The PhilOEsophy. There are only two fundamental molecular descriptors
|
|
|
- Ralf Cross
- 5 years ago
- Views:
Transcription



1 The PhilOEsophy There are only two fundamental molecular descriptors
2 Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment No requirement for (manual) atom matching Pose generation/prediction Matching a binding site Matching a bound ligand
3 Where can we use electrostatics? Lead-hopping Electrostatic analogues are not graph analogues Solvent treatment Continuum and semi-continuum

4 Virtual Screening Protein Preparation Compound Collection Database Preparation Screening Database Structure-based 3D & 2D Ligand-based Ligand Preparation Hybrid & Consensus




 &")
5 Using a protein structure Pose v. protein FRED Score v. protein FRED/SZYBKI Score v. ligand ROCS/EON Pose v. ligand ROCS HYBRID (VS) & POSIT (posing)
6 Virtual Screening with OpenEye QUACPAC tautomers charges FILTER OMEGA Conformations Remove undesirables FRED/HYBRID Posing and SBVS ROCS Shape alignment & scoring
7 OMEGA: Would you like to? Generate high quality conformer ensembles rapidly. Store large ensembles in very compact databases for rapid searching. Calculate useful conformer energetics in a variety of environments.
8 ROCS: Would you like to? Efficiently align molecules by shape and chemical features. Rapidly screen large databases for non-obvious actives. Obtain informative overlays between active and untested compounds.
9 The ROCS GUI: vrocs Generate custom queries
 of molecules in an active site.")
10 FRED: Would you like to? Perform structure-based VS rapidly. Identify binding mode(s) of molecules in an active site. Utilize more information to achieve better results. HYBRID
11 POSIT: Would you like to? Produce good quality predictions of ligand poses with very high frequency. Accurately estimate the probability that a predicted pose is accurate. Automatically determine the best protein structure from a set to pose a molecule against.
12
13 Virtual Screening with OpenEye QUACPAC tautomers charges FILTER OMEGA Conformations Remove undesirables FRED/HYBRID Posing and SBVS ROCS Shape alignment & scoring


14 OMEGA: conformation generation OMEGA Knowledge-based conformation generation Virtual screening Crystal structure reproduction Ensemble properties
15 OMEGA: The best validated conformer generator Carefully selected crystallographic structures PDB and CSD Multiple measures of success Closeness and coverage Rigorous statistical analysis DOI: /ci100031x
16 OMEGA: The process Input molecule (1D, 2D, 3D) Find fragments 3D Fragment library Built-in or custom Assemble fragments -> 3D structure Torsion driving Complete conformer ensemble Torsion library Built-in & extensible Knowledge Base Pruned conformer ensemble
17 Size (MB) The file size problem SD/MOL2 files too large to store large numbers of molecules or conformers OpenEye binary (OEB) much smaller x or more Can we do better? 2000 File size for 22 million conformers How is this done? 0 MOL2 SDF OEB ROC-OEB File Format
18 Rotor-offset compression (ROC) Speeds up downstream tools 10-15% Store one set of coordinates. All other conformers defined by torsion angles.
19 RMSD OMEGA: accuracy on a carefully chosen dataset Mean RMSD: 0.67Å (0.655, 0.688) Median RMSD: 0.53Å Count J. Chem. Inf. Model., 50, 572 (2010).
20 OMEGA: relative accuracy MOE/Import Catalyst/BEST ConfGen/CompMin ConfGen/CombMin OMEGA <0.5 <1 <1.5 <2 Watts et al. J. Chem. Inf. Model. 50, 534 (2010)
21 OMEGA: speed NumConfs Time(s) 30 0 MOE/Import Catalyst/BEST ConfGen/CompMin ConfGen/CombMin OMEGA2 Average OMEGA time = 2.7 secs/molecule J. Chem. Inf. Model. 50, 822 (2010)
22 OMEGA Summary Speed: molecules/sec Fastest of all commercial applications Quality: Excellent reproduction of X-ray poses Best overall at highly precise reproduction (< 0.5Å) Flexibility in generation of conformers Focus/diversity of conformer sets can easily be controlled In vacuo, solution, protein-bound
23
24 Virtual Screening with OpenEye QUACPAC tautomers charges FILTER OMEGA Conformations Remove undesirables FRED/HYBRID Posing and SBVS ROCS Shape alignment & scoring
25 The Shape of Ligand-based Design ROCS ROCS compares molecules by shape & chemistry Rigid overlay of a query conformer(s) with a set of conformers of database molecules Scoring by shape similarity and chemical (color) similarity (in 3D)


26 Per cent actives ROCS: Shape overlay and scoring Effective virtual screening ROCS Per 10 cent 15 screened Identify shared features Molecular alignment Leadhopping
27 Shape similarity & graph similarity are not the same CDK2 inhibitors 10 nm nm ROCS (shape) sim = 0.90 Fingerprint (2D) sim = 0.40
28 ROCS: Overlays + Scores Shape Tanimoto = 0.90 Color Tanimoto = 0.17 TanimotoCombo = 1.07
29
30 VS Comparison from Merck Virtual screening on 11 targets CA, CDK2, COX-2, DHFR, ER, HIV-PR, HIV-RT, NA, PTP- 1B, thrombin, TS Structure-based and ligand-based compared ROCS and docking Same X-ray structures; ligand as query McGaughey et al., J. Chem. Inf. Model., 2007, 47, 1504.
31 E (1%) ROCS is better than docking VS by Merck Mean StdDev Median 0 GLIDE ROCS Application
32 Conclusion Extensive Merck study shows that ROCS is the best overall VS tool available Fast Reliability High hit rate Diverse hit structures Merck no longer uses docking for VS
33 VS against GPCRs Evers et al., J. Med. Chem., 2005, 48, HT2A, A1A, D2, M1 Various 3D techniques Docking to homology models Gold, FlexX-Pharm Ligand-based methods Catalyst, FlexS Compare to ROCS
34 Enrichment Mean of Results GOLD FlexX-Pharm Catalyst FlexS ROCS 2D_MACCS 0 1% 5% 10% Per cent screened FlexS, ROCS: 1 query molecule, 1 computed conformation Catalyst: query molecules -> 1 pharmacophore
35 ROCS Summary Powerful VS application Frequently outperforms docking Success does NOT require a bioactive conformation for the query Only low database conformational sampling required confs/molecule Fast Up to 40 molecules/second conformers/second
36 The ROCS GUI: vrocs Generate queries from molecules Customized queries Multi-molecule queries


")


37 Why vrocs? Enhanced Virtual Screening Active Compound(s) Query Creation vrocs Query Editing Query Validation ROCS
38
39 Virtual Screening with OpenEye QUACPAC tautomers charges FILTER OMEGA Conformations Remove undesirables FRED/HYBRID Posing and SBVS ROCS Shape alignment & scoring
40 How does FRED work? Build/customize receptor model GUI fred_receptor Input conformer database Optimized with OMEGA Exhaustive posing Structure-based & ligand-based scoring FRED Consensus pose selection

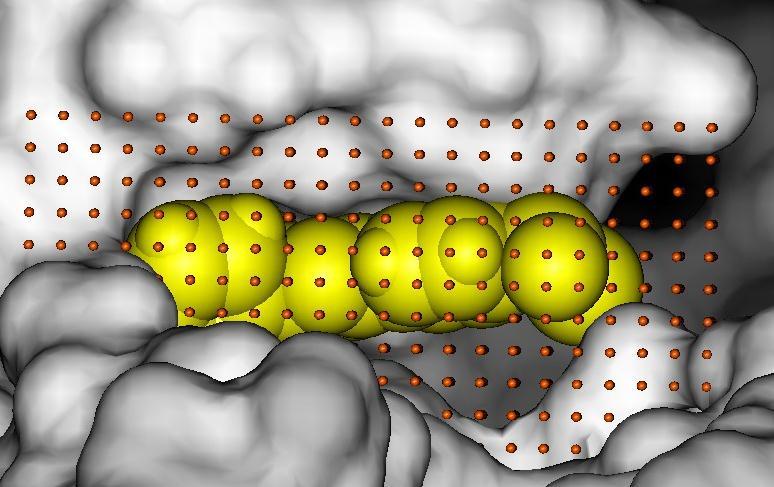
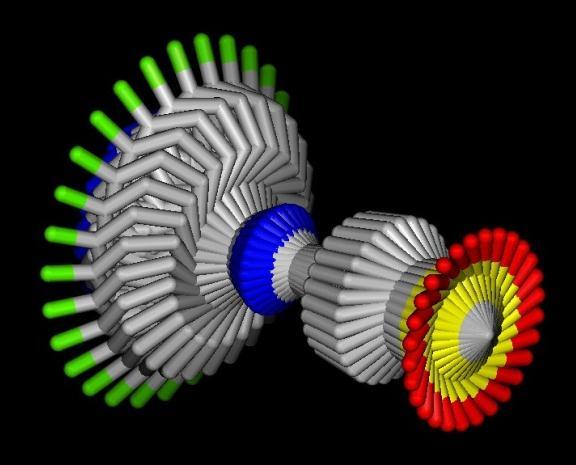
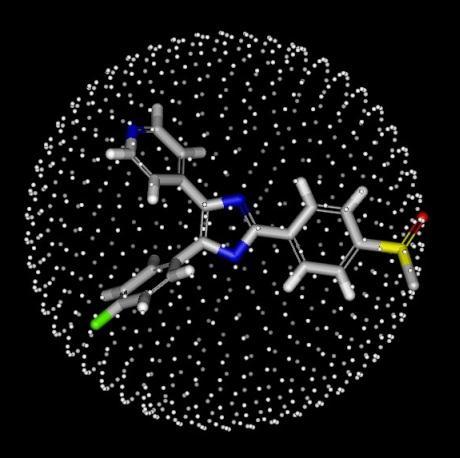

41 Global Exhaustive Search Systematic Rotations Systematic Translations Poses for scoring X Filtering of clashing poses
42 Scoring Operate on best poses from Exhaustive Search Protein-based scoring PLP, ChemScore, ScreenScore ChemGauss3, ShapeGauss PB (electrostatic interactions) Ligand-based scoring CGO Consensus
43
44 Fraction FRED: Self Docking Results RMSD (Å) Top Scoring Pose Top 5 Top 10 Top 20 This is a completely irrelevant problem.
45 Cross-docking is difficult Average self-docking success Average cross-docking success J. Chem. Inf. Model. 50, 1432 (2010)
46 VS Comparison from Merck Virtual screening on 11 targets CA, CDK2, COX-2, DHFR, ER, HIV-PR, HIV-RT, NA, PTP-1B, thrombin, TS Structure-based and ligand-based compared McGaughey et al., J. Chem. Inf. Model., 2007, 47, 1504.
47 Enrichment (1%) FRED = GLIDE for VS Mean StdDev Median FRED - Lower standard deviation, higher consistency 0 FRED Application GLIDE Best indicator of future performance
48 FRED - Summary Does well at posing Cross-docking is very difficult Virtual screening performance is good Reliable Can we do better?
49 Is a co-crystal structure available? Yes Use docking No Ligand-based (2D & 3D) Docking to apo structure is risky Best answer - use BOTH
50 Hybrid Docking: Using what you know Docking e.g. FRED Hybrid Docking Ligand- Based Design e.g. ROCS Bound ligand structure guides docking

51 FRED Hybrid vs. Standard Docking
52 Hybrid docking: speed Docking Time per Compound (one CPU 2.4 GHz Xeon) FRED 2.2 Standard Docking HYBRID 5sec 1sec
53 Hybrid docking: Posing 100% 90% 80% 70% 60% 50% 40% 30% 20% 10% 0% Success Rate at 2Å Hybrid FRED Top20 Top10 Top5 Best HYBRID: 2Å FRED: 2Å

54 Virtual Screening Cross et al., J. Chem. Inf. Model., 49, 1455 (2010); McGann ibid. 51, 578 (2011). Results from DUD dataset Thick bars are the 95% confidence interval for the true average AUC Whiskers are the 95% confidence interval for the result of a single trial 95% confidence interval for the true average AUC. 95% confidence interval for the result of a single trial Performance of docking tools is very variable. Is it possible to show statistically meaningful differences between tools?
55 Virtual Screening Comparison Probability that the mean performance of HYBRID is better than FRED Probability that HYBRID will do better than FRED on one system 93% 62% Use more information Better results
56 FRED Summary Efficient virtual screening 3-5 sec/molecule Good pose prediction 70% < 2Å RMSD Variety of scoring Unique ligand-based Using more information gives better results Hybrid docking
57

58 Why structure-based design? Pose Prediction POSIT Virtual screening FRED/HYBRID Binding affinity prediction
59 Using a protein structure Pose v. protein FRED Score v. protein FRED/SZYBKI Score v. ligand ROCS/EON Pose v. ligand ROCS HYBRID (VS) & POSIT (posing)
60 Structure-based design with OpenEye POSIT Ligand-based posing SZMAP Solvent mapping FRED/HYBRID Posing and SBVS SZYBKI MMFF94 optimisation BROOD ROCS Shape alignment & scoring EON Electrostatic similarity Fragment replacement


61 Count POSIT: Accurate and reliable analogue posing Flexibly fit a new molecule to shape of a known ligand > 90% 0-0.5Å RMSD RMSD
62 Cross-docking pose prediction J. Chem. Inf. Model. 50, 1432 (2010) Average self-docking success Average cross-docking success How can we improve? Predict reliability? Identify likely failure cases?
63 POSIT: analogue posing CDK2 inhibitors Shape analogues are not obvious graph analogues. BUT Obvious graph analogues ARE shape analogues. Shape Tanimoto = Fingerprint Tanimoto = 0.45
64 Molecular Similarity in 3D: How POSIT defines an analogue Shape Tanimoto = 0.90 Color Tanimoto = 0.17 TanimotoCombo = 1.07

65 How to use what you know: POSIT X-ray structure of known ligand New molecule Pose for new molecule using known ligand. Score AND estimate quality with TanimotoCombo.
 Goal Match shape and chemistry of bound ligand Maximize interaction with the protein E POSIT = 1 λ E Overlay + λe MMFF λ =")
66 What POSIT knows Ligand Information Protein Structure Symbol E Overlay E MMFF Potential TanimotoCombo Merck Molecular Force Field (MMFF94) Goal Match shape and chemistry of bound ligand Maximize interaction with the protein E POSIT = 1 λ E Overlay + λe MMFF λ = scaling factor
67 Cross-Docking: the real problem Percent < 2.0 Angstrom Posit* Gold_PLP Glide_PLP AD4 Fred X-ray v. Fit ligands Tanimoto Combo Tuccinardi, et al. J. Chem. Inf. Model., 2010, 50, * Currently validating that the same data set was used
 TC > 1.")
68 Prospective Results POSIT predicted crystal structures versus X-ray Active projects at Abbott Labs 17/20 (85%) TC > 1.4 RMSD <= 2Å TanimotoCombo Pose to Crystallographic Ligand
69 POSIT Summary POSIT gives a pose, a score and a CONFIDENCE POSIT knows when to fail Pose quality can be accurately predicted in POSIT Docking scores cannot predict quality POSIT works PROSPECTIVELY Fast: sec/molecule Reliable: pose + confidence Accurate: 98% poses < 2Å; 90% < 0.5Å

70 POSIT
71
72 The PhilOEsophy There are two fundamental molecular descriptors
73 >omega2 in my_filtered_molecules.ism out my_dbase.oeb.gz >rocs query my_query.oeb dbase my_dbase.oeb.gz besthits 500 prefix my_rocs_results(.sdf) (virtual screening) >fred rec my_receptor.oeb.gz dbase my_dbase.oeb.gz - prefix my_fred_results oformat sdf.gz num_alt_poses 4 (hybrid) >fred rec my_receptor.oeb.gz dbase my_dbase.oeb.gz prefix my_hybrid_results oformat sdf.gz exhaustive_scoring cgo opt Chemgauss3 chemgauss3 true num_alt_poses 4
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today
 est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Pose and affinity prediction by ICM in D3R GC3. Max Totrov Molsoft
 Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
What is Protein-Ligand Docking?
 MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Conformational Sampling of Druglike Molecules with MOE and Catalyst: Implications for Pharmacophore Modeling and Virtual Screening
 J. Chem. Inf. Model. 2008, 48, 1773 1791 1773 Conformational Sampling of Druglike Molecules with MOE and Catalyst: Implications for Pharmacophore Modeling and Virtual Screening I-Jen Chen* and Nicolas
J. Chem. Inf. Model. 2008, 48, 1773 1791 1773 Conformational Sampling of Druglike Molecules with MOE and Catalyst: Implications for Pharmacophore Modeling and Virtual Screening I-Jen Chen* and Nicolas
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
GC and CELPP: Workflows and Insights
 GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Conformational Searching using MacroModel and ConfGen. John Shelley Schrödinger Fellow
 Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Analyzing Molecular Conformations Using the Cambridge Structural Database. Jason Cole Cambridge Crystallographic Data Centre
 Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
BioSolveIT. A Combinatorial Approach for Handling of Protonation and Tautomer Ambiguities in Docking Experiments
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
The Conformation Search Problem
 Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors
 MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
Fragment based drug discovery in teams of medicinal and computational chemists. Carsten Detering
 Fragment based drug discovery in teams of medicinal and computational chemists Carsten Detering BioSolveIT Quick Facts Founded in 2001 by the developers of FlexX ~20 people Core expertise: docking, screening,
Fragment based drug discovery in teams of medicinal and computational chemists Carsten Detering BioSolveIT Quick Facts Founded in 2001 by the developers of FlexX ~20 people Core expertise: docking, screening,
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
BioSolveIT. A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Conformational sampling of macrocycles in solution and in the solid state
 Conformational sampling of macrocycles in solution and in the solid state Paul Hawkins, Ph.D. Head of Scientific Solutions Stanislaw Wlodek, Ph.D. Senior Scientific Developer 6/6/2018 https://berkonomics.com/?p=2437
Conformational sampling of macrocycles in solution and in the solid state Paul Hawkins, Ph.D. Head of Scientific Solutions Stanislaw Wlodek, Ph.D. Senior Scientific Developer 6/6/2018 https://berkonomics.com/?p=2437
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors
 A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
Protein Structure Prediction and Protein-Ligand Docking
 Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score. Protein Structures: The Starting Point for New Drugs 2
 The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score Protein Structures: The Starting Point for New Drugs 2 Matthias Rarey Stefan Bietz Nadine Schneider Sascha Urbaczek University
The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score Protein Structures: The Starting Point for New Drugs 2 Matthias Rarey Stefan Bietz Nadine Schneider Sascha Urbaczek University
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows
 MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
Protein-Ligand Docking Evaluations
 Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
PharmDock: A Pharmacophore-Based Docking Program
 Purdue University Purdue e-pubs Department of Medicinal Chemistry and Molecular Pharmacology Faculty Publications Department of Medicinal Chemistry and Molecular Pharmacology 4-16-2014 PharmDock: A Pharmacophore-Based
Purdue University Purdue e-pubs Department of Medicinal Chemistry and Molecular Pharmacology Faculty Publications Department of Medicinal Chemistry and Molecular Pharmacology 4-16-2014 PharmDock: A Pharmacophore-Based
BUDE. A General Purpose Molecular Docking Program Using OpenCL. Richard B Sessions
 BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
Cross Discipline Analysis made possible with Data Pipelining. J.R. Tozer SciTegic
 Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Structure based drug design and LIE models for GPCRs
 Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Virtual Screening. Anna Linusson. Department of Chemistry Umeå University Sweden
 Virtual Screening Anna Linusson Department of Chemistry Umeå University Sweden Virtual screening N N NH 2 N Cl N N H H H N N N S N HN N H S N N S S N N Free energy of binding + 0 0 G RT ln K A H T S 0
Virtual Screening Anna Linusson Department of Chemistry Umeå University Sweden Virtual screening N N NH 2 N Cl N N H H H N N N S N HN N H S N N S S N N Free energy of binding + 0 0 G RT ln K A H T S 0
On Evaluating Molecular-Docking Methods for Pose Prediction and Enrichment Factors
 J. Chem. Inf. Model. 2006, 46, 401-415 401 On Evaluating Molecular-Docking Methods for Pose Prediction and Enrichment Factors Hongming Chen,*, Paul D. Lyne, Fabrizio Giordanetto, Timothy Lovell,*,, and
J. Chem. Inf. Model. 2006, 46, 401-415 401 On Evaluating Molecular-Docking Methods for Pose Prediction and Enrichment Factors Hongming Chen,*, Paul D. Lyne, Fabrizio Giordanetto, Timothy Lovell,*,, and
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
LS-align: an atom-level, flexible ligand structural alignment algorithm for high-throughput virtual screening
 Bioinformatics, YYYY, 0 0 doi: 10.1093/bioinformatics/xxxxx Advance Access Publication Date: DD Month YYYY Original Paper Structural bioinformatics LS-align: an atom-level, flexible ligand structural alignment
Bioinformatics, YYYY, 0 0 doi: 10.1093/bioinformatics/xxxxx Advance Access Publication Date: DD Month YYYY Original Paper Structural bioinformatics LS-align: an atom-level, flexible ligand structural alignment
SHAFTS: A Hybrid Approach for 3D Molecular Similarity Calculation. 1. Method and Assessment of Virtual Screening
 pubs.acs.org/jcim SHAFTS: A Hybrid Approach for 3D Molecular Similarity Calculation. 1. Method and Assessment of Virtual Screening Xiaofeng Liu,, Hualiang Jiang, and Honglin Li*, State Key Laboratory of
pubs.acs.org/jcim SHAFTS: A Hybrid Approach for 3D Molecular Similarity Calculation. 1. Method and Assessment of Virtual Screening Xiaofeng Liu,, Hualiang Jiang, and Honglin Li*, State Key Laboratory of
PL-PatchSurfer: A Novel Molecular Local Surface-Based Method for Exploring Protein-Ligand Interactions
 Int. J. Mol. Sci. 2014, 15, 15122-15145; doi:10.3390/ijms150915122 Article OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms PL-PatchSurfer: A Novel Molecular
Int. J. Mol. Sci. 2014, 15, 15122-15145; doi:10.3390/ijms150915122 Article OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms PL-PatchSurfer: A Novel Molecular
Part 6. 3D Pharmacophore Modeling
 279 Part 6 3D Pharmacophore Modeling 281 20 3D Pharmacophore Modeling Techniques in Computer Aided Molecular Design Using LigandScout Thomas Seidel, Sharon D. Bryant, Gökhan Ibis, Giulio Poli, and Thierry
279 Part 6 3D Pharmacophore Modeling 281 20 3D Pharmacophore Modeling Techniques in Computer Aided Molecular Design Using LigandScout Thomas Seidel, Sharon D. Bryant, Gökhan Ibis, Giulio Poli, and Thierry
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Introduction to Spark
 1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
KNIME-based scoring functions in Muse 3.0. KNIME User Group Meeting 2013 Fabian Bös
 KIME-based scoring functions in Muse 3.0 KIME User Group Meeting 2013 Fabian Bös Certara Mission: End-to-End Model-Based Drug Development Certara was formed by acquiring and integrating Tripos, Pharsight,
KIME-based scoring functions in Muse 3.0 KIME User Group Meeting 2013 Fabian Bös Certara Mission: End-to-End Model-Based Drug Development Certara was formed by acquiring and integrating Tripos, Pharsight,
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Machine learning for ligand-based virtual screening and chemogenomics!
 Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
Ultra High Throughput Screening using THINK on the Internet
 Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
Comparative Analysis of Pharmacophore Screening Tools
 pubs.acs.org/jcim Comparative Analysis of Pharmacophore Screening Tools Marijn P. A. Sanders,,# Armeńio J. M. Barbosa, Barbara Zarzycka, Gerry A.F. Nicolaes, Jan P.G. Klomp, Jacob de Vlieg,, and Alberto
pubs.acs.org/jcim Comparative Analysis of Pharmacophore Screening Tools Marijn P. A. Sanders,,# Armeńio J. M. Barbosa, Barbara Zarzycka, Gerry A.F. Nicolaes, Jan P.G. Klomp, Jacob de Vlieg,, and Alberto
Identifying Interaction Hot Spots with SuperStar
 Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Virtual Screening in Drug Discovery
 Virtual Screening in Drug Discovery edited by Juan Alvarez and Brian Shoichet Taylor &. Francis Taylor & Francis Group Boca Raton London New York Singapore A CRC title, part of the Taylor & Francis imprint,
Virtual Screening in Drug Discovery edited by Juan Alvarez and Brian Shoichet Taylor &. Francis Taylor & Francis Group Boca Raton London New York Singapore A CRC title, part of the Taylor & Francis imprint,
POSIT: Flexible Shape-Guided Docking For Pose Prediction
 This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jcim POSIT:
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jcim POSIT:
Author Index Volume
 Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
High Throughput In-Silico Screening Against Flexible Protein Receptors
 John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
Toward an Understanding of GPCR-ligand Interactions. Alexander Heifetz
 Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Flexibility and Constraints in GOLD
 Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
SCISSORS: A Linear-Algebraical Technique to Rapidly Approximate Chemical Similarities
 J. Chem. Inf. Model. XXXX, xxx, 000 A SCISSORS: A Linear-Algebraical Technique to Rapidly Approximate Chemical Similarities Imran S. Haque and Vijay S. Pande*,, Department of Computer Science and Department
J. Chem. Inf. Model. XXXX, xxx, 000 A SCISSORS: A Linear-Algebraical Technique to Rapidly Approximate Chemical Similarities Imran S. Haque and Vijay S. Pande*,, Department of Computer Science and Department
Molecular Similarity Searching Using Inference Network
 Molecular Similarity Searching Using Inference Network Ammar Abdo, Naomie Salim* Faculty of Computer Science & Information Systems Universiti Teknologi Malaysia Molecular Similarity Searching Search for
Molecular Similarity Searching Using Inference Network Ammar Abdo, Naomie Salim* Faculty of Computer Science & Information Systems Universiti Teknologi Malaysia Molecular Similarity Searching Search for
MM-PBSA Validation Study. Trent E. Balius Department of Applied Mathematics and Statistics AMS
 MM-PBSA Validation Study Trent. Balius Department of Applied Mathematics and Statistics AMS 535 11-26-2008 Overview MM-PBSA Introduction MD ensembles one snap-shots relaxed structures nrichment Computational
MM-PBSA Validation Study Trent. Balius Department of Applied Mathematics and Statistics AMS 535 11-26-2008 Overview MM-PBSA Introduction MD ensembles one snap-shots relaxed structures nrichment Computational
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery
 21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,..
 3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
Supporting Information
 Supporting Information COMPUTATIONAL DISCOVERY AND EXPERIMENTAL VALIDATION OF INHIBITORS OF THE HUMAN INTESTINAL TRANSPORTER, OATP2B1 Natalia Khuri 1,2,#, Arik A. Zur 2,#, Matthias B. Wittwer 2, Lawrence
Supporting Information COMPUTATIONAL DISCOVERY AND EXPERIMENTAL VALIDATION OF INHIBITORS OF THE HUMAN INTESTINAL TRANSPORTER, OATP2B1 Natalia Khuri 1,2,#, Arik A. Zur 2,#, Matthias B. Wittwer 2, Lawrence
Characterization of Pharmacophore Multiplet Fingerprints as Molecular Descriptors. Robert D. Clark 2004 Tripos, Inc.
 Characterization of Pharmacophore Multiplet Fingerprints as Molecular Descriptors Robert D. Clark Tripos, Inc. bclark@tripos.com 2004 Tripos, Inc. Outline Background o history o mechanics Finding appropriate
Characterization of Pharmacophore Multiplet Fingerprints as Molecular Descriptors Robert D. Clark Tripos, Inc. bclark@tripos.com 2004 Tripos, Inc. Outline Background o history o mechanics Finding appropriate
The reuse of structural data for fragment binding site prediction
 The reuse of structural data for fragment binding site prediction Richard Hall 1 Motivation many examples of fragments binding in a phenyl shaped pocket or a kinase slot good shape complementarity between
The reuse of structural data for fragment binding site prediction Richard Hall 1 Motivation many examples of fragments binding in a phenyl shaped pocket or a kinase slot good shape complementarity between
Molecular Shape and Medicinal Chemistry: A Perspective
 3862 J. Med. Chem. 2010, 53, 3862 3886 DOI: 10.1021/jm900818s Molecular Shape and Medicinal Chemistry: A Perspective Anthony Nicholls,*, Georgia B. McGaughey, Robert P. Sheridan, Andrew C. Good, Gregory
3862 J. Med. Chem. 2010, 53, 3862 3886 DOI: 10.1021/jm900818s Molecular Shape and Medicinal Chemistry: A Perspective Anthony Nicholls,*, Georgia B. McGaughey, Robert P. Sheridan, Andrew C. Good, Gregory
ESPRESSO (Extremely Speedy PRE-Screening method with Segmented compounds) 1
 1") Vol.2016-MPS-108 o.18 Vol.2016-BI-46 o.18 ESPRESS 1,4,a) 2,4 2,4 1,3 1,3,4 1,3,4 - ESPRESS (Extremely Speedy PRE-Screening method with Segmented cmpounds) 1 Glide HTVS ESPRESS 2,900 200 ESPRESS: An ultrafast
Vol.2016-MPS-108 o.18 Vol.2016-BI-46 o.18 ESPRESS 1,4,a) 2,4 2,4 1,3 1,3,4 1,3,4 - ESPRESS (Extremely Speedy PRE-Screening method with Segmented cmpounds) 1 Glide HTVS ESPRESS 2,900 200 ESPRESS: An ultrafast
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Thermodynamic Integration with Enhanced Sampling (TIES)
") Thermodynamic Integration with Enhanced Sampling (TIES) A. P. Bhati, S. Wan, D. W. Wright and P. V. Coveney agastya.bhati.14@ucl.ac.uk Centre for Computational Science Department of Chemistry University
Thermodynamic Integration with Enhanced Sampling (TIES) A. P. Bhati, S. Wan, D. W. Wright and P. V. Coveney agastya.bhati.14@ucl.ac.uk Centre for Computational Science Department of Chemistry University
Focusing Conformational Ensembles on Bioactive-Like Conformations
 Focusing Conformational Ensembles on Bioactive-Like Conformations Hannah H. Avgy, Boaz Musafia, Hanoch Senderowitz Department of Chemistry Bar-Ilan University 3rd Strasbourg Summer School on Chemoinformatics,
Focusing Conformational Ensembles on Bioactive-Like Conformations Hannah H. Avgy, Boaz Musafia, Hanoch Senderowitz Department of Chemistry Bar-Ilan University 3rd Strasbourg Summer School on Chemoinformatics,
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification
 Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation
 Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation Charles Blundell charles.blundell@c4xdiscovery.com www.c4xdiscovery.com Rigid: single
Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation Charles Blundell charles.blundell@c4xdiscovery.com www.c4xdiscovery.com Rigid: single
Fast similarity searching making the virtual real. Stephen Pickett, GSK
 Fast similarity searching making the virtual real Stephen Pickett, GSK Introduction Introduction to similarity searching Use cases Why is speed so crucial? Why MadFast? Some performance stats Implementation
Fast similarity searching making the virtual real Stephen Pickett, GSK Introduction Introduction to similarity searching Use cases Why is speed so crucial? Why MadFast? Some performance stats Implementation