Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
|
|
|
- Amberlynn Malone
- 6 years ago
- Views:
Transcription
1 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
2 Numerous applications
3 Generating conformations MM Agenda ConfGen WhitePaper coming soon Clustering conformations Optimising conformations Scripts QM-MM Mogul CCDC
4 GENERATING CONFORMATIONS
5 Intro to MacroModel, ConfGen Conformational searches are a very important part of 3D-ligand-based design...when we have no crystal structure to guide us we need to generate plausible 3D-structures for our ligands. We may also be interested in conformations of protein-ligandsystems for lead optimization... The Schrödinger suite provides two related tools for this operation: MacroModel The underlying engine for conformational searches Capable of some highly advanced searching ConfGen Part of MacroModeland a separate product. Includes tweaks to the basic force-field to tailor conformations towards those found in protein-bound ligands Prime (complexes and macrocycles) and MM+QM (ligands)
6 Which to use when? MacroModel searches generate all of the possible conformers of a molecule Depending on the settings these will be the gas/solution phase conformers These are different from the bio-active conformers, and the output energy window produces greater number of structures MacroModel conformations are useful when we need to understand a molecule s solution phase behaviour accurately MacroModel is also useful in optimizing PL environments ConfGen is designed to generate bio-active conformations These are useful when we are considering a ligand bound to a protein Docking Generate addiitonal conformations outside of Glide Docking Shape similarity searching Pharmacophores

 MacroModel finds fewer conformers (~150) These all bury the polar functionality, leaving exposed")
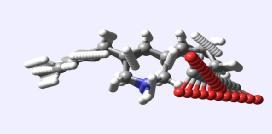
7 MacroModel Conformational Searching MacroModel facilitates conformational searching of large and small systems It has an advanced continuum solvation model that is able to mimic a variety of environments The environment of a system is crucial in obtaining a meaningful set of conformers Conformations of KFGLE-Peptide MacroModelfinds ~230 distinct conformers of KFGLE in water (left) These have minimal intramolecularhydrogenbonding and leave polar groups exposed Conversely in CHCl 3 (right) MacroModel finds fewer conformers (~150) These all bury the polar functionality, leaving exposed hydrophobicity

8 ConfGen Conformational Searching ConfGen attempts to mimic a general protein environment during its conformational searches This penalises compact conformations and rewards more extended forms Careful balancing of intramolecular interactions is the result of considerable parameterisation and research For virtual screening and most ligand-based drug design, ConfGen is the tool of choice Conformations of KFGLE-Peptide ConfGenselects a considerably more open conformation for the KFGLE-Peptide It is assumed that the charged and hydrophobic groups will find partners in the associated host

9 Minima of core rotatable bonds systematically identified and sampled Terminal rotamer groups then sampled Sampling Method in ConfGen No electrostatics Ring template library from Mmod 1-4 interactions to define potential 1. Core rotatable groups 2. Terminal rotamers
10 ConfGen - A New Fast 3D Conformation Generator Process compounds/sec 25x faster then ConfGen (Intermediate mode) Good recovery of bioactive conformations Time Per Compound (sec.) New ConfGen ConfGen Intermediate ConfGen Comprehensive Percent of Compounds Reproduction of Bioactive Conformations New ConfGen ConfGen Intermediate ConfGen Comprehensive <0.5 <0.75 <1.0 <1.25 <1.5 <1.75 <2.0 <2.25 <2.5 <2.75 <3.0 RMSD Cutoff (Angstroms)
11 Old CG Settings ConfGen product page WhitePaper coming soon
12 Simple interface ConfGen GUI Extra option to minimize output conformations While including min, does not improve matching crystal structure (as measured by RMSD), min can eliminate some close contacts
13 Sampling Methods in MacroModel Monte-Carlo Multiple Minima: A MC search of the specified torsional space. Works well for <15 torsions. More than this requires extended runtimes. In practice this is an extremely efficient search methodology which is easy to apply. Systematic Pseudo Monte-Carlo: Modified MC code, drives the search to regions of the PES not normally explored. Most suited to small molecule searches. Low Mode Searching: The eigenvectors of the molecular Hessian matrix give information on concerted motions of the system. These concerted motions are used as coordinates in the conformational search. Large Scale Low Mode Searching: A specially tuned version of the above code for larger systems. Particularly well suited to protein conformational searching. Mixed MCMM/Low Mode Searching: A hybrid technique where specific torsions can be varied outside a standard low mode search. This is an exceptionally powerful methodology for exploring normally difficult questions such as a ligand enclosed within a binding site.


14 MacroModel has a Consistent GUI, irrespective of the task From a simple minimisation to a full conformational search, the panels are consistent
15 CLUSTERING CONFORMATIONS
16
17 Clustering Scripts Clustering of Conformers: A graphical user interface and command-line script to cluster conformations based on Cartesian or torsional RMSD. User can specify the atoms to use (based on the ASL) and the clustering settings. [Script name: conformer_cluster.py (Revision 3.16] [Script type: Maestro/Python] Spectral clustering: A script that implements the Spectral Clustering method as described by Mark Brewer in 'Development of a Spectral Clustering Method for the Analysis of Molecular Data Sets,' J. Chem. Inf. Model, 2007, 47, The cluster properties (cluster membership, cluster contribution and cluster eigenvalue) are added to the project table for each input entry. [Script name: spectral_cluster.py (Revision 3.3)] [Script type: Maestro/Python] [Requires: Canvas]

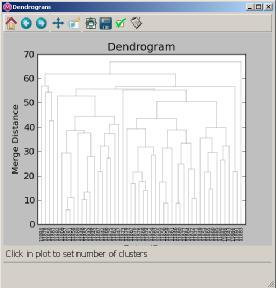
18 Processing with conformer_cluster.py 1. Select atoms (e.g. all ) and calculate RMSD matrix 2. Run clustering 3. Find out optimal number of clusters 4. Apply clustering
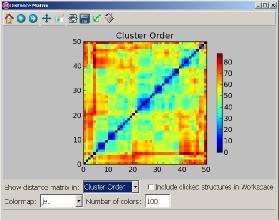
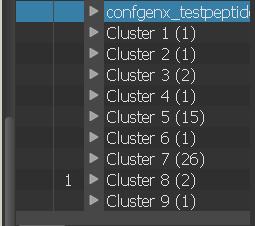
19 Clustering Statistics...
20 OPTIMISING CONFORMATIONS
21 Combining MacroModel with Jaguar Confsearch_jag_min_workflow.py ( Background
22 -h
23 Mogul, CCDC Mogul provides precise information on preferred molecular geometries by enabling access to millions of chemically classified bond lengths, valence angles, acyclic torsion angles, and ring conformations derived from the CSD. Mogul enables you to rapidly validate the complete geometry of a given query structure and identify any unusual features without the need to construct complex search queries, or carry out detailed data analyses. Medicinal Chemists rely on Mogul to Validate conformations (calculated ones, or filtering out PL focking solutions involving unlikely ligand conformation geometry validation, eg checking the molecular dimensions of new crystal structures Search for Mogul in Tasks in Maestro 11
24 Validate Ligand Conformations with CCDC Mogul Check torsions, angle, and bond geometries against structures in the CSD Use local or remote Mogul instance with Schrödinger s JobControl Mogul is separately licensed
25 Multi-Tiered View of Torsion, Angle, and Bond Distributions
26 USING THE INTERFACES
27 WHAT ABOUT PROTEIN-LIGAND COMPLEXES?

28 Protein-Ligand Min /Conformational Search MacroModel provides CS of both ligands and complexes Options for searching over the PE surface of a protein include Low Mode Analysis which is very useful for jumping wells on the PE surface in order to find lowest energy conformations, optimise PL interactions and model induced fit effects MacroModel offers comprehensive tools for defining constraints Below is the DHFR enzyme complexed with methotrexatemm substructure facility, is used to setup varying levels of LP flexibility during CS process Flexible regions in white, semi flexible in orange, frozen regions in magenta

29 MacroModel Substructure tab allows us to easily setup this kind of flexibility
30 MacroModel : Refining and Modelling LigandPoses in Active Site Docking will always find a good, low energy pose for a ligand But this may not be the global minima A more comprehensive search, including the protein may enable the system to find an even better solution Energy G Because of the exponential relationship between free-energy and binding, even a small G can correspond to a large real-world activity change Conformational Space MacroModel Embrace Allows congeneric ideas to be scored in a parameterless fashion -Permits extremely advanced conformational sampling to be performed to model induced fit effects

31 An Example of the Effect of MacroModel Embrace Conformational Searches HIV-Protease Inhibitor Docked to 1QBR Glide leaves the ligand in the cyan pose The napthyl-group is sensibly placed, but non-optimal Similarly a hydroxyl-group is missing out two key interactions The Glide generated pose scores poorly The orange pose is generated by Embrace CSearch, the napthylgroup has been flipped significantly, while the hydroxyl is now making its proper interactions This refined pose scores significantly better
 R 2 = 0.")
32 Scoring the HIV-Protease Ligands: MacroModel Embrace The raw poses generated by Glide do a reasonable job of modelling the variation in potency However, asking Embrace to refine each of the poses by carrying out a conformational sampling of the ligand within the binding site, yields a superior correlation Embrace CSearch Refinement and Rescoring Pure minimisation (x) R 2 = 0.45 Embrace CSearch (ligand only) (o) R 2 = 0.53



33 Setting Up An Embrace Calculation Shows output pose from Docking and Embrace Calculation
34 Summary Together ConfGen and MacroModel can be used to search a wide range of molecules from ligands to protein/ligand complexes and other types of molecules Maestro makes setting up and running these calculations easy Wide range of speed/quality trade offs possible Small subsets of conformers generated by ConfGen can cover conformational space quite well Searching multiple structures in a single calculation automatically is supported Distributing calculations is supported
35 APPLICATION TEASER NEW PHASE
36 Schrödinger Has A Complete Suite of LBDD Solutions Pharmacophoremodeling (Phase) 3D QSAR (Field-Based QSAR) Shape screening (Shape Screening) Conformation generation (ConfGen) ADME property prediction (QikProp) QSAR/QSPR (Canvas, AutoQSAR) Significant ongoing investment in LBDD R&D Validate ligand conformations in Maestro with CCDC Mogul
37 Several Ways to Create Pharmacophore Hypotheses From multiple active ligands From one ligand conformation From apoprotein 2 From protein-ligand complex 1 1 Salam, N., Nuti, R., Sherman, W., J Chem Inf Model, 2009, 49(10), Loving, K., Salam, N., Sherman, W., J Comput-Aid Design, 2009, 23(8), 541.
38 New Phase Interface for Maestro 11 Highly interactive Modify attributes of hypothesis directly from hypothesis Easy to use for experts and non-experts
39 Resources for All Demos




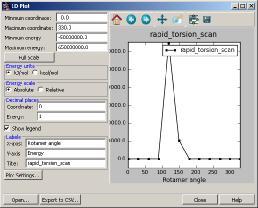
40 1. Two-dimensional Coordinate Scan A contour diagram describing the variation in energy of a molecule with respect to rotation of two dihedral angles Tasks > MacroModel Coordinate Scan Help, documentation index...search for macromodel scan

41 Tasks > MacroModel Minimization 2. Simple Minimization Help, documentation index...search for MacroModel minimization

42 3. CS in MM (Peptide example demo) Tasks > MacroModel conformational search Help, documentation index...search for MacroModel conformational searches
43 3b. CS in ConfGen (Peptide example demo) Simple GUI
44 Combining MacroModel with Jaguar Confsearch_jag_min_workflow.py ( Background

45 MacroModel Embrace Help, documentation index...search for MacroModel embrace


46 Python Scripts ~ 7 scripts associated with MacroModelon our Website
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Conformational Searching using MacroModel and ConfGen. John Shelley Schrödinger Fellow
 Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
CSD. Unlock value from crystal structure information in the CSD
 CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
CSD CSD-System Unlock value from crystal structure information in the CSD The Cambridge Structural Database (CSD) is the world s most comprehensive and up-todate knowledge base of crystal structure data,
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Zurich, February 2014 The Schrödinger extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Zurich, February 2014 The Schrödinger extensions
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Towards Physics-based Models for ADME/Tox. Tyler Day
 Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors
 MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
Analyzing Molecular Conformations Using the Cambridge Structural Database. Jason Cole Cambridge Crystallographic Data Centre
 Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Version 1.2 October 2017 CSD v5.39
 Mogul Geometry Check Table of Contents Introduction... 2 Example 1. Using Mogul to assess intramolecular geometry... 3 Example 2. Using Mogul to explain activity data... 5 Conclusions... 8 Further Exercises...
Mogul Geometry Check Table of Contents Introduction... 2 Example 1. Using Mogul to assess intramolecular geometry... 3 Example 2. Using Mogul to explain activity data... 5 Conclusions... 8 Further Exercises...
CSD Conformer Generator User Guide
 CSD Conformer Generator User Guide 2017 CSD Release Copyright 2016 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The CSD Conformer Generator is copyright work belonging
CSD Conformer Generator User Guide 2017 CSD Release Copyright 2016 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The CSD Conformer Generator is copyright work belonging
CSD Conformer Generator User Guide
 CSD Conformer Generator User Guide 2018 CSD Release Copyright 2017 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The CSD Conformer Generator is copyright work belonging
CSD Conformer Generator User Guide 2018 CSD Release Copyright 2017 Cambridge Crystallographic Data Centre Registered Charity No 800579 Conditions of Use The CSD Conformer Generator is copyright work belonging
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
Practical QSAR and Library Design: Advanced tools for research teams
 DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
A Brief Guide to All Atom/Coarse Grain Simulations 1.1
 A Brief Guide to All Atom/Coarse Grain Simulations 1.1 Contents 1. Introduction 2. AACG simulations 2.1. Capabilities and limitations 2.2. AACG commands 2.3. Visualization and analysis 2.4. AACG simulation
A Brief Guide to All Atom/Coarse Grain Simulations 1.1 Contents 1. Introduction 2. AACG simulations 2.1. Capabilities and limitations 2.2. AACG commands 2.3. Visualization and analysis 2.4. AACG simulation
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression
 APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
APPLICATION NOTE QSAR Modeling of ErbB1 Inhibitors Using Genetic Algorithm-Based Regression GAINING EFFICIENCY IN QUANTITATIVE STRUCTURE ACTIVITY RELATIONSHIPS ErbB1 kinase is the cell-surface receptor
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Flexibility and Constraints in GOLD
 Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
What is Protein-Ligand Docking?
 MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification
 Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Introduction to Structure Preparation and Visualization
 Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Identifying Interaction Hot Spots with SuperStar
 Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Pose and affinity prediction by ICM in D3R GC3. Max Totrov Molsoft
 Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Ultra High Throughput Screening using THINK on the Internet
 Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
CombiGlide 2.8. User Manual. Schrödinger Press
 CombiGlide User Manual CombiGlide 2.8 User Manual Schrödinger Press CombiGlide User Manual Copyright 2012 Schrödinger, LLC. All rights reserved. While care has been taken in the preparation of this publication,
CombiGlide User Manual CombiGlide 2.8 User Manual Schrödinger Press CombiGlide User Manual Copyright 2012 Schrödinger, LLC. All rights reserved. While care has been taken in the preparation of this publication,
tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules
 Software News and Updates tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules DANIEL SEELIGER, BERT L. DE GROOT Computational Biomolecular Dynamics Group, Max-Planck-Institute
Software News and Updates tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules DANIEL SEELIGER, BERT L. DE GROOT Computational Biomolecular Dynamics Group, Max-Planck-Institute
Cheminformatics platform for drug discovery application
 EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
How is molecular dynamics being used in life sciences? Davide Branduardi
 How is molecular dynamics being used in life sciences? Davide Branduardi davide.branduardi@schrodinger.com Exploring molecular processes with MD Drug discovery and design Protein-protein interactions Protein-DNA
How is molecular dynamics being used in life sciences? Davide Branduardi davide.branduardi@schrodinger.com Exploring molecular processes with MD Drug discovery and design Protein-protein interactions Protein-DNA
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today
 est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Principles of Drug Design
 Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Conformational Sampling of Druglike Molecules with MOE and Catalyst: Implications for Pharmacophore Modeling and Virtual Screening
 J. Chem. Inf. Model. 2008, 48, 1773 1791 1773 Conformational Sampling of Druglike Molecules with MOE and Catalyst: Implications for Pharmacophore Modeling and Virtual Screening I-Jen Chen* and Nicolas
J. Chem. Inf. Model. 2008, 48, 1773 1791 1773 Conformational Sampling of Druglike Molecules with MOE and Catalyst: Implications for Pharmacophore Modeling and Virtual Screening I-Jen Chen* and Nicolas
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Protein Structure Prediction and Protein-Ligand Docking
 Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
The Cambridge Structural Database (CSD) a Vital Resource for Structural Chemistry and Biology Stephen Maginn, CCDC, Cambridge, UK
 a Vital Resource for Structural Chemistry and Biology Stephen Maginn, CCDC, Cambridge, UK") The Cambridge Structural Database (CSD) a Vital Resource for Structural Chemistry and Biology Stephen Maginn, CCDC, Cambridge, UK 1 The Cambridge Crystallographic Data Centre The advancement and promotion
The Cambridge Structural Database (CSD) a Vital Resource for Structural Chemistry and Biology Stephen Maginn, CCDC, Cambridge, UK 1 The Cambridge Crystallographic Data Centre The advancement and promotion
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Ranking of HIV-protease inhibitors using AutoDock
 Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Life Science Webinar Series
 Life Science Webinar Series Elegant protein- protein docking in Discovery Studio Francisco Hernandez-Guzman, Ph.D. November 20, 2007 Sr. Solutions Scientist fhernandez@accelrys.com Agenda In silico protein-protein
Life Science Webinar Series Elegant protein- protein docking in Discovery Studio Francisco Hernandez-Guzman, Ph.D. November 20, 2007 Sr. Solutions Scientist fhernandez@accelrys.com Agenda In silico protein-protein
Portal. User Guide Version 1.0. Contributors
 Portal www.dockthor.lncc.br User Guide Version 1.0 Contributors Diogo A. Marinho, Isabella A. Guedes, Eduardo Krempser, Camila S. de Magalhães, Hélio J. C. Barbosa and Laurent E. Dardenne www.gmmsb.lncc.br
Portal www.dockthor.lncc.br User Guide Version 1.0 Contributors Diogo A. Marinho, Isabella A. Guedes, Eduardo Krempser, Camila S. de Magalhães, Hélio J. C. Barbosa and Laurent E. Dardenne www.gmmsb.lncc.br
GC and CELPP: Workflows and Insights
 GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
GC and CELPP: Workflows and Insights Xianjin Xu, Zhiwei Ma, Rui Duan, Xiaoqin Zou Dalton Cardiovascular Research Center, Department of Physics and Astronomy, Department of Biochemistry, & Informatics Institute
In Silico Investigation of Off-Target Effects
 PHARMA & LIFE SCIENCES WHITEPAPER In Silico Investigation of Off-Target Effects STREAMLINING IN SILICO PROFILING In silico techniques require exhaustive data and sophisticated, well-structured informatics
PHARMA & LIFE SCIENCES WHITEPAPER In Silico Investigation of Off-Target Effects STREAMLINING IN SILICO PROFILING In silico techniques require exhaustive data and sophisticated, well-structured informatics
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites. J. Andrew Surface
 Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Docking with Water in the Binding Site using GOLD
 Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
Garib N Murshudov MRC-LMB, Cambridge
 Garib N Murshudov MRC-LMB, Cambridge Contents Introduction AceDRG: two functions Validation of entries in the DB and derived data Generation of new ligand description Jligand for link description Conclusions
Garib N Murshudov MRC-LMB, Cambridge Contents Introduction AceDRG: two functions Validation of entries in the DB and derived data Generation of new ligand description Jligand for link description Conclusions
European Life Science Bootcamp: Case Studies in Enhanced Sampling Methods.
 European Life Science Bootcamp: Case Studies in Enhanced Sampling Methods davide.branduardi@schrodinger.com Outline Limitations of molecular dynamics Overcoming them with Enhanced sampling Pragmatic use
European Life Science Bootcamp: Case Studies in Enhanced Sampling Methods davide.branduardi@schrodinger.com Outline Limitations of molecular dynamics Overcoming them with Enhanced sampling Pragmatic use
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Jaguar DFT Optimizations and Transition State Searches
 Jaguar DFT Optimizations and Transition State Searches Density Functional Theory (DFT) is a quantum mechanical (QM) method that gives results superior to Hartree Fock (HF) in less computational time. A
Jaguar DFT Optimizations and Transition State Searches Density Functional Theory (DFT) is a quantum mechanical (QM) method that gives results superior to Hartree Fock (HF) in less computational time. A
Focusing Conformational Ensembles on Bioactive-Like Conformations
 Focusing Conformational Ensembles on Bioactive-Like Conformations Hannah H. Avgy, Boaz Musafia, Hanoch Senderowitz Department of Chemistry Bar-Ilan University 3rd Strasbourg Summer School on Chemoinformatics,
Focusing Conformational Ensembles on Bioactive-Like Conformations Hannah H. Avgy, Boaz Musafia, Hanoch Senderowitz Department of Chemistry Bar-Ilan University 3rd Strasbourg Summer School on Chemoinformatics,
Build_model v User Guide
 Build_model v.2.0.1 User Guide MolTech Build_model User Guide 2008-2011 Molecular Technologies Ltd. www.moltech.ru Please send your comments and suggestions to contact@moltech.ru. Table of Contents Input
Build_model v.2.0.1 User Guide MolTech Build_model User Guide 2008-2011 Molecular Technologies Ltd. www.moltech.ru Please send your comments and suggestions to contact@moltech.ru. Table of Contents Input
Molecular Modelling for Medicinal Chemistry (F13MMM) Room A36
 Room A36") Molecular Modelling for Medicinal Chemistry (F13MMM) jonathan.hirst@nottingham.ac.uk Room A36 http://comp.chem.nottingham.ac.uk Assisted reading Molecular Modelling: Principles and Applications. Andrew
Molecular Modelling for Medicinal Chemistry (F13MMM) jonathan.hirst@nottingham.ac.uk Room A36 http://comp.chem.nottingham.ac.uk Assisted reading Molecular Modelling: Principles and Applications. Andrew
New approaches to scoring function design for protein-ligand binding affinities. Richard A. Friesner Columbia University
 New approaches to scoring function design for protein-ligand binding affinities Richard A. Friesner Columbia University Overview Brief discussion of advantages of empirical scoring approaches Analysis
New approaches to scoring function design for protein-ligand binding affinities Richard A. Friesner Columbia University Overview Brief discussion of advantages of empirical scoring approaches Analysis
BUDE. A General Purpose Molecular Docking Program Using OpenCL. Richard B Sessions
 BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
ENERGY MINIMIZATION AND CONFORMATION SEARCH ANALYSIS OF TYPE-2 ANTI-DIABETES DRUGS
 Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Int. J. Chem. Sci.: 6(2), 2008, 982-992 EERGY MIIMIZATI AD CFRMATI SEARC AALYSIS F TYPE-2 ATI-DIABETES DRUGS R. PRASAA LAKSMI a, C. ARASIMA KUMAR a, B. VASATA LAKSMI, K. AGA SUDA, K. MAJA, V. JAYA LAKSMI
Type II Kinase Inhibitors Show an Unexpected Inhibition Mode against Parkinson s Disease-Linked LRRK2 Mutant G2019S.
 Type II Kinase Inhibitors Show an Unexpected Inhibition Mode against Parkinson s Disease-Linked LRRK2 Mutant G219S. Min Liu@&*, Samantha A. Bender%*, Gregory D Cuny@, Woody Sherman, Marcie Glicksman@ Soumya
Type II Kinase Inhibitors Show an Unexpected Inhibition Mode against Parkinson s Disease-Linked LRRK2 Mutant G219S. Min Liu@&*, Samantha A. Bender%*, Gregory D Cuny@, Woody Sherman, Marcie Glicksman@ Soumya
Covalent Docking. Schrödinger Software Release Schrödinger Press
 Covalent Docking Covalent Docking Schrödinger Software Release 2015-2 Schrödinger Press Covalent Docking Copyright 2015 Schrödinger, LLC. All rights reserved. While care has been taken in the preparation
Covalent Docking Covalent Docking Schrödinger Software Release 2015-2 Schrödinger Press Covalent Docking Copyright 2015 Schrödinger, LLC. All rights reserved. While care has been taken in the preparation
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,..
 3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
3 Conformational Search Molecular Docking Simulate Annealing Ab Initio QM Molecular Dynamics Graphical Visualization 3-D QSAR Pharmacophore QSAR, COMBINE, Scoring Functions, Homology Modeling,.. Rino Ragno:
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Different conformations of the drugs within the virtual library of FDA approved drugs will be generated.
 Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?!
 for structure refinement from X-ray diffraction data?!") 1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?! Visual analysis: Identification of ligand density from
1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?! Visual analysis: Identification of ligand density from
Assignment 2 Atomic-Level Molecular Modeling
 Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
The Conformation Search Problem
 Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
PDBe TUTORIAL. PDBePISA (Protein Interfaces, Surfaces and Assemblies)
") PDBe TUTORIAL PDBePISA (Protein Interfaces, Surfaces and Assemblies) http://pdbe.org/pisa/ This tutorial introduces the PDBePISA (PISA for short) service, which is a webbased interactive tool offered by
PDBe TUTORIAL PDBePISA (Protein Interfaces, Surfaces and Assemblies) http://pdbe.org/pisa/ This tutorial introduces the PDBePISA (PISA for short) service, which is a webbased interactive tool offered by
Cross Discipline Analysis made possible with Data Pipelining. J.R. Tozer SciTegic
 Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Hologram and Receptor-Guided 3D QSAR Analysis of Anilinobipyridine JNK3 Inhibitors
 3D QSAR Analysis of Anilinobipyridine JK3 Inhibitors Bull. Korean Chem. Soc. 2009, Vol. 30, o. 11 2739 Hologram and Receptor-Guided 3D QSAR Analysis of Anilinobipyridine JK3 Inhibitors Jae Yoon Chung,,
3D QSAR Analysis of Anilinobipyridine JK3 Inhibitors Bull. Korean Chem. Soc. 2009, Vol. 30, o. 11 2739 Hologram and Receptor-Guided 3D QSAR Analysis of Anilinobipyridine JK3 Inhibitors Jae Yoon Chung,,
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Molecular Modelling. Computational Chemistry Demystified. RSC Publishing. Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK
 Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Molecular Modelling Computational Chemistry Demystified Peter Bladon Interprobe Chemical Services, Lenzie, Kirkintilloch, Glasgow, UK John E. Gorton Gorton Systems, Glasgow, UK Robert B. Hammond Institute
Pose Prediction with GOLD
 Pose Prediction with GOLD Version 3.0 November 2018 GOLD v5.7.0 Table of Contents The Purpose of Docking... 2 GOLD s Evolutionary Algorithm... 3 A Checklist for Docking... 3 GOLD and Hermes... 3 Redocking
Pose Prediction with GOLD Version 3.0 November 2018 GOLD v5.7.0 Table of Contents The Purpose of Docking... 2 GOLD s Evolutionary Algorithm... 3 A Checklist for Docking... 3 GOLD and Hermes... 3 Redocking
Supplementary Material. Table of Contents. Evaluation of the test compounds using Lipinski s rule 2
 Supplementary Material Table of Contents Pages Evaluation of the test compounds using Lipinski s rule 2 Characterization of compounds PB3, PB6, PB10, PB11, PB12, PB14, PB15, PB17, PB19 2-6 Docking Methodology
Supplementary Material Table of Contents Pages Evaluation of the test compounds using Lipinski s rule 2 Characterization of compounds PB3, PB6, PB10, PB11, PB12, PB14, PB15, PB17, PB19 2-6 Docking Methodology
Preparing a PDB File
 Figure 1: Schematic view of the ligand-binding domain from the vitamin D receptor (PDB file 1IE9). The crystallographic waters are shown as small spheres and the bound ligand is shown as a CPK model. HO
Figure 1: Schematic view of the ligand-binding domain from the vitamin D receptor (PDB file 1IE9). The crystallographic waters are shown as small spheres and the bound ligand is shown as a CPK model. HO
Schrödinger Workshop 2013
 Schrödinger Workshop 2013 Structure Based Virtual Screening -Various Approaches Jas Bhachoo Schrodinger Senior Applications Scientist Your Files for Today 4 main directories Open the latest *prjzip file
Schrödinger Workshop 2013 Structure Based Virtual Screening -Various Approaches Jas Bhachoo Schrodinger Senior Applications Scientist Your Files for Today 4 main directories Open the latest *prjzip file