Structural biology and drug design: An overview
|
|
|
- Shana Morrison
- 5 years ago
- Views:
Transcription

1 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu
2
3



4 Drug discovery
 or inhibition (antagonist) to a biological target (protein, receptor, enzymes, cells ).")
5 Drug and drug design A drug is a key molecule involved in a particular metabolic or signaling pathway that is specific to a disease condition or pathology. Action of activation (agonist) or inhibition (antagonist) to a biological target (protein, receptor, enzymes, cells ). Drug design is the approach of finding drugs by design, based on their biological targets. An important part of drug design is the prediction of small molecules binding to a target protein (pharmacophore, docking, QSAR, )

6 Virtual screening- Chemoinformatics Exploit any knowledge of target(s) and/or active ligand(s) and/or gene family. It involves computational technique for a rapid assessment of large libraries of chemical structures in order to guide the selection of likely drug candidates. Structure based Docking Ligand based QSAR, similarity search VS Pharmacophore based knowledge based Chemical-Biology network
 5 If two or more of these rules are violated, the compound might have problems with oral bioavailability. (Lipinski et al., Adv. Drug Delivery Rev., 23, 1997, 3.")
7 Drug-likeness with a simple counting method, rule of five ctanol-water partition coefficient (logp) 5 Molecular weight 500 No. hydrogen bond acceptors (HBA) 10 No. hydrogen bond donors (HBD) 5 If two or more of these rules are violated, the compound might have problems with oral bioavailability. (Lipinski et al., Adv. Drug Delivery Rev., 23, 1997, 3.) Rules have always exception. (antibiotics, antibacterials and antimicrobials, )
8 What the chemist sees. Aspirin Sildenafil Viagra!!!
9 1D structure, line notation SMILES CC(=)C1=CC=CC=C1C(=) CCCC1=NN(C2=C1NC(=NC2=)C3=C(C= CC(=C3)S(=)(=)N4CCN(CC4)C)CC)C SMILES Simplified Molecular Line Entry System



10 Aspirin What a protein sees. Sildenafil Electrostatic fields (red are negative and blue positive)


11 Aspirin What a protein sees. Sildenafil Green are hydrophilic area and red are hydrophobic areas
12 Ligand based- QSAR Based on a set of experimental data (biological activity, solubility, toxicity, permeability, ) one tries to correlate these data with some structural descriptors. Method used are usually, PLS, SVM, NN, K-means.


13 Descriptors 1D descriptors: MW, number of features, MACCS key 2D descriptors: Topological, physichochemical, BCUT,
14 Descriptors QSAR based on 3 D interaction energies (GRID, CoMFA...) GRID: Determines a total interaction energy. Etot = Evdw + Eelec + Ehb Structural model Molecular interaction field (GRID) PCA and PLS model Score Plot 000 PC PC1 DRY N1 H2


15 Blue areas represent the favorable electronegative region and red the unfavorable electronegative regions (based on CoMFA) Green areas represent the favorable steric region and yellow the unfavorable steric regions (based on CoMFA).

16 Concept for scaffold hopping De novo design-scaffold hopping Protein- ligand complex
17 De novo design: Scaffold hopping Scaffold removed
 H-donor H-Bond Donor description")
 E (K c a l / m o l ) 0-0,2 0 2 4 6 8 10 12 14 16-0,4-0,6-0,8-1 -1,2-1,4-1,6-1,8")
 Energy (kcal/mol) E ( K cal / m o l ) 0-1 0 2 4 6 8 10 12 14 16-2 -3-4 -5-6 -7-8")
18 De novo design: Scaffold hopping physicochemical description Hydrophobic H-bond donor H-bond acceptor hydrophobic Hydrophobic-Protein description descriptor (anchor 1) H-donor H-Bond Donor description -Protein descriptor( anchor 1) H-acceptor H-Bond Acceptor description -Protein descriptor( anchor 1) Energy (kcal/mol) E (K c a l / m o l ) 0-0, ,4-0,6-0,8-1 -1,2-1,4-1,6-1,8 Distance Distance (A) Energy (kcal/mol) E (K c a l /m o l) Distance (A) Energy (kcal/mol) E ( K cal / m o l ) Distance (A)

19 De novo design: Scaffold hopping New scaffold insert



20 Scaffold hopping: Example with HIV protease N S N A N S N A H H Query Scaffold Definition A H A H Scaffold Hopping N H P Synthetic Feasibiltiy N H P New Compound Building A N A H A P A ASP25 S1 ASP25 ASP25 S1 ASP25 S1 S1 S2 S2 S2 S2 ILE50 ILE50 ILE50 ILE50 N N 2 Br H 2 N TBMS N N H P H P Bergmann R. et al., j. med. Chem (11):
21 Pharmacophore Definition A pharmacophore is the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target structure and to trigger (or to block) its biological response. A pharmacophore does not represent a real molecule or a real association of functional groups, but a purely abstract concept that accounts for the common molecular interaction capacities of a group of compounds towards their target structure.
22 Pharmacophore Pharmacophore: chemical features The chemical features can be hydrogen bonds acceptors, hydrogen bond donors, charge interactions, hydrophobic areas, aromatic rings, positive or negative ionizable group.) The shape or volume is also considered. Hyd Acc Acc Aro Acc & Don Start to be complex!!! Pharmacophores represent chemical functions, valid not only for the curretly bound, but also unknown molecules. The steric hindrance may explain lack of activity.


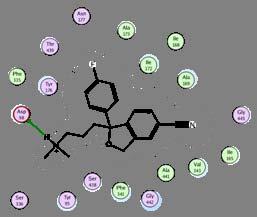
23 Pharmacophore Example 1 Atom is acceptor if it s a nitrogen, oxygen or sulfur and not an amide nitrogen, aniline nitrogen and sulfonyl sulfur and nitro group nitrogen Acceptor Acceptor Donor Aro ring center Don Acc Acc Acc & Don Acc & Don Acc & Don Acc & Don Acc Acc


 Apomorphine")

24 Pharmacophore Example 2 with 3 inhibitors Agonist at D2 receptor Dopamine (2 rotations and 2 H groups) Apomorphine (no rotations) 5-H DPAT (one H group and many rotation)


25 Pharmacophore Example 2 Active agonists define important groups: -Aromatic ring -meta H group -N atom, righ distance from aromatic ring -other molecular scaffolding does NT get in the way at the receptor
26 Pharmacophore Effect of ph Acceptor ph = 7 Donor ph = 1
 But most drugs have some conformational flexibility, and can have different shapes (e.")
27 Pharmacophore Which conformation? Some drugs are rigid (e.g, strychnine, a Glycine receptor antagonist) But most drugs have some conformational flexibility, and can have different shapes (e.g, sildenafil) Pharmacophore should be presented only by high energy conformation (Xray, NMR, minimisation, stochastic search)
28 Pharmacophore Pharmacophore flexibility A pharmacophore is usually obtained by connecting the average spatial positions of the pharmacophoric points of all the molecules. But sometime, several binding site or flexible binding site.

29 Pharmacophore Binding site activity can be flexible
30 Pharmacophore
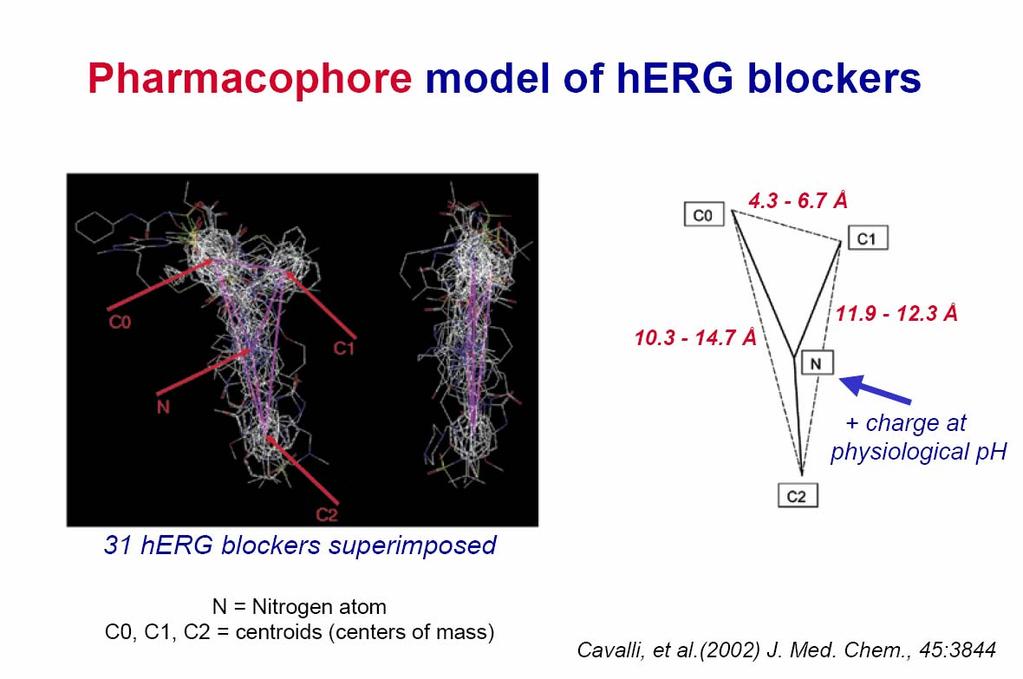
31 Pharmacophore

32 Are features of the site unique to herg?
33
34 What is a good pharmacophore? As many protein structures are described as sets of points, pharmacophore identification is commonly reduced directly to the problem of finding points common to all functional ligand conformations Pharmacophore identification From X-ray crystallography measure X-ray structure with drug at the active site (can sometime be done) or infer binding by measuring distance between likely binding groups. From comparison of active compounds The traditional way to identify binding groups. Automatic identification of pharmacophores (GALAHAD, Pharmacophore elucidation )

35 Structural based design: Docking

36 Docking


37 Structural based design: Docking Comparison of some of the docking tools. Kellenberger E. et al. Proteins 2004, 57(2):
38 Structural based design: Docking Induced fit docking Substrate (ligand) + Enzyme Substrate (receptor) + (ligand) Enzyme (receptor) Lock and Key Induced Fit
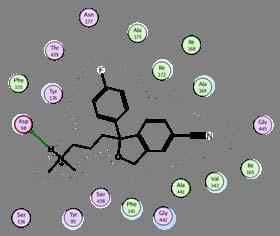
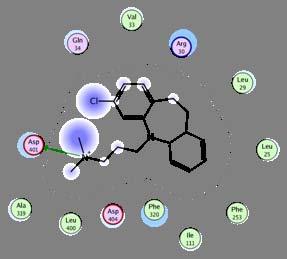
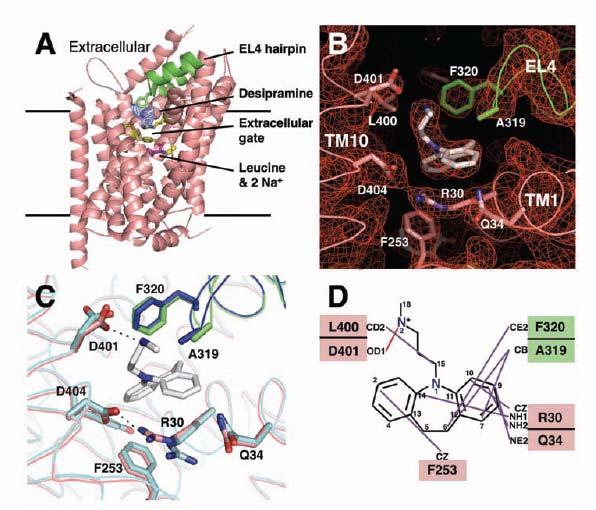
39 Structural based design: an example with antidepressant Zhou, Sciences 2007

40 Chemogenomics and Pharmacogenomics Chemogenomics: Studied the biological effect of a wide array of small molecules on a wide array of macromolecular targets. Pharmacogenomics: Design drugs according of the genetic variation.
41 Chemical network: example with neurotransmitter transporters
42 Drug efficacy-mutation: an example with citalopram A441G CIT: 2.5x gain of function CIT with ears : 2x gain of function A441G H 2 N V343S V343N V343S CIT: 4.5x gain of function Des-CN: 4.9x gain of function [Des-F: 7.4x gain of function] V343N CIT: 3.5x gain of function Des-CN: 35.5x gain of function [Des-F: 3.8x gain of function] A173S A173S CIT: 4.5x gain of function Des-F: 16.5x gain of function S438T S438T CIT: 175x loss of function Monomethyl: 3.5x loss of function
43 Chemogenomics and Pharmacogenomics: an example with citalopram A441G CIT: 2.5x gain of function CIT with ears : 2x gain of function A441G H 2 N V343S V343N V343S CIT: 4.5x gain of function Des-CN: 4.9x gain of function [Des-F: 7.4x gain of function] V343N CIT: 3.5x gain of function Des-CN: 35.5x gain of function [Des-F: 3.8x gain of function] A173S A173S CIT: 4.5x gain of function Des-F: 16.5x gain of function S438T S438T CIT: 175x loss of function Monomethyl: 3.5x loss of function
44 Conclusion According of the information you have, different strategies can be used. If you can develop different strategies which come to the same conclusion, that will reinforce your hypothesis. The most information (experimental) you have, the better your validation will be.
Plan. Day 2: Exercise on MHC molecules.
 Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Plan. Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics.
 and its used in Chemoinformatics.") Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Early Stages of Drug Discovery in the Pharmaceutical Industry
 Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Early Stages of Drug Discovery in the Pharmaceutical Industry Daniel Seeliger / Jan Kriegl, Discovery Research, Boehringer Ingelheim September 29, 2016 Historical Drug Discovery From Accidential Discovery
Advanced Medicinal Chemistry SLIDES B
 Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Nonlinear QSAR and 3D QSAR
 onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Ping-Chiang Lyu. Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University.
 Pharmacophore-based Drug design Ping-Chiang Lyu Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University 96/08/07 Outline Part I: Analysis The analytical
Pharmacophore-based Drug design Ping-Chiang Lyu Institute of Bioinformatics and Structural Biology, Department of Life Science, National Tsing Hua University 96/08/07 Outline Part I: Analysis The analytical
Structure-Activity Modeling - QSAR. Uwe Koch
 Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
ASSESSING THE DRUG ABILITY OF CHALCONES USING IN- SILICO TOOLS
 Page109 IJPBS Volume 5 Issue 1 JAN-MAR 2015 109-114 Research Article Pharmaceutical Sciences ASSESSING THE DRUG ABILITY OF CHALCONES USING IN- SILICO TOOLS Department of Pharmaceutical Chemistry, Institute
Page109 IJPBS Volume 5 Issue 1 JAN-MAR 2015 109-114 Research Article Pharmaceutical Sciences ASSESSING THE DRUG ABILITY OF CHALCONES USING IN- SILICO TOOLS Department of Pharmaceutical Chemistry, Institute
Targeting protein-protein interactions: A hot topic in drug discovery
 Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Drug Discovery. Zainab Al Kharusi Office: 33-10
 Drug Discovery Zainab Al Kharusi Office: 33-10 Objectives: To understand the processes of Drug Development To be able to develop a plan for drug discovery Reference: Patrick G L. An Introduction to Medicinal
Drug Discovery Zainab Al Kharusi Office: 33-10 Objectives: To understand the processes of Drug Development To be able to develop a plan for drug discovery Reference: Patrick G L. An Introduction to Medicinal
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Principles of Drug Design
 Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Hydrogen Bonding & Molecular Design Peter
 Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
Structure based drug design and LIE models for GPCRs
 Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
Navigation in Chemical Space Towards Biological Activity. Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland
 Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Molecular docking, 3D-QSAR studies of indole hydrazone as Staphylococcus aureus pyruvate kinase inhibitor
 World Journal of Pharmaceutical Sciences ISSN (Print): 2321-3310; ISSN (Online): 2321-3086 Published by Atom and Cell Publishers All Rights Reserved Available online at: http://www.wjpsonline.org/ Original
World Journal of Pharmaceutical Sciences ISSN (Print): 2321-3310; ISSN (Online): 2321-3086 Published by Atom and Cell Publishers All Rights Reserved Available online at: http://www.wjpsonline.org/ Original
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Keywords: anti-coagulants, factor Xa, QSAR, Thrombosis. Introduction
 PostDoc Journal Vol. 2, No. 3, March 2014 Journal of Postdoctoral Research www.postdocjournal.com QSAR Study of Thiophene-Anthranilamides Based Factor Xa Direct Inhibitors Preetpal S. Sidhu Department
PostDoc Journal Vol. 2, No. 3, March 2014 Journal of Postdoctoral Research www.postdocjournal.com QSAR Study of Thiophene-Anthranilamides Based Factor Xa Direct Inhibitors Preetpal S. Sidhu Department
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Chemical library design
 Chemical library design Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery Today,
Chemical library design Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery Today,
BioSolveIT. A Combinatorial Approach for Handling of Protonation and Tautomer Ambiguities in Docking Experiments
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
Ligand-receptor interactions
 University of Silesia, Katowice, Poland 11 22 March 2013 Ligand-receptor interactions Dr. Pavel Polishchuk A.V. Bogatsky Physico-Chemical Institute of National Academy of Sciences of Ukraine Odessa, Ukraine
University of Silesia, Katowice, Poland 11 22 March 2013 Ligand-receptor interactions Dr. Pavel Polishchuk A.V. Bogatsky Physico-Chemical Institute of National Academy of Sciences of Ukraine Odessa, Ukraine
Virtual screening for drug discovery. Markus Lill Purdue University
 Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Quantum Mechanical Models of P450 Metabolism to Guide Optimization of Metabolic Stability
 Quantum Mechanical Models of P450 Metabolism to Guide Optimization of Metabolic Stability Optibrium Webinar 2015, June 17 2015 Jonathan Tyzack, Matthew Segall, Peter Hunt Optibrium, StarDrop, Auto-Modeller
Quantum Mechanical Models of P450 Metabolism to Guide Optimization of Metabolic Stability Optibrium Webinar 2015, June 17 2015 Jonathan Tyzack, Matthew Segall, Peter Hunt Optibrium, StarDrop, Auto-Modeller
Overview. Descriptors. Definition. Descriptors. Overview 2D-QSAR. Number Vector Function. Physicochemical property (log P) Atom
 Atom") verview D-QSAR Definition Examples Features counts Topological indices D fingerprints and fragment counts R-group descriptors ow good are D descriptors in practice? Summary Peter Gedeck ovartis Institutes
verview D-QSAR Definition Examples Features counts Topological indices D fingerprints and fragment counts R-group descriptors ow good are D descriptors in practice? Summary Peter Gedeck ovartis Institutes
Different conformations of the drugs within the virtual library of FDA approved drugs will be generated.
 Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Computational Methods and Drug-Likeness. Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004
 Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
FRAUNHOFER IME SCREENINGPORT
 FRAUNHOFER IME SCREENINGPORT Design of screening projects General remarks Introduction Screening is done to identify new chemical substances against molecular mechanisms of a disease It is a question of
FRAUNHOFER IME SCREENINGPORT Design of screening projects General remarks Introduction Screening is done to identify new chemical substances against molecular mechanisms of a disease It is a question of
Principles of Drug Design
 (16:663:502) Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu For more current information, please check WebCT at https://webct.rutgers.edu
(16:663:502) Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu For more current information, please check WebCT at https://webct.rutgers.edu
MOLECULAR DRUG TARGETS
 MOLECULAR DRUG TARGETS LEARNING OUTCOMES At the end of this session student shall be able to: List different types of druggable targets Describe forces involved in drug-receptor interactions Describe theories
MOLECULAR DRUG TARGETS LEARNING OUTCOMES At the end of this session student shall be able to: List different types of druggable targets Describe forces involved in drug-receptor interactions Describe theories
1. (18) Multiple choice questions. Please place your answer on the line preceding each question.
 Multiple choice questions. Please place your answer on the line preceding each question.") CEM 5720 ame KEY Exam 2 ctober 21, 2015 Read all questions carefully and attempt those questions you are sure of first. Remember to proof your work; art work carries as much importance as written responses.
CEM 5720 ame KEY Exam 2 ctober 21, 2015 Read all questions carefully and attempt those questions you are sure of first. Remember to proof your work; art work carries as much importance as written responses.
Ignasi Belda, PhD CEO. HPC Advisory Council Spain Conference 2015
 Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Medicinal Chemistry/ CHEM 458/658 Chapter 3- SAR and QSAR
 Medicinal Chemistry/ CHEM 458/658 Chapter 3- SAR and QSAR Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Introduction Structure-Activity Relationship (SAR) - similar
Medicinal Chemistry/ CHEM 458/658 Chapter 3- SAR and QSAR Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Introduction Structure-Activity Relationship (SAR) - similar
Structure-Based Drug Discovery An Overview
 Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
Structure-Based Drug Discovery An Overview Edited by Roderick E. Hubbard University of York, Heslington, York, UK and Vernalis (R&D) Ltd, Abington, Cambridge, UK RSC Publishing Contents Chapter 1 3D Structure
Supporting Information (Part II) for ACS Combinatorial Science
 for ACS Combinatorial Science") Supporting Information (Part II) for ACS Combinatorial Science Application of 6,7Indole Aryne Cycloaddition and Pd(0)Catalyzed SuzukiMiyaura and BuchwaldHartwig CrossCoupling Reactions for the Preparation
Supporting Information (Part II) for ACS Combinatorial Science Application of 6,7Indole Aryne Cycloaddition and Pd(0)Catalyzed SuzukiMiyaura and BuchwaldHartwig CrossCoupling Reactions for the Preparation
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
Author Index Volume
 Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Machine Learning Concepts in Chemoinformatics
 Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
Synthetic organic compounds
 Synthetic organic compounds for research and drug discovery Compounds for TS Fragment libraries Target-focused libraries Chemical building blocks Custom synthesis Drug discovery services Contract research
Synthetic organic compounds for research and drug discovery Compounds for TS Fragment libraries Target-focused libraries Chemical building blocks Custom synthesis Drug discovery services Contract research
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors
 A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites. J. Andrew Surface
 Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
MEDICINAL CHEMISTRY Homework 2 - ANSWERS
 MEDICIAL CEMISTY omework 2 - ASWES 1. eceptor binding curves. Make a plot of %drug-bound versus -log[drug] for a drug whose K D is 5 x 10-6 M. Make a plot of %drug-bound vs [drug] for this same system.
MEDICIAL CEMISTY omework 2 - ASWES 1. eceptor binding curves. Make a plot of %drug-bound versus -log[drug] for a drug whose K D is 5 x 10-6 M. Make a plot of %drug-bound vs [drug] for this same system.
Solved and Unsolved Problems in Chemoinformatics
 Solved and Unsolved Problems in Chemoinformatics Johann Gasteiger Computer-Chemie-Centrum University of Erlangen-Nürnberg D-91052 Erlangen, Germany Johann.Gasteiger@fau.de Overview objectives of lecture
Solved and Unsolved Problems in Chemoinformatics Johann Gasteiger Computer-Chemie-Centrum University of Erlangen-Nürnberg D-91052 Erlangen, Germany Johann.Gasteiger@fau.de Overview objectives of lecture
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Relative Drug Likelihood: Going beyond Drug-Likeness
 Relative Drug Likelihood: Going beyond Drug-Likeness ACS Fall National Meeting, August 23rd 2012 Matthew Segall, Iskander Yusof Optibrium, StarDrop, Auto-Modeller and Glowing Molecule are trademarks of
Relative Drug Likelihood: Going beyond Drug-Likeness ACS Fall National Meeting, August 23rd 2012 Matthew Segall, Iskander Yusof Optibrium, StarDrop, Auto-Modeller and Glowing Molecule are trademarks of
Patrick: An Introduction to Medicinal Chemistry 5e Chapter 01
 Questions Patrick: An Introduction to Medicinal Chemistry 5e 01) Which of the following molecules is a phospholipid? a. i b. ii c. iii d. iv 02) Which of the following statements is false regarding the
Questions Patrick: An Introduction to Medicinal Chemistry 5e 01) Which of the following molecules is a phospholipid? a. i b. ii c. iii d. iv 02) Which of the following statements is false regarding the
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Chapter 8: Introduction to QSAR
 : Introduction to 8) Chapter 8: 181 8.1 Introduction to 181 8.2 Objectives of 181 8.3 Historical development of 182 8.4 Molecular descriptors used in 183 8.5 Methods of 185 8.5.1 2D methods 186 8.6 Introduction
: Introduction to 8) Chapter 8: 181 8.1 Introduction to 181 8.2 Objectives of 181 8.3 Historical development of 182 8.4 Molecular descriptors used in 183 8.5 Methods of 185 8.5.1 2D methods 186 8.6 Introduction
Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,..
 3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
SCULPT 3.0. Using SCULPT to Gain Competitive Insights. Brings 3D Visualization to the Lab Bench SPECIAL REPORT. 4 Molecular Connection Fall 1999
 SPECIAL REPORT SCULPT 3.0 Brings 3D Visualization to the Lab Bench ith the acquisition of Interactive Simulations, Inc., last April, MDL added to its arsenal of discovery informatics tools a powerful analysis
SPECIAL REPORT SCULPT 3.0 Brings 3D Visualization to the Lab Bench ith the acquisition of Interactive Simulations, Inc., last April, MDL added to its arsenal of discovery informatics tools a powerful analysis
Life Sciences 1a Lecture Slides Set 10 Fall Prof. David R. Liu. Lecture Readings. Required: Lecture Notes McMurray p , O NH
 Life ciences 1a Lecture lides et 10 Fall 2006-2007 Prof. David R. Liu Lectures 17-18: The molecular basis of drug-protein binding: IV protease inhibitors 1. Drug development and its impact on IV-infected
Life ciences 1a Lecture lides et 10 Fall 2006-2007 Prof. David R. Liu Lectures 17-18: The molecular basis of drug-protein binding: IV protease inhibitors 1. Drug development and its impact on IV-infected
Open PHACTS Explorer: Compound by Name
 Open PHACTS Explorer: Compound by Name This document is a tutorial for obtaining compound information in Open PHACTS Explorer (explorer.openphacts.org). Features: One-click access to integrated compound
Open PHACTS Explorer: Compound by Name This document is a tutorial for obtaining compound information in Open PHACTS Explorer (explorer.openphacts.org). Features: One-click access to integrated compound
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses
 Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
CHAPTER 3. Pharmacophore modelling studies:
 CHAPTER 3 Pharmacophore modelling studies: 3.1 Introduction: According to Langer & Wolber (2004), the key goal of computer-aided molecular design methods in medicinal chemistry is to reduce the time and
CHAPTER 3 Pharmacophore modelling studies: 3.1 Introduction: According to Langer & Wolber (2004), the key goal of computer-aided molecular design methods in medicinal chemistry is to reduce the time and
RATIONAL DRUG DESIGN
 RATIOAL DRUG DESIG Drug Design & Discovery: Introduction Drugs: Targets: atural sources Synthetic sources Ideal Drug 1) target: bio-molecule,involved in signaling or metabolic pathways, that are specific
RATIOAL DRUG DESIG Drug Design & Discovery: Introduction Drugs: Targets: atural sources Synthetic sources Ideal Drug 1) target: bio-molecule,involved in signaling or metabolic pathways, that are specific
Welcome to Week 5. Chapter 9 - Binding, Structure, and Diversity. 9.1 Intermolecular Forces. Starting week five video. Introduction to Chapter 9
 Welcome to Week 5 Starting week five video Please watch the online video (49 seconds). Chapter 9 - Binding, Structure, and Diversity Introduction to Chapter 9 Chapter 9 contains six subsections. Intermolecular
Welcome to Week 5 Starting week five video Please watch the online video (49 seconds). Chapter 9 - Binding, Structure, and Diversity Introduction to Chapter 9 Chapter 9 contains six subsections. Intermolecular
Practical QSAR and Library Design: Advanced tools for research teams
 DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
* Author to whom correspondence should be addressed; Tel.: ; Fax:
 Int. J. Mol. Sci. 2011, 12, 946-970; doi:10.3390/ijms12020946 OPEN ACCESS Article International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Structural Determination of Three
Int. J. Mol. Sci. 2011, 12, 946-970; doi:10.3390/ijms12020946 OPEN ACCESS Article International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Structural Determination of Three
Data Mining in the Chemical Industry. Overview of presentation
 Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
Data Mining in the Chemical Industry Glenn J. Myatt, Ph.D. Partner, Myatt & Johnson, Inc. glenn.myatt@gmail.com verview of presentation verview of the chemical industry Example of the pharmaceutical industry
3D-QSAR Studies on Angiotensin-Converting Enzyme (ACE) Inhibitors: a Molecular Design in Hypertensive Agents
 Inhibitors: a Molecular Design in Hypertensive Agents") 952 Bull. Korean Chem. Soc. 2005, Vol. 26, No. 6 Amor A. San Juan and Seung Joo Cho 3D-QSAR Studies on Angiotensin-Converting Enzyme (ACE) Inhibitors: a Molecular Design in Hypertensive Agents Amor A.
952 Bull. Korean Chem. Soc. 2005, Vol. 26, No. 6 Amor A. San Juan and Seung Joo Cho 3D-QSAR Studies on Angiotensin-Converting Enzyme (ACE) Inhibitors: a Molecular Design in Hypertensive Agents Amor A.
Saba Al Fayoumi. Tamer Barakat. Dr. Mamoun Ahram + Dr. Diala Abu-Hassan
 1 Saba Al Fayoumi Tamer Barakat Dr. Mamoun Ahram + Dr. Diala Abu-Hassan What is BIOCHEMISTRY??? Biochemistry = understanding life Chemical reactions are what makes an organism (An organism is simply atoms
1 Saba Al Fayoumi Tamer Barakat Dr. Mamoun Ahram + Dr. Diala Abu-Hassan What is BIOCHEMISTRY??? Biochemistry = understanding life Chemical reactions are what makes an organism (An organism is simply atoms
Toward an Understanding of GPCR-ligand Interactions. Alexander Heifetz
 Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
COMPUTER AIDED DRUG DESIGN (CADD) AND DEVELOPMENT METHODS
 AND DEVELOPMENT METHODS") COMPUTER AIDED DRUG DESIGN (CADD) AND DEVELOPMENT METHODS DRUG DEVELOPMENT Drug development is a challenging path Today, the causes of many diseases (rheumatoid arthritis, cancer, mental diseases, etc.)
COMPUTER AIDED DRUG DESIGN (CADD) AND DEVELOPMENT METHODS DRUG DEVELOPMENT Drug development is a challenging path Today, the causes of many diseases (rheumatoid arthritis, cancer, mental diseases, etc.)
György M. Keserű H2020 FRAGNET Network Hungarian Academy of Sciences
 Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
schematic diagram; EGF binding, dimerization, phosphorylation, Grb2 binding, etc.
 Lecture 1: Noncovalent Biomolecular Interactions Bioengineering and Modeling of biological processes -e.g. tissue engineering, cancer, autoimmune disease Example: RTK signaling, e.g. EGFR Growth responses
Lecture 1: Noncovalent Biomolecular Interactions Bioengineering and Modeling of biological processes -e.g. tissue engineering, cancer, autoimmune disease Example: RTK signaling, e.g. EGFR Growth responses
Introduction to medicinal chemisry
 2017 1 st edition Introduction to medicinal chemisry Med Chem I Mohammed Nooraldeen (PhD) كيمياء دوائية )1 ) محمد نورالدين محمود Introduction to medicinal chemistry Medicinal Chemistry Biology Medicine
2017 1 st edition Introduction to medicinal chemisry Med Chem I Mohammed Nooraldeen (PhD) كيمياء دوائية )1 ) محمد نورالدين محمود Introduction to medicinal chemistry Medicinal Chemistry Biology Medicine
FORCES THAT DETERMINE LIGAND-RECEPTOR INTERACTIONS
 Corpora non agunt nisi fixata Paul Ehrlich Address in Pathology on Chemotherapeutics: Scientific Principles, Methods, and Results Lancet II, 445 (1913) FRCES TAT DETERMIE LIGAD-RECEPTR ITERACTIS Favourable
Corpora non agunt nisi fixata Paul Ehrlich Address in Pathology on Chemotherapeutics: Scientific Principles, Methods, and Results Lancet II, 445 (1913) FRCES TAT DETERMIE LIGAD-RECEPTR ITERACTIS Favourable
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Drug Informatics for Chemical Genomics...
 Drug Informatics for Chemical Genomics... An Overview First Annual ChemGen IGERT Retreat Sept 2005 Drug Informatics for Chemical Genomics... p. Topics ChemGen Informatics The ChemMine Project Library Comparison
Drug Informatics for Chemical Genomics... An Overview First Annual ChemGen IGERT Retreat Sept 2005 Drug Informatics for Chemical Genomics... p. Topics ChemGen Informatics The ChemMine Project Library Comparison
Design and Synthesis of the Comprehensive Fragment Library
 YOUR INNOVATIVE CHEMISTRY PARTNER IN DRUG DISCOVERY Design and Synthesis of the Comprehensive Fragment Library A 3D Enabled Library for Medicinal Chemistry Discovery Warren S Wade 1, Kuei-Lin Chang 1,
YOUR INNOVATIVE CHEMISTRY PARTNER IN DRUG DISCOVERY Design and Synthesis of the Comprehensive Fragment Library A 3D Enabled Library for Medicinal Chemistry Discovery Warren S Wade 1, Kuei-Lin Chang 1,
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
CHEM 4170 Problem Set #1
 CHEM 4170 Problem Set #1 0. Work problems 1-7 at the end of Chapter ne and problems 1, 3, 4, 5, 8, 10, 12, 17, 18, 19, 22, 24, and 25 at the end of Chapter Two and problem 1 at the end of Chapter Three
CHEM 4170 Problem Set #1 0. Work problems 1-7 at the end of Chapter ne and problems 1, 3, 4, 5, 8, 10, 12, 17, 18, 19, 22, 24, and 25 at the end of Chapter Two and problem 1 at the end of Chapter Three
Alkane/water partition coefficients and hydrogen bonding. Peter Kenny
 Alkane/water partition coefficients and hydrogen bonding Peter Kenny (pwk.pub.2008@gmail.com) Neglect of hydrogen bond strength: A recurring theme in medicinal chemistry Rule of 5 Rule of 3 Scoring functions
Alkane/water partition coefficients and hydrogen bonding Peter Kenny (pwk.pub.2008@gmail.com) Neglect of hydrogen bond strength: A recurring theme in medicinal chemistry Rule of 5 Rule of 3 Scoring functions
Notes of Dr. Anil Mishra at 1
 Introduction Quantitative Structure-Activity Relationships QSPR Quantitative Structure-Property Relationships What is? is a mathematical relationship between a biological activity of a molecular system
Introduction Quantitative Structure-Activity Relationships QSPR Quantitative Structure-Property Relationships What is? is a mathematical relationship between a biological activity of a molecular system
EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS
 EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS PETER GUND Pharmacopeia Inc., CN 5350 Princeton, NJ 08543, USA pgund@pharmacop.com Empirical and theoretical approaches to drug discovery have often
EMPIRICAL VS. RATIONAL METHODS OF DISCOVERING NEW DRUGS PETER GUND Pharmacopeia Inc., CN 5350 Princeton, NJ 08543, USA pgund@pharmacop.com Empirical and theoretical approaches to drug discovery have often
Synthetic organic compounds
 Synthetic organic compounds for research and drug discovery chemicals Compounds for TS Fragment libraries Target-focused libraries Chemical building blocks Custom synthesis Drug discovery services Contract
Synthetic organic compounds for research and drug discovery chemicals Compounds for TS Fragment libraries Target-focused libraries Chemical building blocks Custom synthesis Drug discovery services Contract
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification
 Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
THERMODYNAMICS IN DRUG DESIGN. HIGH AFFINITY AND SELECTIVITY
 1 The Chemical Theatre of Biological Systems, May 24 th - 28 th, 2004, Bozen, Italy THERMODYNAMICS IN DRUG DESIGN. HIGH AFFINITY AND SELECTIVITY ERNESTO FREIRE Johns Hopkins University, Department of Biology,
1 The Chemical Theatre of Biological Systems, May 24 th - 28 th, 2004, Bozen, Italy THERMODYNAMICS IN DRUG DESIGN. HIGH AFFINITY AND SELECTIVITY ERNESTO FREIRE Johns Hopkins University, Department of Biology,
QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression
 APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor