Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification
|
|
|
- Annabella Rodgers
- 5 years ago
- Views:
Transcription
1 Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux radoux@ccdc.cam.ac.uk 1
2 Introduction Hotspots Strongly attractive to organic molecules Organic molecules cluster at hotspots in multiple solvent crystal structures Capable of binding fragments Overcome limited binding interface Disproportionately large contribution to binding Analogous to PPIs 2
3 Hotspots and Fragments Chosen definition: The minimum binding site that will bind a fragment, maintaining the fragment binding position once it has been elaborated Fragment crystal structures to be used as a gold standard 3
4 Hotspots, Unhappy Waters and Fragments Unhappy waters (Young 2007): a strongly hydrophobic cavity that encloses multiple water molecules [or] one to three hydrogen bonds with the protein by the ligand, where the remainder of the local environment is hydrophobically enclosed Fragments (Ichihara 2014): Using WaterMap fragment hits tend to displace water molecules with notably unfavorable excess entropies configurationally constrained water molecules likely to be caused by confinement in hydrophobic pockets or a combination of hydrophobic enclosure with hydrogen bonds. Hotspots (Kozakov 2015):.concave topology combined with a mosaic-like pattern of hydrophobic and polar functionality 4
5 Hotspots, Unhappy Waters and Fragments Unhappy waters (Young 2007): a strongly hydrophobic cavity that encloses multiple water molecules [or] one to three hydrogen bonds with the protein by the ligand, where the remainder of the local environment is hydrophobically enclosed Fragments (Ichihara 2014):. confinement in hydrophobic pockets or a combination of hydrophobic enclosure with hydrogen bonds Using WaterMap fragment hits tend to displace water molecules with notably unfavorable excess entropies configurationally constrained water molecules likely to be caused by confinement in hydrophobic pockets or a combination of hydrophobic enclosure with hydrogen bonds. Hotspots (Kozakov 2015):.concave topology combined with a mosaic-like pattern of hydrophobic and polar functionality 5

6 Fragment Hotspot Maps Look for the environments that cause Hotspots Knowledge-based Faster calculations Aims: Start from a global search of an apo protein Find fragment binding sites Predict the interactions made by fragments 6
7 Representing Hotspot Environments Hydrogen bonds plus hydrophobic enclosure Or just Hydrophobic enclosure Use molecular probes that reflect hotspot environments 7
8 Atomic Propensities from SuperStar SuperStar 1 used to generate atomic propensities Red: Acceptor Blue: Donor Yellow: Hydrophobe Each grid point contains A propensity for each probe type A buriedness score 1. M. L. Verdonk, J. C. Cole and R. Taylor, J. Mol. Biol., 289, ,
9 Introducing Enclosure Fragment Hotspot Map method from this point onward Hydrogen bonds plus hydrophobic enclosure Multiply each propensity by the buriedness to represent enclosure Buriedness calculated using LIGSITE 1 Weighted propensity 1. Hendlich, M.; Rippmann, F.; Barnickel, G. J. Mol. Graph. Model. 1997, 15 (6), ,
10 Sampling Example: Acceptor probe Translate probe to acceptor grid points with score >= rotations Weighted propensity assigned to each atom Probe score = Geometric mean of atom scores = G.M
11 Creating Fragment Hotspot Maps Probe scores assigned to nearest grid point High scoring regions: High propensity for the polar atom (if applicable) Surrounding environment is hydrophobic Environment is enclosed Probe size eliminates pockets too small for binding 5-10 minutes on a desktop 12
12 Displaying the Output Contoured at: 0 13

13 Displaying the Output Contoured at: 10 14
14 Displaying the Output Contoured at: 11 15
15 Displaying the Output Contoured at: 12 16
16 Displaying the Output Contoured at: 13 17
17 Displaying the Output Contoured at: 14 18
18 Displaying the Output Contoured at: 15 19
19 Displaying the Output Contoured at: 16 20
20 Displaying the Output Contoured at: 17 21
21 Displaying the Output Contoured at: 18 22
22 Displaying the Output Contoured at: 19 23

23 Validation Dataset previously published by Ichihara and colleagues 1 Set of fragment- and leadbound protein crystal structures Fragment binding position maintained in lead Perfect dataset for testing hotspot prediction The minimum binding site that will bind a fragment, maintaining the fragment binding position once it has been elaborated 1. O. Ichihara, Y. Shimada and D. Yoshidome, ChemMedChem, 2014, 9,


24 Validation 21 fragment and lead-bound protein crystal structures We added corresponding apo structures Maps calculated for apo structures Scores assigned to ligand atoms from their corresponding maps 25 C. J. Radoux, T. S. G. Olsson, W. R. Pitt, C. R. Groom and T. L. Blundell, J. Med. Chem., 2016, 59,

25 Results Across whole dataset Fragment atoms consistently in the highest scoring regions Beyond binding site prediction Where can fragments bind? Which interactions will the fragments make? Fragment Lead 26

26 Current Limitation No charged probes, charged interactions often missed Previously implemented, but gave unsatisfactory results Polar probes do a poor job of predicting these interactions Two types of hotspot? Ichihara 1 and colleagues suggested fragments displace two types of unhappy water Looking into handling charged probes differently 1. O. Ichihara, Y. Shimada and D. Yoshidome, ChemMedChem, 2014, 9,


27 Ligandability Assessment Pocket or subpocket ligandability assessment Scores comparable between proteins Calibrated using fragment scores 17+ Strong hotspot May be able to bind fragments <14 Unlikely to lead to fragment binding 28

28 fragment-hotspot-maps.ccdc.cam.ac.uk 29

29 fragment-hotspot-maps.ccdc.cam.ac.uk 30
30 fragment-hotspot-maps.ccdc.cam.ac.uk 31
 results = h.from_file('1hcl.")
31 Integration with CSD Python API Hotspot calculations run from a ccdc protein object or a PBD file Returned results object has functions for using the hotspot maps: Score ligand Score protein Write pharmacophore file Assign protein H bond constraints for GOLD Output PyMOL file from fragment_hotspots import Hotspots # Calculate hotspots h = Hotspots() results = h.from_file('1hcl.pdb', smooth=true) # Write out a CrossMiner pharmacophore file results.write_pharmacophore_file(output_format='crossminer') # Visualise with PyMOL results.output_pymol_file(pharmacophores=true) 32
 Difficult to dock 11 non-terminal rotatable bonds Partially open pocket")
32 Application to Docking with GOLD Docking S- adenosylhomocysteine (SAH) to mixed lineage leukaemia (mll1) Difficult to dock 11 non-terminal rotatable bonds Partially open pocket 33
 prot = Protein.from_file('2w5y_protein_prepared.mol2') native_mol = io.moleculereader('sah_native.")
33 Application to Docking with GOLD Histidine N gives highest scoring interaction Adding constraint leads to correct result from fragment_hotspots import Hotspots from ccdc_internal.docking import Docker from ccdc.protein import Protein from ccdc import io h = Hotspots() prot = Protein.from_file('2w5y_protein_prepared.mol2') native_mol = io.moleculereader('sah_native.mol2')[0] # Calculate hotspots hotspot_results = h.from_protein(prot) # Set up docking s = Docker.Settings() s.add_protein_file('2w5y_protein_prepared.mol2') s.add_ligand_file('sah_native.mol2', ndocks=15) s.reference_ligand_file = 'SAH_native.mol2' s.binding_site = s.bindingsitefromligand(prot, native_mol) # Add constraints to Docker.settings object hotspot_results.predict_protein_hbond_constraints(s) # Run docking docker = Docker(settings=s) results = docker.dock() hotspot_results.output_pymol_file(prot_file=prot_file) 34

34 Hotspot Derived Pharmacophores Discrete hotspot pharmacophores Convert maps into discrete fragment sized hotspots Represent hotspot as a pharmacophore Search for molecules that match pharmacophore 35

35 Guiding Hit to Lead A. W. Hung, H. L. Silvestre, S. Wen, A. Ciulli, T. L. Blundell and C. Abell, Angew. Chem. Int. Ed. Engl., 2009, 48,
36 Guiding Hit to Lead A. W. Hung, H. L. Silvestre, S. Wen, G. P. C. George, J. Boland, T. L. Blundell, A. Ciulli and C. Abell, ChemMedChem, 2016, 11,
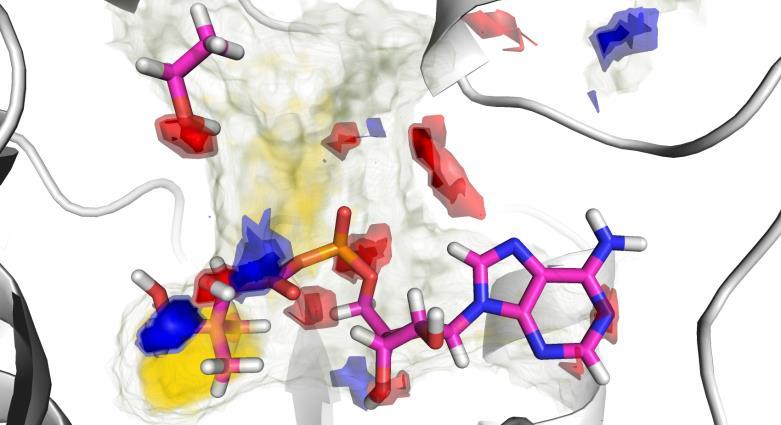

37 38

38 MD and Hotspots What about flexibility? Example Bcl-xl Able to process 6000 structures in ~48 hours Visualise dynamic maps Summary maps over trajectory Average Maximum 39

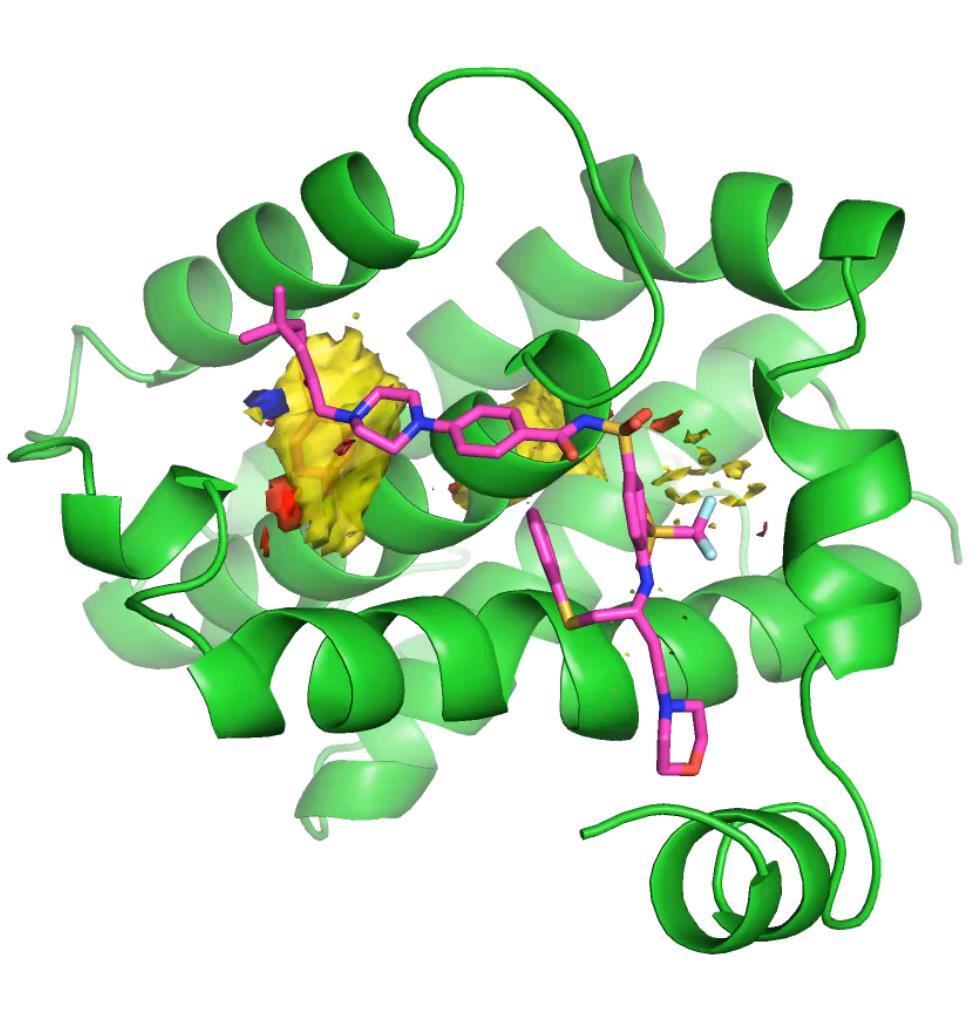
39 MD and Hotspots 40
40 Summary Fragment Hotspot Maps can add knowledge at several stages of the early drug discovery process Target tractability assessment Presence of a hotspot indicates a ligandable target Evaluate suitability of a pocket for fragment-based approaches Hit ID Identification important interactions can be used to guide docking, or other virtual screening method. Hit to lead Can be used as a visual guide to direct medicinal chemists Can highlight sub optimal interactions 41
 Will Pitt")
 Blundell group")

41 Acknowledgements Supervisors Colin Groom (CCDC) Will Pitt (UCB) Tom Blundell (Biochemistry) Blundell group CCDC Life sciences team UCB 42
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
Identifying Interaction Hot Spots with SuperStar
 Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
CSD. Unlock value from crystal structure information in the CSD
 CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
Detection of Protein Binding Sites II
 Detection of Protein Binding Sites II Goal: Given a protein structure, predict where a ligand might bind Thomas Funkhouser Princeton University CS597A, Fall 2007 1hld Geometric, chemical, evolutionary
Detection of Protein Binding Sites II Goal: Given a protein structure, predict where a ligand might bind Thomas Funkhouser Princeton University CS597A, Fall 2007 1hld Geometric, chemical, evolutionary
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Flexibility and Constraints in GOLD
 Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
The reuse of structural data for fragment binding site prediction
 The reuse of structural data for fragment binding site prediction Richard Hall 1 Motivation many examples of fragments binding in a phenyl shaped pocket or a kinase slot good shape complementarity between
The reuse of structural data for fragment binding site prediction Richard Hall 1 Motivation many examples of fragments binding in a phenyl shaped pocket or a kinase slot good shape complementarity between
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
György M. Keserű H2020 FRAGNET Network Hungarian Academy of Sciences
 Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Targeting protein-protein interactions: A hot topic in drug discovery
 Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites. J. Andrew Surface
 Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
SuperStar User Guide and Tutorials
 SuperStar User Guide and Tutorials 2.1.3 Release Copyright 2015 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use SuperStar is a back-end release of SuperStar. If you
SuperStar User Guide and Tutorials 2.1.3 Release Copyright 2015 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use SuperStar is a back-end release of SuperStar. If you
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
GOLD User Guide. 5.5 Release. To access our new format tutorials please
 GOLD User Guide A Component of the GOLD Suite 5.5 Release Copyright 2016 Cambridge Crystallographic Data Centre Registered Charity No 800579 To access our new format tutorials please visit the GOLD web
GOLD User Guide A Component of the GOLD Suite 5.5 Release Copyright 2016 Cambridge Crystallographic Data Centre Registered Charity No 800579 To access our new format tutorials please visit the GOLD web
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Pose Prediction with GOLD
 Pose Prediction with GOLD Version 3.0 November 2018 GOLD v5.7.0 Table of Contents The Purpose of Docking... 2 GOLD s Evolutionary Algorithm... 3 A Checklist for Docking... 3 GOLD and Hermes... 3 Redocking
Pose Prediction with GOLD Version 3.0 November 2018 GOLD v5.7.0 Table of Contents The Purpose of Docking... 2 GOLD s Evolutionary Algorithm... 3 A Checklist for Docking... 3 GOLD and Hermes... 3 Redocking
11. IN SILICO DOCKING ANALYSIS
 11. IN SILICO DOCKING ANALYSIS 11.1 Molecular docking studies of major active constituents of ethanolic extract of GP The major active constituents are identified from the ethanolic extract of Glycosmis
11. IN SILICO DOCKING ANALYSIS 11.1 Molecular docking studies of major active constituents of ethanolic extract of GP The major active constituents are identified from the ethanolic extract of Glycosmis
The Cambridge Structural Database (CSD) a Vital Resource for Structural Chemistry and Biology Stephen Maginn, CCDC, Cambridge, UK
 a Vital Resource for Structural Chemistry and Biology Stephen Maginn, CCDC, Cambridge, UK") The Cambridge Structural Database (CSD) a Vital Resource for Structural Chemistry and Biology Stephen Maginn, CCDC, Cambridge, UK 1 The Cambridge Crystallographic Data Centre The advancement and promotion
The Cambridge Structural Database (CSD) a Vital Resource for Structural Chemistry and Biology Stephen Maginn, CCDC, Cambridge, UK 1 The Cambridge Crystallographic Data Centre The advancement and promotion
Protein-Ligand Docking Evaluations
 Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Advanced Medicinal Chemistry SLIDES B
 Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Fragment based drug discovery in teams of medicinal and computational chemists. Carsten Detering
 Fragment based drug discovery in teams of medicinal and computational chemists Carsten Detering BioSolveIT Quick Facts Founded in 2001 by the developers of FlexX ~20 people Core expertise: docking, screening,
Fragment based drug discovery in teams of medicinal and computational chemists Carsten Detering BioSolveIT Quick Facts Founded in 2001 by the developers of FlexX ~20 people Core expertise: docking, screening,
Plan. Day 2: Exercise on MHC molecules.
 Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Viewing and Analyzing Proteins, Ligands and their Complexes 2
 2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Docking with Water in the Binding Site using GOLD
 Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
A Brief Guide to All Atom/Coarse Grain Simulations 1.1
 A Brief Guide to All Atom/Coarse Grain Simulations 1.1 Contents 1. Introduction 2. AACG simulations 2.1. Capabilities and limitations 2.2. AACG commands 2.3. Visualization and analysis 2.4. AACG simulation
A Brief Guide to All Atom/Coarse Grain Simulations 1.1 Contents 1. Introduction 2. AACG simulations 2.1. Capabilities and limitations 2.2. AACG commands 2.3. Visualization and analysis 2.4. AACG simulation
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Analyzing Molecular Conformations Using the Cambridge Structural Database. Jason Cole Cambridge Crystallographic Data Centre
 Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery
 AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
Other Cells. Hormones. Viruses. Toxins. Cell. Bacteria
 Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors
 MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Introduction to Structure Preparation and Visualization
 Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
GOLD Configuration File User Guide
 GOLD Configuration File User Guide A Component of the GOLD Suite 5.6.1 GOLDSuite Release Copyright 2018 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The GOLD suite
GOLD Configuration File User Guide A Component of the GOLD Suite 5.6.1 GOLDSuite Release Copyright 2018 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The GOLD suite
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
The Conformation Search Problem
 Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Version 1.2 October 2017 CSD v5.39
 Mogul Geometry Check Table of Contents Introduction... 2 Example 1. Using Mogul to assess intramolecular geometry... 3 Example 2. Using Mogul to explain activity data... 5 Conclusions... 8 Further Exercises...
Mogul Geometry Check Table of Contents Introduction... 2 Example 1. Using Mogul to assess intramolecular geometry... 3 Example 2. Using Mogul to explain activity data... 5 Conclusions... 8 Further Exercises...
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Chemoinformatics and information management. Peter Willett, University of Sheffield, UK
 Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Chemoinformatics and information management Peter Willett, University of Sheffield, UK verview What is chemoinformatics and why is it necessary Managing structural information Typical facilities in chemoinformatics
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Biological Mass Spectrometry
 Biochemistry 412 Biological Mass Spectrometry February 13 th, 2007 Proteomics The study of the complete complement of proteins found in an organism Degrees of Freedom for Protein Variability Covalent Modifications
Biochemistry 412 Biological Mass Spectrometry February 13 th, 2007 Proteomics The study of the complete complement of proteins found in an organism Degrees of Freedom for Protein Variability Covalent Modifications
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Tutorial: Structural Analysis of a Protein-Protein Complex
 Molecular Modeling Section (MMS) Department of Pharmaceutical and Pharmacological Sciences University of Padova Via Marzolo 5-35131 Padova (IT) @contact: stefano.moro@unipd.it Tutorial: Structural Analysis
Molecular Modeling Section (MMS) Department of Pharmaceutical and Pharmacological Sciences University of Padova Via Marzolo 5-35131 Padova (IT) @contact: stefano.moro@unipd.it Tutorial: Structural Analysis
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
New approaches to scoring function design for protein-ligand binding affinities. Richard A. Friesner Columbia University
 New approaches to scoring function design for protein-ligand binding affinities Richard A. Friesner Columbia University Overview Brief discussion of advantages of empirical scoring approaches Analysis
New approaches to scoring function design for protein-ligand binding affinities Richard A. Friesner Columbia University Overview Brief discussion of advantages of empirical scoring approaches Analysis
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Protein-Ligand Interactions* and Energy Evaluation Methods
 Protein-Ligand Interactions* and Energy Evaluation Methods *with a revealing look at roles of water Glen E. Kellogg Department of Medicinal Chemistry Institute for Structural Biology, Drug Discovery &
Protein-Ligand Interactions* and Energy Evaluation Methods *with a revealing look at roles of water Glen E. Kellogg Department of Medicinal Chemistry Institute for Structural Biology, Drug Discovery &
Validation of Experimental Crystal Structures
 Validation of Experimental Crystal Structures Aim This use case focuses on the subject of validating crystal structures using tools to analyse both molecular geometry and intermolecular packing. Introduction
Validation of Experimental Crystal Structures Aim This use case focuses on the subject of validating crystal structures using tools to analyse both molecular geometry and intermolecular packing. Introduction
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses
 Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules
 Software News and Updates tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules DANIEL SEELIGER, BERT L. DE GROOT Computational Biomolecular Dynamics Group, Max-Planck-Institute
Software News and Updates tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules DANIEL SEELIGER, BERT L. DE GROOT Computational Biomolecular Dynamics Group, Max-Planck-Institute
Plan. Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics.
 and its used in Chemoinformatics.") Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Toward an Understanding of GPCR-ligand Interactions. Alexander Heifetz
 Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Bioisosteres in Medicinal Chemistry
 Edited by Nathan Brown Bioisosteres in Medicinal Chemistry VCH Verlag GmbH & Co. KGaA Contents List of Contributors Preface XV A Personal Foreword XI XVII Part One Principles 1 Bioisosterism in Medicinal
Edited by Nathan Brown Bioisosteres in Medicinal Chemistry VCH Verlag GmbH & Co. KGaA Contents List of Contributors Preface XV A Personal Foreword XI XVII Part One Principles 1 Bioisosterism in Medicinal
Practical QSAR and Library Design: Advanced tools for research teams
 DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
Tutorial: Molecular docking
 Drug Design Tutorial: Molecular docking RNDr. Karel Berka, PhD RNDr. Jindřich Fanfrlík, PhD RNDr. Martin Lepšík, PhD Dpt. Physical Chemistry, RCPTM, Faculty of Science, Palacky University in Olomouc Tutorial
Drug Design Tutorial: Molecular docking RNDr. Karel Berka, PhD RNDr. Jindřich Fanfrlík, PhD RNDr. Martin Lepšík, PhD Dpt. Physical Chemistry, RCPTM, Faculty of Science, Palacky University in Olomouc Tutorial
Structure-based maximal affinity model predicts small-molecule druggability
 Structure-based maximal affinity model predicts small-molecule druggability Alan Cheng alan.cheng@amgen.com IMA Workshop (Jan 17, 2008) Druggability prediction Introduction Affinity model Some results
Structure-based maximal affinity model predicts small-molecule druggability Alan Cheng alan.cheng@amgen.com IMA Workshop (Jan 17, 2008) Druggability prediction Introduction Affinity model Some results
Bridging the Dimensions:
 Bridging the Dimensions: Seamless Integration of 3D Structure-based Design and 2D Structure-activity Relationships to Guide Medicinal Chemistry ACS Spring National Meeting. COMP, March 13 th 2016 Marcus
Bridging the Dimensions: Seamless Integration of 3D Structure-based Design and 2D Structure-activity Relationships to Guide Medicinal Chemistry ACS Spring National Meeting. COMP, March 13 th 2016 Marcus
Autodock tutorial VINA with UCSF Chimera
 Autodock tutorial VINA with UCSF Chimera José R. Valverde CNB/CSIC jrvalverde@cnb.csic.es José R. Valverde, 2014 CC-BY-NC-SA Loading the receptor Open UCSF Chimera and then load the protein: File Open
Autodock tutorial VINA with UCSF Chimera José R. Valverde CNB/CSIC jrvalverde@cnb.csic.es José R. Valverde, 2014 CC-BY-NC-SA Loading the receptor Open UCSF Chimera and then load the protein: File Open
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Hydrogen Bonding & Molecular Design Peter
 Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
Chem 253. Tutorial for Materials Studio
 Chem 253 Tutorial for Materials Studio This tutorial is designed to introduce Materials Studio 7.0, which is a program used for modeling and simulating materials for predicting and rationalizing structure
Chem 253 Tutorial for Materials Studio This tutorial is designed to introduce Materials Studio 7.0, which is a program used for modeling and simulating materials for predicting and rationalizing structure
Molecular dynamics simulations of anti-aggregation effect of ibuprofen. Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov
 Biophysical Journal, Volume 98 Supporting Material Molecular dynamics simulations of anti-aggregation effect of ibuprofen Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov Supplemental
Biophysical Journal, Volume 98 Supporting Material Molecular dynamics simulations of anti-aggregation effect of ibuprofen Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov Supplemental
Machine-learning scoring functions for docking
 Machine-learning scoring functions for docking Dr Pedro J Ballester MRC Methodology Research Fellow EMBL-EBI, Cambridge, United Kingdom EBI is an Outstation of the European Molecular Biology Laboratory.
Machine-learning scoring functions for docking Dr Pedro J Ballester MRC Methodology Research Fellow EMBL-EBI, Cambridge, United Kingdom EBI is an Outstation of the European Molecular Biology Laboratory.
SUPPLEMENTARY INFORMATION
 Figure S1. Secondary structure of CAP (in the camp 2 -bound state) 10. α-helices are shown as cylinders and β- strands as arrows. Labeling of secondary structure is indicated. CDB, DBD and the hinge are
Figure S1. Secondary structure of CAP (in the camp 2 -bound state) 10. α-helices are shown as cylinders and β- strands as arrows. Labeling of secondary structure is indicated. CDB, DBD and the hinge are
Ranking of HIV-protease inhibitors using AutoDock
 Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Dock Ligands from a 2D Molecule Sketch
 Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
Structure Investigation of Fam20C, a Golgi Casein Kinase
 Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Protein-Ligand Docking
 Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows
 MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
Microcalorimetry for the Life Sciences
 Microcalorimetry for the Life Sciences Why Microcalorimetry? Microcalorimetry is universal detector Heat is generated or absorbed in every chemical process In-solution No molecular weight limitations Label-free
Microcalorimetry for the Life Sciences Why Microcalorimetry? Microcalorimetry is universal detector Heat is generated or absorbed in every chemical process In-solution No molecular weight limitations Label-free
BUDE. A General Purpose Molecular Docking Program Using OpenCL. Richard B Sessions
 BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
Cross Discipline Analysis made possible with Data Pipelining. J.R. Tozer SciTegic
 Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Electronic Supplementary Information Effective lead optimization targeted for displacing bridging water molecule
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Electronic Supplementary Information Effective lead optimization targeted for displacing
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Electronic Supplementary Information Effective lead optimization targeted for displacing
Implementation of novel tools to facilitate fragment-based drug discovery by NMR:
 Implementation of novel tools to facilitate fragment-based drug discovery by NMR: Automated analysis of large sets of ligand-observed NMR binding data and 19 F methods Andreas Lingel Global Discovery Chemistry
Implementation of novel tools to facilitate fragment-based drug discovery by NMR: Automated analysis of large sets of ligand-observed NMR binding data and 19 F methods Andreas Lingel Global Discovery Chemistry