Bridging the Dimensions:
|
|
|
- Marybeth Sullivan
- 5 years ago
- Views:
Transcription


1 Bridging the Dimensions: Seamless Integration of 3D Structure-based Design and 2D Structure-activity Relationships to Guide Medicinal Chemistry ACS Spring National Meeting. COMP, March 13 th 2016 Marcus Gastreich, Matthew Segall, Carsten Detering, Ed Champness, Christian Lemmen Optibrium, StarDrop, Auto-Modeller, Card View and Glowing Molecule are trademarks of Optibrium Ltd.
2 Overview 2-dimensional (2D) structure-activity relationships (SAR) Qualitative: Activity cliffs, matched molecular pair analysis Quantitative: QSAR models 3-dimensional (3D) structure-based design Scoring/affinity prediction Understanding 3D SAR Linking 2D and 3D SAR to guide design Conclusions 2
3 2D Structure-Activity Relationships
4 Qualitative SAR Many methods are routinely used for analysis of data to reveal patterns and trends to guide compound optimisation, e.g. Clustering Group similar compounds to identify series with interesting SAR Activity cliff detection Small changes in structure that cause a large change in activity Matched molecular pair analysis Pairs of compounds that are identical except for one small change at the same position 4



 Drug Discov.")
5 Visualising 2D SAR Card View Freedom from the constraints of chemical spreadsheets Represent compound relationships Work the way you think Cards: Display key compound data Stacks: Summarise and compare data for groups of compounds Links: Highlight compound relationships Intuitive visualisation of analyses Clustering, activity cliffs, matched molecular pairs Quickly identify optimisation strategies M.D. Segall et al. (2015) Drug Discov. Today 20(9) pp
 Drug Discov. Today 20(9) pp.")
6 Activity Neighbourhood Activity Cliff Visualisation M.D. Segall et al. (2015) Drug Discov. Today 20(9) pp
 Drug Discov. Today 20(9) pp.")
7 Matched Molecular Pair Analysis M.D. Segall et al. (2015) Drug Discov. Today 20(9) pp
8 Quantitative Structure-Activity Relationships Principles Data y = f(x 1, x 2, x 3, ) Statistical uncertainty Quality data is essential Public data needs very careful curation (and may not be good enough) Descriptors, e.g. Whole molecule properties, e.g. logp, MW, PSA Structural descriptors, SMARTS, fingerprints Statistical fitting or machine learning method, e.g. Partial least squares, artificial neural networks, support vector machines, random forest, Gaussian processes Widely applied to prediction of ADME and physicochemical properties Please see talk: COMP Wed. 8.40am Room 25C 8
 Project specific scoring profile Compounds ranked by likelihood of success Histograms for quick visual guide to compound properties M.D. Segall (2012) Curr. Pharm. Des. 18(9) pp.")
9 Multi-Parameter Optimisation Probabilistic Scoring Integrated assessment of data against project criteria Accounts for the uncertainties in all compound-related data (experimental or calculated) Project specific scoring profile Compounds ranked by likelihood of success Histograms for quick visual guide to compound properties M.D. Segall (2012) Curr. Pharm. Des. 18(9) pp


 Chem. & Biodiv.")
10 Interactive Redesign QSAR models provide estimates of compounds properties Instant feedback on how properties are likely to change Explore strategies for redesign But, important questions Why is a property value predicted? Where can I change this property? Glowing Molecule : Visual indication of structural influences on predicted properties M.D. Segall et al. (2009) Chem. & Biodiv. 6(11) p

11 3D Structure-Based Design 2016 BioSolveIT 11

12 Visual Understanding of 3D Affinity Data 2016 BioSolveIT 12

13 HYDE: A Different Energetics* Physics only No calibration to complexes Relates to real Free Energies *CINF 9: Christian Lemmen, Predicting binding affinity doesn't work, or does it? Next talk! 2016 BioSolveIT 13




14 HYDE Scoring Function Concept* G H-bond G dehydration G i HYDE= atom i G i dehydration + G i H bond *Reulecke et al., ChemMedChem BioSolveIT




15 HYDE color code: + G contribution - G contribution no G contribution Hyde - Visual Affinities receptor carbonyl oxygen ligand aromatic oxygen total desolvation cost Nr. 8.2 kj/mol 2.4 kj/mol 10.6 kj/mol receptor aromatic carbons -5.2 kj/mol N C H O N7 O8 C6 ligand aromatic carbon total desolvation gain receptor amide N dehydrat interaction energy ligand aromatic N dehydrat -2.0 kj/mol -7.2 kj/mol 6.3 kj/mol -7.4 kj/mol 6.4 kj/mol interaction energy -7.5 kj/mol total H-bond energy -2.2 kj/mol 2016 BioSolveIT
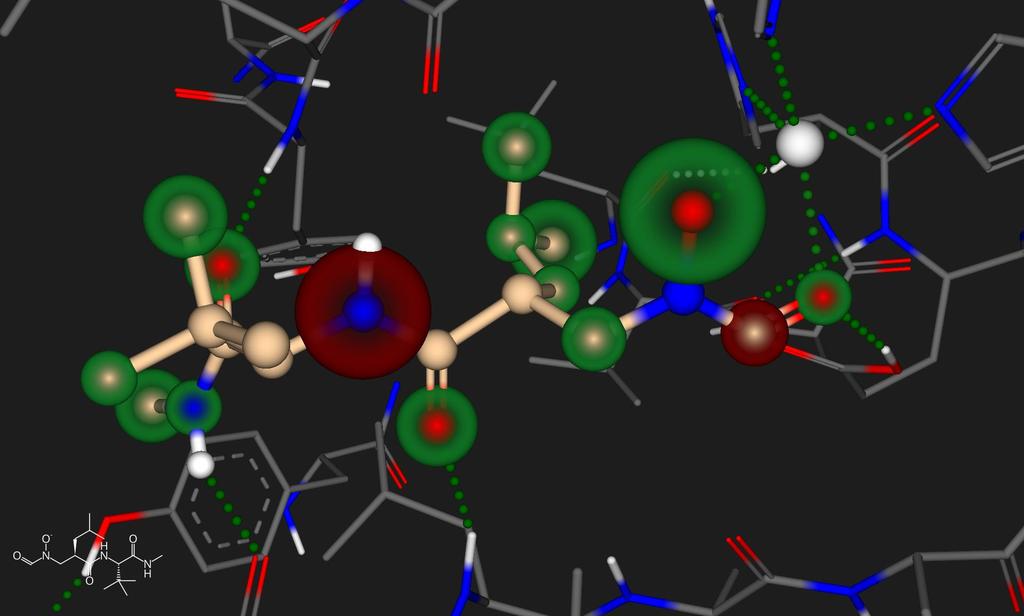


16 Visual Understanding of 3D Affinity Data PDB: 1GKC 2016 BioSolveIT

17 Linking 2D and 3D SAR to Guide Design
18 Understanding Activity Cliffs in 3D PPAR γ PDB 4EMA 18
19 Understanding Activity Cliffs in 3D PPAR γ PDB 4EMA 19

20 Understanding Activity Cliffs in 3D PPAR γ PDB 4EMA 20

21 Understanding Activity Cliffs in 3D PPAR γ PDB 4EMA 21
22 Understanding Activity Cliffs in 3D PPAR γ PDB 4EMA 22

23 Exploration of Virtual Screening Results HSP90 Crystal structure PDB ref. 2XJX Virtual library generated using STORM workflow in KNIME Amide substitution using Schotten Baumann reaction on beta resorcylic core Building blocks from vendor catalogues Tail of molecule not contributing to affinity Resulting library docked with FlexX Scored using SeeSAR and HYDE to estimate pk i 23

24 Matched Molecular Pair Analysis 24

25 Matched Molecular Pair Analysis 25
26 Matched Molecular Pair Analysis 26
27 Matched Molecular Pair Analysis 27



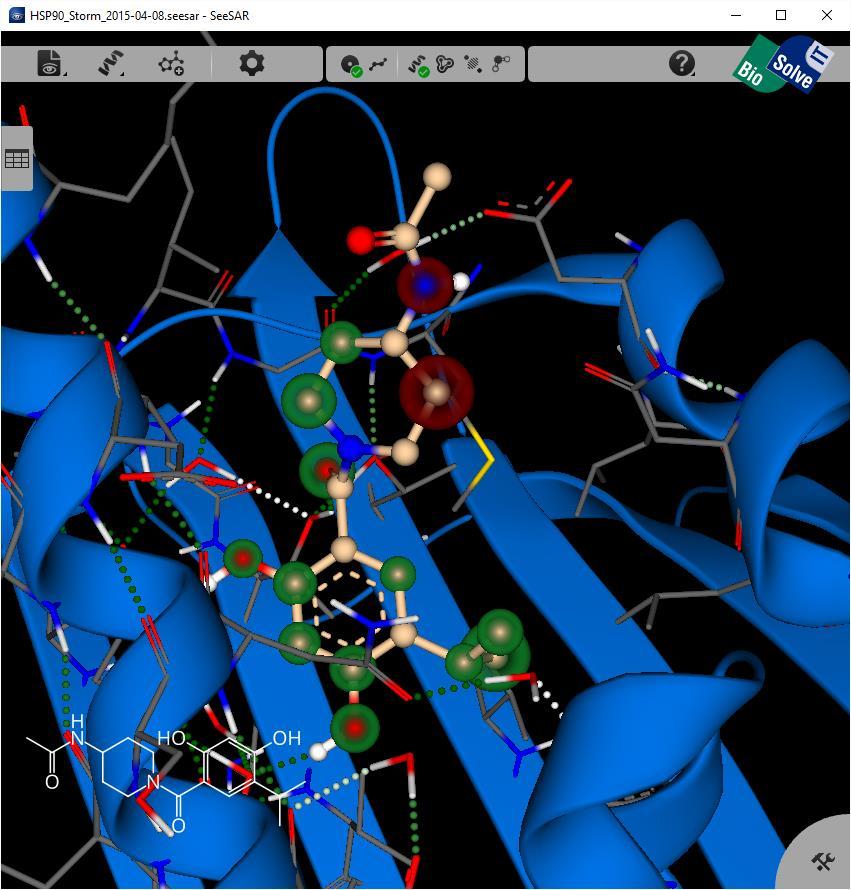
28 HYDE Analysis in SeeSAR 28

29 Combine with 2D QSAR Predictions Multi-parameter optimisation 29

30 3D View... Optimisation opportunities 30

31 HYDE Analysis in SeeSAR 31

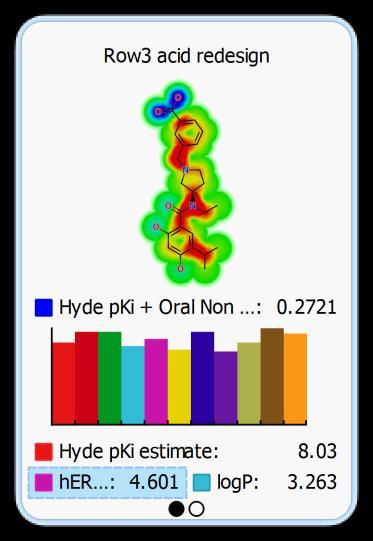
32 Optimisation Idea Modify pka of Nitrogen by amide substitution 32
33 Optimisation Idea Add polar group to phenyl ring 33

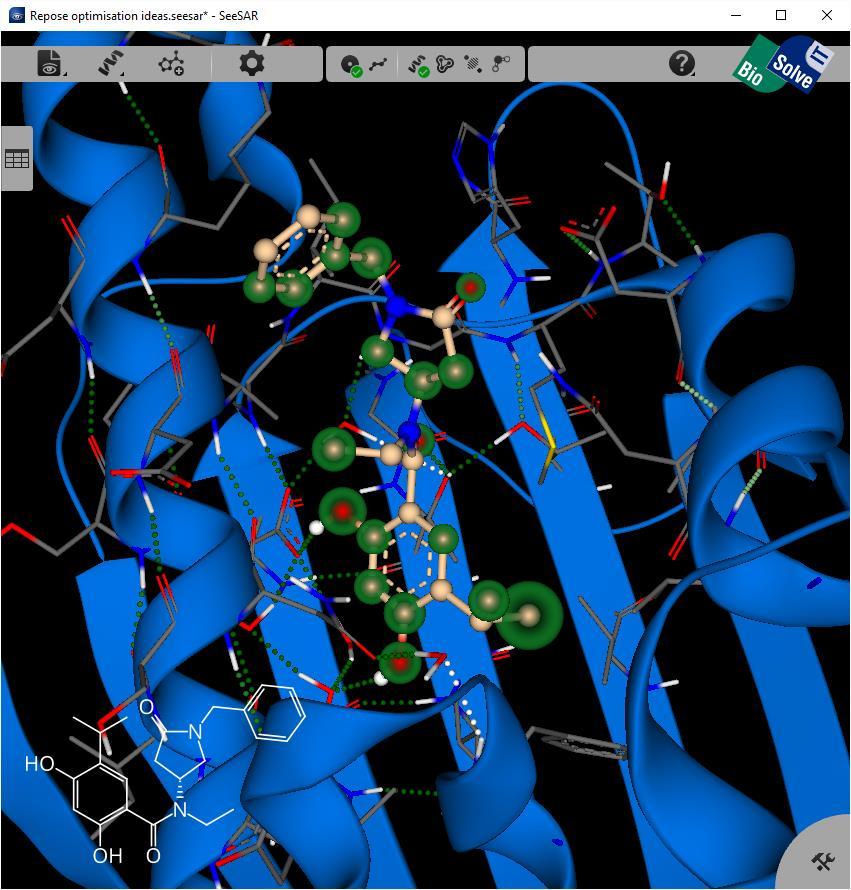
34 Repose in SeeSAR 34


35 Conclusions Both 2D and 3D information are important to interpret SAR and guide design A seamless combination between these two views of the chemical world maximises the benefits that they bring Understanding SAR from experimental data Analysis of virtual screening/docking results Multi-parameter optimisation of potency, physicochemical and ADMET properties For more information: and Optibrium: Booth 1227 or outside of room 6E (MEDI) 35



36 Free Hands-On Workshop Seamless Integration of 2D and 3D SAR to Guide MPO Where: San Diego Convention Center, Room 15B When: Monday 3:30pm to 6pm Practical examples with SeeSAR and StarDrop Spaces are limited, so please register at Booth #1227 in the Exhibition Hall 36
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Exploring the chemical space of screening results
 Exploring the chemical space of screening results Edmund Champness, Matthew Segall, Chris Leeding, James Chisholm, Iskander Yusof, Nick Foster, Hector Martinez ACS Spring 2013, 7 th April 2013 Optibrium,
Exploring the chemical space of screening results Edmund Champness, Matthew Segall, Chris Leeding, James Chisholm, Iskander Yusof, Nick Foster, Hector Martinez ACS Spring 2013, 7 th April 2013 Optibrium,
Translating Methods from Pharma to Flavours & Fragrances
 Translating Methods from Pharma to Flavours & Fragrances CINF 27: ACS National Meeting, New Orleans, LA - 18 th March 2018 Peter Hunt, Edmund Champness, Nicholas Foster, Tamsin Mansley & Matthew Segall
Translating Methods from Pharma to Flavours & Fragrances CINF 27: ACS National Meeting, New Orleans, LA - 18 th March 2018 Peter Hunt, Edmund Champness, Nicholas Foster, Tamsin Mansley & Matthew Segall
Gaussian Processes: We demand rigorously defined areas of uncertainty and doubt
 Gaussian Processes: We demand rigorously defined areas of uncertainty and doubt ACS Spring National Meeting. COMP, March 16 th 2016 Matthew Segall, Peter Hunt, Ed Champness matt.segall@optibrium.com Optibrium,
Gaussian Processes: We demand rigorously defined areas of uncertainty and doubt ACS Spring National Meeting. COMP, March 16 th 2016 Matthew Segall, Peter Hunt, Ed Champness matt.segall@optibrium.com Optibrium,
Relative Drug Likelihood: Going beyond Drug-Likeness
 Relative Drug Likelihood: Going beyond Drug-Likeness ACS Fall National Meeting, August 23rd 2012 Matthew Segall, Iskander Yusof Optibrium, StarDrop, Auto-Modeller and Glowing Molecule are trademarks of
Relative Drug Likelihood: Going beyond Drug-Likeness ACS Fall National Meeting, August 23rd 2012 Matthew Segall, Iskander Yusof Optibrium, StarDrop, Auto-Modeller and Glowing Molecule are trademarks of
Quantum Mechanical Models of P450 Metabolism to Guide Optimization of Metabolic Stability
 Quantum Mechanical Models of P450 Metabolism to Guide Optimization of Metabolic Stability Optibrium Webinar 2015, June 17 2015 Jonathan Tyzack, Matthew Segall, Peter Hunt Optibrium, StarDrop, Auto-Modeller
Quantum Mechanical Models of P450 Metabolism to Guide Optimization of Metabolic Stability Optibrium Webinar 2015, June 17 2015 Jonathan Tyzack, Matthew Segall, Peter Hunt Optibrium, StarDrop, Auto-Modeller
Applying Bioisosteric Transformations to Predict Novel, High Quality Compounds
 Applying Bioisosteric Transformations to Predict Novel, High Quality Compounds Dr James Chisholm,* Dr John Barnard, Dr Julian Hayward, Dr Matthew Segall*, Mr Edmund Champness*, Dr Chris Leeding,* Mr Hector
Applying Bioisosteric Transformations to Predict Novel, High Quality Compounds Dr James Chisholm,* Dr John Barnard, Dr Julian Hayward, Dr Matthew Segall*, Mr Edmund Champness*, Dr Chris Leeding,* Mr Hector
2009 Optibrium Ltd. Optibrium, STarDrop, Auto-Modeler and Glowing Molecule are trademarks of Optibrium Ltd.
 Enabling Interactive Multi-Objective Optimization for Drug Discovery Scientists Matthew Segall matt.segall@optibrium.com Lorentz Workshop Optimizing Drug Design 2009 Optibrium Ltd. Optibrium, STarDrop,
Enabling Interactive Multi-Objective Optimization for Drug Discovery Scientists Matthew Segall matt.segall@optibrium.com Lorentz Workshop Optimizing Drug Design 2009 Optibrium Ltd. Optibrium, STarDrop,
Development of a Structure Generator to Explore Target Areas on Chemical Space
 Development of a Structure Generator to Explore Target Areas on Chemical Space Kimito Funatsu Department of Chemical System Engineering, This materials will be published on Molecular Informatics Drug Development
Development of a Structure Generator to Explore Target Areas on Chemical Space Kimito Funatsu Department of Chemical System Engineering, This materials will be published on Molecular Informatics Drug Development
Fragment based drug discovery in teams of medicinal and computational chemists. Carsten Detering
 Fragment based drug discovery in teams of medicinal and computational chemists Carsten Detering BioSolveIT Quick Facts Founded in 2001 by the developers of FlexX ~20 people Core expertise: docking, screening,
Fragment based drug discovery in teams of medicinal and computational chemists Carsten Detering BioSolveIT Quick Facts Founded in 2001 by the developers of FlexX ~20 people Core expertise: docking, screening,
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
BioSolveIT. A Combinatorial Approach for Handling of Protonation and Tautomer Ambiguities in Docking Experiments
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
N- and S-Oxidation Model of the Flavin-containing MonoOxygenases
 N- and S-Oxidation Model of the Flavin-containing MonoOxygenases Peter J. Walton, Mario Ӧeren, Peter Hunt, Matthew Segall Optibrium, StarDrop, Auto-Modeller, Card View, Glowing Molecule and Augmented Chemistry
N- and S-Oxidation Model of the Flavin-containing MonoOxygenases Peter J. Walton, Mario Ӧeren, Peter Hunt, Matthew Segall Optibrium, StarDrop, Auto-Modeller, Card View, Glowing Molecule and Augmented Chemistry
BioSolveIT. A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Rules for drug discovery: can simple property criteria help you to find a drug?
 Rules for drug discovery: can simple property criteria help you to find a drug? The years since the publication of Lipinski s Rule of Five (Ro5) 1 in 1997 have seen the growth of a minor industry, dedicated
Rules for drug discovery: can simple property criteria help you to find a drug? The years since the publication of Lipinski s Rule of Five (Ro5) 1 in 1997 have seen the growth of a minor industry, dedicated
Building innovative drug discovery alliances. Just in KNIME: Successful Process Driven Drug Discovery
 Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
PROVIDING CHEMINFORMATICS SOLUTIONS TO SUPPORT DRUG DISCOVERY DECISIONS
 179 Molecular Informatics: Confronting Complexity, May 13 th - 16 th 2002, Bozen, Italy PROVIDING CHEMINFORMATICS SOLUTIONS TO SUPPORT DRUG DISCOVERY DECISIONS CARLETON R. SAGE, KEVIN R. HOLME, NIANISH
179 Molecular Informatics: Confronting Complexity, May 13 th - 16 th 2002, Bozen, Italy PROVIDING CHEMINFORMATICS SOLUTIONS TO SUPPORT DRUG DISCOVERY DECISIONS CARLETON R. SAGE, KEVIN R. HOLME, NIANISH
SeeSAR 7.1 Beginners Guide. June 2017
 SeeSAR 7.1 Beginners Guide June 2017 Part 1: Basics 1 Type a pdb code and press return or Load your own protein or already existing project, or Just load molecules To begin, let s type 2zff and download
SeeSAR 7.1 Beginners Guide June 2017 Part 1: Basics 1 Type a pdb code and press return or Load your own protein or already existing project, or Just load molecules To begin, let s type 2zff and download
The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score. Protein Structures: The Starting Point for New Drugs 2
 The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score Protein Structures: The Starting Point for New Drugs 2 Matthias Rarey Stefan Bietz Nadine Schneider Sascha Urbaczek University
The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score Protein Structures: The Starting Point for New Drugs 2 Matthias Rarey Stefan Bietz Nadine Schneider Sascha Urbaczek University
Computational Methods and Drug-Likeness. Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004
 Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Practical QSAR and Library Design: Advanced tools for research teams
 DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
CSD. Unlock value from crystal structure information in the CSD
 CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
Towards Physics-based Models for ADME/Tox. Tyler Day
 Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Artificial Intelligence Methods for Molecular Property Prediction. Evan N. Feinberg Pande Lab
 Artificial Intelligence Methods for Molecular Property Prediction Evan N. Feinberg Pande Lab AI Drug Discovery trends.google.com https://g.co/trends/3y4uf MNIST Performance over Time https://srconstantin.wordpress.com/2017/01/28/performance-trends-in-ai/
Artificial Intelligence Methods for Molecular Property Prediction Evan N. Feinberg Pande Lab AI Drug Discovery trends.google.com https://g.co/trends/3y4uf MNIST Performance over Time https://srconstantin.wordpress.com/2017/01/28/performance-trends-in-ai/
A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery
 AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
Hydrogen Bonding & Molecular Design Peter
 Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
QSAR Modeling of Human Liver Microsomal Stability Alexey Zakharov
 QSAR Modeling of Human Liver Microsomal Stability Alexey Zakharov CADD Group Chemical Biology Laboratory Frederick National Laboratory for Cancer Research National Cancer Institute, National Institutes
QSAR Modeling of Human Liver Microsomal Stability Alexey Zakharov CADD Group Chemical Biology Laboratory Frederick National Laboratory for Cancer Research National Cancer Institute, National Institutes
Patrick: An Introduction to Medicinal Chemistry 5e Chapter 01
 Questions Patrick: An Introduction to Medicinal Chemistry 5e 01) Which of the following molecules is a phospholipid? a. i b. ii c. iii d. iv 02) Which of the following statements is false regarding the
Questions Patrick: An Introduction to Medicinal Chemistry 5e 01) Which of the following molecules is a phospholipid? a. i b. ii c. iii d. iv 02) Which of the following statements is false regarding the
 ADMET property estimation, oral bioavailability predictions, SAR elucidation, & QSAR model building software www.simulations-plus.com +1-661-723-7723 What is? is an advanced computer program that enables
ADMET property estimation, oral bioavailability predictions, SAR elucidation, & QSAR model building software www.simulations-plus.com +1-661-723-7723 What is? is an advanced computer program that enables
The use of Design of Experiments to develop Efficient Arrays for SAR and Property Exploration
 The use of Design of Experiments to develop Efficient Arrays for SAR and Property Exploration Chris Luscombe, Computational Chemistry GlaxoSmithKline Summary of Talk Traditional approaches SAR Free-Wilson
The use of Design of Experiments to develop Efficient Arrays for SAR and Property Exploration Chris Luscombe, Computational Chemistry GlaxoSmithKline Summary of Talk Traditional approaches SAR Free-Wilson
Introduction to FBDD Fragment screening methods and library design
 Introduction to FBDD Fragment screening methods and library design Samantha Hughes, PhD Fragments 2013 RSC BMCS Workshop 3 rd March 2013 Copyright 2013 Galapagos NV Why fragment screening methods? Guess
Introduction to FBDD Fragment screening methods and library design Samantha Hughes, PhD Fragments 2013 RSC BMCS Workshop 3 rd March 2013 Copyright 2013 Galapagos NV Why fragment screening methods? Guess
How IJC is Adding Value to a Molecular Design Business
 How IJC is Adding Value to a Molecular Design Business James Mills Sexis LLP ChemAxon TechTalk Stevenage, ov 2012 james.mills@sexis.co.uk Overview Introduction to Sexis Sexis IJC use cases Data visualisation
How IJC is Adding Value to a Molecular Design Business James Mills Sexis LLP ChemAxon TechTalk Stevenage, ov 2012 james.mills@sexis.co.uk Overview Introduction to Sexis Sexis IJC use cases Data visualisation
Dock Ligands from a 2D Molecule Sketch
 Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
What is a property-based similarity?
 What is a property-based similarity? Igor V. Tetko (1) GSF - ational Centre for Environment and Health, Institute for Bioinformatics, Ingolstaedter Landstrasse 1, euherberg, 85764, Germany, (2) Institute
What is a property-based similarity? Igor V. Tetko (1) GSF - ational Centre for Environment and Health, Institute for Bioinformatics, Ingolstaedter Landstrasse 1, euherberg, 85764, Germany, (2) Institute
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Considering the Impact of Drug-like Properties on the Chance of Success
 Considering the Impact of Drug-like Properties on the Chance of Success Iskander Yusof and Matthew D. Segall Optibrium Ltd., 7221 Cambridge Research Park, Beach Drive, Cambridge, CB25 9TL Abstract Many
Considering the Impact of Drug-like Properties on the Chance of Success Iskander Yusof and Matthew D. Segall Optibrium Ltd., 7221 Cambridge Research Park, Beach Drive, Cambridge, CB25 9TL Abstract Many
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
Molecular Modelling for Medicinal Chemistry (F13MMM) Room A36
 Room A36") Molecular Modelling for Medicinal Chemistry (F13MMM) jonathan.hirst@nottingham.ac.uk Room A36 http://comp.chem.nottingham.ac.uk Assisted reading Molecular Modelling: Principles and Applications. Andrew
Molecular Modelling for Medicinal Chemistry (F13MMM) jonathan.hirst@nottingham.ac.uk Room A36 http://comp.chem.nottingham.ac.uk Assisted reading Molecular Modelling: Principles and Applications. Andrew
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today
 est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Alkane/water partition coefficients and hydrogen bonding. Peter Kenny
 Alkane/water partition coefficients and hydrogen bonding Peter Kenny (pwk.pub.2008@gmail.com) Neglect of hydrogen bond strength: A recurring theme in medicinal chemistry Rule of 5 Rule of 3 Scoring functions
Alkane/water partition coefficients and hydrogen bonding Peter Kenny (pwk.pub.2008@gmail.com) Neglect of hydrogen bond strength: A recurring theme in medicinal chemistry Rule of 5 Rule of 3 Scoring functions
Machine learning for ligand-based virtual screening and chemogenomics!
 Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Overview. Descriptors. Definition. Descriptors. Overview 2D-QSAR. Number Vector Function. Physicochemical property (log P) Atom
 Atom") verview D-QSAR Definition Examples Features counts Topological indices D fingerprints and fragment counts R-group descriptors ow good are D descriptors in practice? Summary Peter Gedeck ovartis Institutes
verview D-QSAR Definition Examples Features counts Topological indices D fingerprints and fragment counts R-group descriptors ow good are D descriptors in practice? Summary Peter Gedeck ovartis Institutes
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
CH MEDICINAL CHEMISTRY
 CH 458 - MEDICINAL CHEMISTRY SPRING 2011 M: 5:15pm-8 pm Sci-1-089 Prerequisite: Organic Chemistry II (Chem 254 or Chem 252, or equivalent transfer course) Instructor: Dr. Bela Torok Room S-1-132, Science
CH 458 - MEDICINAL CHEMISTRY SPRING 2011 M: 5:15pm-8 pm Sci-1-089 Prerequisite: Organic Chemistry II (Chem 254 or Chem 252, or equivalent transfer course) Instructor: Dr. Bela Torok Room S-1-132, Science
Structure-Activity Modeling - QSAR. Uwe Koch
 Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
Medicinal Chemist s Relationship with Additivity: Are we Taking the Fundamentals for Granted?
 Medicinal Chemist s Relationship with Additivity: Are we Taking the Fundamentals for Granted? J. Guy Breitenbucher Streamlining Drug Discovery Conference San Francisco, CA ct. 25, 2018 Additivity as the
Medicinal Chemist s Relationship with Additivity: Are we Taking the Fundamentals for Granted? J. Guy Breitenbucher Streamlining Drug Discovery Conference San Francisco, CA ct. 25, 2018 Additivity as the
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Statistical concepts in QSAR.
 Statistical concepts in QSAR. Computational chemistry represents molecular structures as a numerical models and simulates their behavior with the equations of quantum and classical physics. Available programs
Statistical concepts in QSAR. Computational chemistry represents molecular structures as a numerical models and simulates their behavior with the equations of quantum and classical physics. Available programs
Notes of Dr. Anil Mishra at 1
 Introduction Quantitative Structure-Activity Relationships QSPR Quantitative Structure-Property Relationships What is? is a mathematical relationship between a biological activity of a molecular system
Introduction Quantitative Structure-Activity Relationships QSPR Quantitative Structure-Property Relationships What is? is a mathematical relationship between a biological activity of a molecular system
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
MultiscaleMaterialsDesignUsingInformatics. S. R. Kalidindi, A. Agrawal, A. Choudhary, V. Sundararaghavan AFOSR-FA
 MultiscaleMaterialsDesignUsingInformatics S. R. Kalidindi, A. Agrawal, A. Choudhary, V. Sundararaghavan AFOSR-FA9550-12-1-0458 1 Hierarchical Material Structure Kalidindi and DeGraef, ARMS, 2015 Main Challenges
MultiscaleMaterialsDesignUsingInformatics S. R. Kalidindi, A. Agrawal, A. Choudhary, V. Sundararaghavan AFOSR-FA9550-12-1-0458 1 Hierarchical Material Structure Kalidindi and DeGraef, ARMS, 2015 Main Challenges
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Plan. Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics.
 and its used in Chemoinformatics.") Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Assignment 1: Molecular Mechanics (PART 1 25 points)
") Chemistry 380.37 Fall 2015 Dr. Jean M. Standard August 19, 2015 Assignment 1: Molecular Mechanics (PART 1 25 points) In this assignment, you will perform some molecular mechanics calculations using the
Chemistry 380.37 Fall 2015 Dr. Jean M. Standard August 19, 2015 Assignment 1: Molecular Mechanics (PART 1 25 points) In this assignment, you will perform some molecular mechanics calculations using the
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Pipeline Pilot Integration
 Scientific & technical Presentation Pipeline Pilot Integration Szilárd Dóránt July 2009 The Component Collection: Quick facts Provides access to ChemAxon tools from Pipeline Pilot Free of charge Open source
Scientific & technical Presentation Pipeline Pilot Integration Szilárd Dóránt July 2009 The Component Collection: Quick facts Provides access to ChemAxon tools from Pipeline Pilot Free of charge Open source
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Ranking of HIV-protease inhibitors using AutoDock
 Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Navigation in Chemical Space Towards Biological Activity. Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland
 Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Cheminformatics platform for drug discovery application
 EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
New approaches to scoring function design for protein-ligand binding affinities. Richard A. Friesner Columbia University
 New approaches to scoring function design for protein-ligand binding affinities Richard A. Friesner Columbia University Overview Brief discussion of advantages of empirical scoring approaches Analysis
New approaches to scoring function design for protein-ligand binding affinities Richard A. Friesner Columbia University Overview Brief discussion of advantages of empirical scoring approaches Analysis
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
Advances in Multi-parameter Optimisation Methods for de Novo Drug Design
 Advances in Multi-parameter Optimisation Methods for de Novo Drug Design Abstract Introduction A high quality drug must achieve a balance of physicochemical and ADME properties, safety and potency against
Advances in Multi-parameter Optimisation Methods for de Novo Drug Design Abstract Introduction A high quality drug must achieve a balance of physicochemical and ADME properties, safety and potency against
Machine Learning Concepts in Chemoinformatics
 Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
Advanced Medicinal Chemistry SLIDES B
 Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Introduction to Structure Preparation and Visualization
 Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Chapter 24. Computational Chemistry Research. Computational Chemistry for Chemistry Educators - Gotwals/Sendlinger Copyright 2007 All Rights Reserved
 Chapter 24 Computational Chemistry Research 1 Contents Choosing a Research Problem Applying for a research account Choosing a Model Chemistry The Computational Chemistry Notebook Presenting Results Sample
Chapter 24 Computational Chemistry Research 1 Contents Choosing a Research Problem Applying for a research account Choosing a Model Chemistry The Computational Chemistry Notebook Presenting Results Sample
Quantitative Structure-Activity Relationship (QSAR) computational-drug-design.html
 computational-drug-design.html") Quantitative Structure-Activity Relationship (QSAR) http://www.biophys.mpg.de/en/theoretical-biophysics/ computational-drug-design.html 07.11.2017 Ahmad Reza Mehdipour 07.11.2017 Course Outline 1. 1.Ligand-
Quantitative Structure-Activity Relationship (QSAR) http://www.biophys.mpg.de/en/theoretical-biophysics/ computational-drug-design.html 07.11.2017 Ahmad Reza Mehdipour 07.11.2017 Course Outline 1. 1.Ligand-
Enamine Golden Fragment Library
 Enamine Golden Fragment Library 14 March 216 1794 compounds deliverable as entire set or as selected items. Fragment Based Drug Discovery (FBDD) [1,2] demonstrates remarkable results: more than 3 compounds
Enamine Golden Fragment Library 14 March 216 1794 compounds deliverable as entire set or as selected items. Fragment Based Drug Discovery (FBDD) [1,2] demonstrates remarkable results: more than 3 compounds
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
OPEN LESSON SAMPLE LESSONS FOR THE CLASSROOM FROM LAYING THE FOUNDATION
 OPEN LESSON SAMPLE LESSONS FOR THE CLASSROOM FROM LAYING THE FOUNDATION Middle Grades Science Sugar and Salt Solutions Exploring Common Substances Using a PhET Simulation About this Lesson This activity
OPEN LESSON SAMPLE LESSONS FOR THE CLASSROOM FROM LAYING THE FOUNDATION Middle Grades Science Sugar and Salt Solutions Exploring Common Substances Using a PhET Simulation About this Lesson This activity
Structure Investigation of Fam20C, a Golgi Casein Kinase
 Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Validation of Experimental Crystal Structures
 Validation of Experimental Crystal Structures Aim This use case focuses on the subject of validating crystal structures using tools to analyse both molecular geometry and intermolecular packing. Introduction
Validation of Experimental Crystal Structures Aim This use case focuses on the subject of validating crystal structures using tools to analyse both molecular geometry and intermolecular packing. Introduction
CHEM 347 Organic Chemistry II (for Majors) Instructor: Paul J. Bracher. Quiz # 4. Due in Monsanto Hall 103 by: Friday, April 4 th, 2014, 7:00 p.m.
 Instructor: Paul J. Bracher. Quiz # 4. Due in Monsanto Hall 103 by: Friday, April 4 th, 2014, 7:00 p.m.") CHEM 347 Quiz # 4 Spring 2014 Page 1 of 9 CHEM 347 Organic Chemistry II (for Majors) Instructor: Paul J. Bracher Quiz # 4 Due in Monsanto Hall 103 by: Friday, April 4 th, 2014, 7:00 p.m. Student Name (Printed)
CHEM 347 Quiz # 4 Spring 2014 Page 1 of 9 CHEM 347 Organic Chemistry II (for Majors) Instructor: Paul J. Bracher Quiz # 4 Due in Monsanto Hall 103 by: Friday, April 4 th, 2014, 7:00 p.m. Student Name (Printed)
Learning Organic Chemistry
 Objective 1 Represent organic molecules with chemical formulas, expanded formulas, Lewis structures, skeletal structures. Determine shape (VSEPR), bond polarity, and molecule polarity. Identify functional
Objective 1 Represent organic molecules with chemical formulas, expanded formulas, Lewis structures, skeletal structures. Determine shape (VSEPR), bond polarity, and molecule polarity. Identify functional
Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,..
 3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
Plan. Day 2: Exercise on MHC molecules.
 Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Analysis of a Large Structure/Biological Activity. Data Set Using Recursive Partitioning and. Simulated Annealing
 Analysis of a Large Structure/Biological Activity Data Set Using Recursive Partitioning and Simulated Annealing Student: Ke Zhang MBMA Committee: Dr. Charles E. Smith (Chair) Dr. Jacqueline M. Hughes-Oliver
Analysis of a Large Structure/Biological Activity Data Set Using Recursive Partitioning and Simulated Annealing Student: Ke Zhang MBMA Committee: Dr. Charles E. Smith (Chair) Dr. Jacqueline M. Hughes-Oliver
CHEM 4170 Problem Set #1
 CHEM 4170 Problem Set #1 0. Work problems 1-7 at the end of Chapter ne and problems 1, 3, 4, 5, 8, 10, 12, 17, 18, 19, 22, 24, and 25 at the end of Chapter Two and problem 1 at the end of Chapter Three
CHEM 4170 Problem Set #1 0. Work problems 1-7 at the end of Chapter ne and problems 1, 3, 4, 5, 8, 10, 12, 17, 18, 19, 22, 24, and 25 at the end of Chapter Two and problem 1 at the end of Chapter Three
An Integrated Approach to in-silico
 An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification
 Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment