Steindl
Efficient overlay of molecular 3-D pharmacophores
 Efficient overlay of molecular 3D pharmacophores Gerhard Wolber*, Alois A. Dornhofer & Thierry Langer * EMail: wolber@inteligand.com Superposition of molecules 1 Alignment: Outline Scope, design goals
Efficient overlay of molecular 3D pharmacophores Gerhard Wolber*, Alois A. Dornhofer & Thierry Langer * EMail: wolber@inteligand.com Superposition of molecules 1 Alignment: Outline Scope, design goals
Pharmacophore Approaches In Drug Discovery
 Pharmacophore Approaches In Drug Discovery Prof. Thierry Langer Prestwick Chemical, Inc. Boulevard Gonthier d Andernach 67400 Strasbourg-Illkirch, France Contents Introduction - Non HTS Hit Recognition
Pharmacophore Approaches In Drug Discovery Prof. Thierry Langer Prestwick Chemical, Inc. Boulevard Gonthier d Andernach 67400 Strasbourg-Illkirch, France Contents Introduction - Non HTS Hit Recognition
The Conformation Search Problem
 Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Cross Discipline Analysis made possible with Data Pipelining. J.R. Tozer SciTegic
 Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Part 6. 3D Pharmacophore Modeling
 279 Part 6 3D Pharmacophore Modeling 281 20 3D Pharmacophore Modeling Techniques in Computer Aided Molecular Design Using LigandScout Thomas Seidel, Sharon D. Bryant, Gökhan Ibis, Giulio Poli, and Thierry
279 Part 6 3D Pharmacophore Modeling 281 20 3D Pharmacophore Modeling Techniques in Computer Aided Molecular Design Using LigandScout Thomas Seidel, Sharon D. Bryant, Gökhan Ibis, Giulio Poli, and Thierry
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Pharmacophores and Pharmacophore Searches
 Pharmacophores and Pharmacophore Searches Edited by Thierry Longer and Remy D. Hoffmann WILEY- VCH WILEY-VCH Verlag GmbH &. Co. KGaA Contents Preface XIII A Personal Foreword List of Contributors XV XVII
Pharmacophores and Pharmacophore Searches Edited by Thierry Longer and Remy D. Hoffmann WILEY- VCH WILEY-VCH Verlag GmbH &. Co. KGaA Contents Preface XIII A Personal Foreword List of Contributors XV XVII
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Ignasi Belda, PhD CEO. HPC Advisory Council Spain Conference 2015
 Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Marvin. Sketching, viewing and predicting properties with Marvin - features, tips and tricks. Gyorgy Pirok. Solutions for Cheminformatics
 Marvin Sketching, viewing and predicting properties with Marvin - features, tips and tricks Gyorgy Pirok Solutions for Cheminformatics The Marvin family The Marvin toolkit provides web-enabled components
Marvin Sketching, viewing and predicting properties with Marvin - features, tips and tricks Gyorgy Pirok Solutions for Cheminformatics The Marvin family The Marvin toolkit provides web-enabled components
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
CHAPTER 3. Pharmacophore modelling studies:
 CHAPTER 3 Pharmacophore modelling studies: 3.1 Introduction: According to Langer & Wolber (2004), the key goal of computer-aided molecular design methods in medicinal chemistry is to reduce the time and
CHAPTER 3 Pharmacophore modelling studies: 3.1 Introduction: According to Langer & Wolber (2004), the key goal of computer-aided molecular design methods in medicinal chemistry is to reduce the time and
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Plan. Day 2: Exercise on MHC molecules.
 Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Navigation in Chemical Space Towards Biological Activity. Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland
 Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,..
 3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
GC and CELPP: Workflows and Insights
 GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
Identifying Interaction Hot Spots with SuperStar
 Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
BioSolveIT. A Combinatorial Approach for Handling of Protonation and Tautomer Ambiguities in Docking Experiments
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
Molecular Modelling. Computational Chemistry Demystified. RSC Publishing. Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK
 Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
PATTERN RECOGNITION AND DISTRIBUTED COMPUTING
 49 Molecular Informatics: Confronting Complexity, May 13 th - 16 th 2002, Bozen, Italy PATTERN RECOGNITION AND DISTRIBUTED COMPUTING IN DRUG DESIGN W. GRAHAM RICHARDS Department of Chemistry, University
49 Molecular Informatics: Confronting Complexity, May 13 th - 16 th 2002, Bozen, Italy PATTERN RECOGNITION AND DISTRIBUTED COMPUTING IN DRUG DESIGN W. GRAHAM RICHARDS Department of Chemistry, University
Supporting Information
 Supporting Information Discovery of ovel CB Receptor Ligands by a Pharmacophore-based Virtual Screening Workflow Patrick Markt, a emens Feldmann, a Judith Maria Rollinger, b Stefan Raduner, c Daniela Schuster,
Supporting Information Discovery of ovel CB Receptor Ligands by a Pharmacophore-based Virtual Screening Workflow Patrick Markt, a emens Feldmann, a Judith Maria Rollinger, b Stefan Raduner, c Daniela Schuster,
Different conformations of the drugs within the virtual library of FDA approved drugs will be generated.
 Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites. J. Andrew Surface
 Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Targeting protein-protein interactions: A hot topic in drug discovery
 Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Ultra High Throughput Screening using THINK on the Internet
 Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Chemoinformatics and information management. Peter Willett, University of Sheffield, UK
 Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Journal of Pharmacology and Experimental Therapy-JPET#172536
 A NEW NON-PEPTIDIC INHIBITOR OF THE 14-3-3 DOCKING SITE INDUCES APOPTOTIC CELL DEATH IN CHRONIC MYELOID LEUKEMIA SENSITIVE OR RESISTANT TO IMATINIB Manuela Mancini, Valentina Corradi, Sara Petta, Enza
A NEW NON-PEPTIDIC INHIBITOR OF THE 14-3-3 DOCKING SITE INDUCES APOPTOTIC CELL DEATH IN CHRONIC MYELOID LEUKEMIA SENSITIVE OR RESISTANT TO IMATINIB Manuela Mancini, Valentina Corradi, Sara Petta, Enza
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
ENERGY MINIMIZATION AND CONFORMATION SEARCH ANALYSIS OF TYPE-2 ANTI-DIABETES DRUGS
 Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
ICM-Chemist How-To Guide. Version 3.6-1g Last Updated 12/01/2009
 ICM-Chemist How-To Guide Version 3.6-1g Last Updated 12/01/2009 ICM-Chemist HOW TO IMPORT, SKETCH AND EDIT CHEMICALS How to access the ICM Molecular Editor. 1. Click here 2. Start sketching How to sketch
ICM-Chemist How-To Guide Version 3.6-1g Last Updated 12/01/2009 ICM-Chemist HOW TO IMPORT, SKETCH AND EDIT CHEMICALS How to access the ICM Molecular Editor. 1. Click here 2. Start sketching How to sketch
tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules
 Software News and Updates tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules DANIEL SEELIGER, BERT L. DE GROOT Computational Biomolecular Dynamics Group, Max-Planck-Institute
Software News and Updates tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules DANIEL SEELIGER, BERT L. DE GROOT Computational Biomolecular Dynamics Group, Max-Planck-Institute
Canonical Line Notations
 Canonical Line otations InChI vs SMILES Krisztina Boda verview Compound naming InChI SMILES Molecular equivalency Isomorphism Kekule Tautomers Finding duplicates What s Your ame? 1. Unique numbers CAS
Canonical Line otations InChI vs SMILES Krisztina Boda verview Compound naming InChI SMILES Molecular equivalency Isomorphism Kekule Tautomers Finding duplicates What s Your ame? 1. Unique numbers CAS
Drug Informatics for Chemical Genomics...
 Drug Informatics for Chemical Genomics... An Overview First Annual ChemGen IGERT Retreat Sept 2005 Drug Informatics for Chemical Genomics... p. Topics ChemGen Informatics The ChemMine Project Library Comparison
Drug Informatics for Chemical Genomics... An Overview First Annual ChemGen IGERT Retreat Sept 2005 Drug Informatics for Chemical Genomics... p. Topics ChemGen Informatics The ChemMine Project Library Comparison
CHAPTER-2. Drug discovery is a comprehensive approach wherein several disciplines
 36 CAPTER-2 Molecular Modeling Analog Based Studies Drug discovery is a comprehensive approach wherein several disciplines are used to design or discover the drugs. The R&D expenditure incurred to bring
36 CAPTER-2 Molecular Modeling Analog Based Studies Drug discovery is a comprehensive approach wherein several disciplines are used to design or discover the drugs. The R&D expenditure incurred to bring
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Conformational Searching using MacroModel and ConfGen. John Shelley Schrödinger Fellow
 Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Self-Organizing Superimposition Algorithm for Conformational Sampling
 Self-Organizing Superimposition Algorithm for Conformational Sampling FANGQIANG ZHU, DIMITRIS K. AGRAFIOTIS Johnson & Johnson Pharmaceutical Research & Development, L.L.C., 665 Stockton Drive, Exton, Pennsylvania
Self-Organizing Superimposition Algorithm for Conformational Sampling FANGQIANG ZHU, DIMITRIS K. AGRAFIOTIS Johnson & Johnson Pharmaceutical Research & Development, L.L.C., 665 Stockton Drive, Exton, Pennsylvania
Data Mining in the Chemical Industry. Overview of presentation
 Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Introduction to Structure Preparation and Visualization
 Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Docking with Water in the Binding Site using GOLD
 Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
Scoring functions for of protein-ligand docking: New routes towards old goals
 3nd Strasbourg Summer School on Chemoinformatics Strasbourg, June 25-29, 2012 Scoring functions for of protein-ligand docking: New routes towards old goals Christoph Sotriffer Institute of Pharmacy and
3nd Strasbourg Summer School on Chemoinformatics Strasbourg, June 25-29, 2012 Scoring functions for of protein-ligand docking: New routes towards old goals Christoph Sotriffer Institute of Pharmacy and
JCICS Major Research Areas
 JCICS Major Research Areas Chemical Information Text Searching Structure and Substructure Searching Databases Patents George W.A. Milne C571 Lecture Fall 2002 1 JCICS Major Research Areas Chemical Computation
JCICS Major Research Areas Chemical Information Text Searching Structure and Substructure Searching Databases Patents George W.A. Milne C571 Lecture Fall 2002 1 JCICS Major Research Areas Chemical Computation
What is Protein-Ligand Docking?
 MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
Tautomerism in chemical information management systems
 Tautomerism in chemical information management systems Dr. Wendy A. Warr http://www.warr.com Tautomerism in chemical information management systems Author: Wendy A. Warr DOI: 10.1007/s10822-010-9338-4
Tautomerism in chemical information management systems Dr. Wendy A. Warr http://www.warr.com Tautomerism in chemical information management systems Author: Wendy A. Warr DOI: 10.1007/s10822-010-9338-4
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Plan. Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics.
 and its used in Chemoinformatics.") Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Machine learning for ligand-based virtual screening and chemogenomics!
 Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
BioSolveIT. A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit
 October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
October 6 University Faculty of pharmacy Computer Aided Drug Design Unit CADD@O6U.edu.eg CADD Computer-Aided Drug Design Unit The development of new drugs is no longer a process of trial and error or strokes
The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score. Protein Structures: The Starting Point for New Drugs 2
 The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score Protein Structures: The Starting Point for New Drugs 2 Matthias Rarey Stefan Bietz Nadine Schneider Sascha Urbaczek University
The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score Protein Structures: The Starting Point for New Drugs 2 Matthias Rarey Stefan Bietz Nadine Schneider Sascha Urbaczek University
An Integrated Approach to in-silico
 An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
Pipeline Pilot Integration
 Scientific & technical Presentation Pipeline Pilot Integration Szilárd Dóránt July 2009 The Component Collection: Quick facts Provides access to ChemAxon tools from Pipeline Pilot Free of charge Open source
Scientific & technical Presentation Pipeline Pilot Integration Szilárd Dóránt July 2009 The Component Collection: Quick facts Provides access to ChemAxon tools from Pipeline Pilot Free of charge Open source
Protein-Ligand Docking
 Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
ChemAxon. Content. By György Pirok. D Standardization D Virtual Reactions. D Fragmentation. ChemAxon European UGM Visegrad 2008
 Transformers f off ChemAxon By György Pirok Content Standardization Virtual Reactions Metabolism M b li P Prediction di i Fragmentation 2 1 Standardization http://www.chemaxon.com/jchem/doc/user/standardizer.html
Transformers f off ChemAxon By György Pirok Content Standardization Virtual Reactions Metabolism M b li P Prediction di i Fragmentation 2 1 Standardization http://www.chemaxon.com/jchem/doc/user/standardizer.html
Practical QSAR and Library Design: Advanced tools for research teams
 DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
BIOINF Drug Design 2. Jens Krüger and Philipp Thiel Summer Lecture 5: 3D Structure Comparison Part 1: Rigid Superposition, Pharmacophores
 BIOINF 472 Drug Design 2 Jens Krüger and Philipp Thiel Summer 2014 Lecture 5: D Structure Comparison Part 1: Rigid Superposition, Pharmacophores Overview Comparison of D structures Rigid superposition
BIOINF 472 Drug Design 2 Jens Krüger and Philipp Thiel Summer 2014 Lecture 5: D Structure Comparison Part 1: Rigid Superposition, Pharmacophores Overview Comparison of D structures Rigid superposition
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification
 Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery
 21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
Computational analysis of the activity of pongachalcone I against highly resistant bacteria Pseudomonas putida
 Computational analysis of the activity of pongachalcone I against highly resistant bacteria Pseudomonas putida Satya B. Paul 1, Sudip Choudhury 2,* 1 Department of Chemistry, Assam University, Silchar,
Computational analysis of the activity of pongachalcone I against highly resistant bacteria Pseudomonas putida Satya B. Paul 1, Sudip Choudhury 2,* 1 Department of Chemistry, Assam University, Silchar,
STRUCTURAL BIOINFORMATICS II. Spring 2018
 STRUCTURAL BIOINFORMATICS II Spring 2018 Syllabus Course Number - Classification: Chemistry 5412 Class Schedule: Monday 5:30-7:50 PM, SERC Room 456 (4 th floor) Instructors: Ronald Levy, SERC 718 (ronlevy@temple.edu)
STRUCTURAL BIOINFORMATICS II Spring 2018 Syllabus Course Number - Classification: Chemistry 5412 Class Schedule: Monday 5:30-7:50 PM, SERC Room 456 (4 th floor) Instructors: Ronald Levy, SERC 718 (ronlevy@temple.edu)
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Analyzing Molecular Conformations Using the Cambridge Structural Database. Jason Cole Cambridge Crystallographic Data Centre
 Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Pose and affinity prediction by ICM in D3R GC3. Max Totrov Molsoft
 Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
SCULPT 3.0. Using SCULPT to Gain Competitive Insights. Brings 3D Visualization to the Lab Bench SPECIAL REPORT. 4 Molecular Connection Fall 1999
 SPECIAL REPORT SCULPT 3.0 Brings 3D Visualization to the Lab Bench ith the acquisition of Interactive Simulations, Inc., last April, MDL added to its arsenal of discovery informatics tools a powerful analysis
SPECIAL REPORT SCULPT 3.0 Brings 3D Visualization to the Lab Bench ith the acquisition of Interactive Simulations, Inc., last April, MDL added to its arsenal of discovery informatics tools a powerful analysis
Pharmaceutical e-learning at the University of Innsbruck
 Universität Innsbruck Pharmaceutical e-learning at the University of Innsbruck Dr. Daniela Schuster Institute of Pharmacy / Pharmaceutical Chemistry Outline How it all began: PharmXplorer Current applications
Universität Innsbruck Pharmaceutical e-learning at the University of Innsbruck Dr. Daniela Schuster Institute of Pharmacy / Pharmaceutical Chemistry Outline How it all began: PharmXplorer Current applications
Structure-Based Drug Discovery An Overview
 Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure