The reuse of structural data for fragment binding site prediction
|
|
|
- Helena Hicks
- 5 years ago
- Views:
Transcription


1 The reuse of structural data for fragment binding site prediction Richard Hall 1
2 Motivation many examples of fragments binding in a phenyl shaped pocket or a kinase slot good shape complementarity between the ligand and receptor can we ascertain the shapes of pockets filled by fragments use the wealth of structural data from the Astex database can we generate a descriptor for this kind of pocket 2

3 The Phenyl Shaped Pocket 3


4 Astex Diverse Set Fragments 1N1M: Dipeptidyl Peptidase IV 1W1P: Chitinase B 1N2V: TGT 1Q41: GSK-3beta 1LRH: Auxin binding protein 1 1SG0: Quinone Reductase 2 4
5 Other Pocket Finding Algorithms Are Available Pérot, S., Sperandio, O., Miteva, M.A., Camproux, A.-C., Villoutreix, B.O., Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery, Drug Discovery Today (2010), doi: /j.drudis lists 30 algorithms to search for binding pockets we are particularly interested in areas where fragments are likely to bind 5
6 Ligsite ligsite is a simple algorithm for finding pockets in receptor structures create an excluded volume grid at each point on the grid count number of vectors along which an atom hits the surface each point scores between 0 (very exposed) and 7 (very enclosed) 6
7 ADS Fragments 7

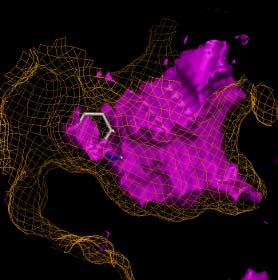
8 ADS Ligsite 8
9 Digsite: Distant Dependant Ligsite digsite evolved from the ligsite algorithm added to the number of vectors (additional 6), reducing rotational variance bin each distance (0-6 Å in 0.5A bins, plus one mop bucket) from ligand atoms to the excluded volume of the receptor generate a fingerprint spectrum smooth spectrum using a 25:50:25 function # mop r 9
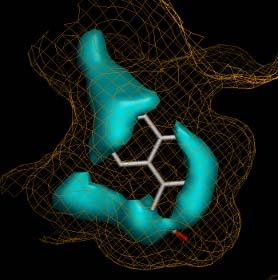
10 Digsite Training Set we selected good binding fragments for 17 Astex targets (including 6 kinase, 5 protease) HAC <= 15 MW <= 300 ClogP <= 3 NDon <= 3 NAcc <= 3 ranked by LE diverse 127 complexes average HAC = 11.7 calculated the spectrum for each atom 1492 spectra 10
11 Clustering The Spectra spectra were clustered in R (Euclidean distance, Wards method) dendrogram was cut to produce 15 clusters 11
12 Cluster Pruning we averaged the spectra of each cluster average for each target and again for kinases we wanted to find descriptors for tight binding any average cluster spectrum with >2 distances in the mop bucket was ignored rank average cluster spectra by a consensus count of distances in the low distance bins top three clusters used as prototype spectra prototype spectra # distance Spectrum 1 Spectrum 2 Spectrum 3 12
13 Digsite Grid Generation at each grid point outside the excluded volume: generate a digsite spectrum calculate the minimum average z score to each of the three prototype spectra transform scores to max(2-zscore, 0) expand/smooth resulting grids 13
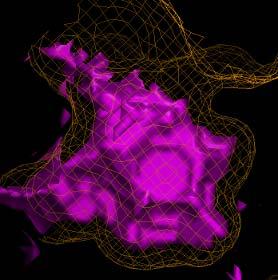
14 ADS Fragments 14
15 ADS Ligsite 15


16 ADS Digsite 16

17 A Poor Digsite Score 17


18 A Confession when we started this project, the idea was to weight scoring functions so as to bias docking towards fragment binding regions... it didn t improve results 18
19 Fragment Binding Site Prediction build a digsite or ligsite grid for the entire protein look at how many good scoring points (digsite > 1.2, ligsite >6) overlap with fragment volume(s) (VdW+1Å) do not count any point in the receptor excluded volume (<VdW + 1Å) or distant (>VdW+5Å) from the receptor how well do we perform for pdb and astex structures tn fp fn tp fp tp fn 19
20 PDB Fragments the astex diverse set are 85 diverse protein ligand targets that are relevant to drug discovery ligands are drug like but only 7/85 are fragment like there are other pdb structures that have good sequence identity with the astex diverse set CLASP database clusters where sequence similarity > 0.7 ligands extracted and stored separately can we find examples of fragment ligands bound to these similar structures not rigorously qc ed as per the original 85 20
21 PDB Fragment Selection similar fragment binding site within 5A of the centroid of the original ads ligand only organic atoms, >2 carbon atoms rule of three compliant 6 <= HAC <= 15 found 114 fragments 80 unique smiles 27/85 clusters have at least one fragment bound choose one pdb code to represent each cluster conserve original ADS fragments 21
22 PDB Fragment Analysis use a confusion matrix to quantify success for each complex true +ve false +ve true +ve false +ve false ve 1074 true ve false ve 1085 true ve ligsite digsite compare precision, (true +ve)/(true +ve and false +ve) above example ligsite 0.08, digsite 0.36 (best case, 3KGP) can be misleading when ligsite true+ve >> digsite true+ve compare false +ve ratio for the two methods 22

23 Precision Explanation 23
24 PDB Fragment Results both methods find a ligand binding site in 100% of cases digsite is more precise than ligsite average ratio 1.7, median 1.5 ligsite produces far more false positives than digsite average ratio 7.4, median
25 Astex Fragments 67 reference structures (23 targets, 724 fragments) all fragment ligands aligned to reference structure and used to mark true binding regions HAC <= 15 atoms metrics computed for ligsite and digsite 91% of digsite grids found true positives, 96% ligsite two targets were not amenable to digsite open nature of binding sites 25

26 Astex Fragment Results digsite is more precise than ligsite average ratio 1.7, median 1.2 ligsite produces far more false positives than digsite average ratio 12.1, median
27 Conclusions we used our database of crystal structures to derive a descriptor that can be used to highlight fragment binding sites a retrospective analysis of PDB and Astex data has shown that digsite can be used to find fragment binding sites digsite grids are much more focussed than ligsite grids 27
28 Future Work more extensive pdb evaluation compare digsite to other pocket finding methods eg ASP grids, pocketome method of An et al perform a prospective analysis of in house structures, to ensure we have found all potential binding sites look at island counts, rather than precision/false positives consider a coloured digsite spectra (metals, donors, acceptors, etc) 28
29 Acknowledgements Fred Ludlow Ilenia Giangreco Marcel Verdonk Astex CCI CCDC 29


30 Thank you
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification
 Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Towards Physics-based Models for ADME/Tox. Tyler Day
 Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Pose and affinity prediction by ICM in D3R GC3. Max Totrov Molsoft
 Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Detection of Protein Binding Sites II
 Detection of Protein Binding Sites II Goal: Given a protein structure, predict where a ligand might bind Thomas Funkhouser Princeton University CS597A, Fall 2007 1hld Geometric, chemical, evolutionary
Detection of Protein Binding Sites II Goal: Given a protein structure, predict where a ligand might bind Thomas Funkhouser Princeton University CS597A, Fall 2007 1hld Geometric, chemical, evolutionary
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Introduction to FBDD Fragment screening methods and library design
 Introduction to FBDD Fragment screening methods and library design Samantha Hughes, PhD Fragments 2013 RSC BMCS Workshop 3 rd March 2013 Copyright 2013 Galapagos NV Why fragment screening methods? Guess
Introduction to FBDD Fragment screening methods and library design Samantha Hughes, PhD Fragments 2013 RSC BMCS Workshop 3 rd March 2013 Copyright 2013 Galapagos NV Why fragment screening methods? Guess
György M. Keserű H2020 FRAGNET Network Hungarian Academy of Sciences
 Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Machine Learning Concepts in Chemoinformatics
 Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
Machine Learning Concepts in Chemoinformatics Martin Vogt B-IT Life Science Informatics Rheinische Friedrich-Wilhelms-Universität Bonn BigChem Winter School 2017 25. October Data Mining in Chemoinformatics
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Functional Group Fingerprints CNS Chemistry Wilmington, USA
 Functional Group Fingerprints CS Chemistry Wilmington, USA James R. Arnold Charles L. Lerman William F. Michne James R. Damewood American Chemical Society ational Meeting August, 2004 Philadelphia, PA
Functional Group Fingerprints CS Chemistry Wilmington, USA James R. Arnold Charles L. Lerman William F. Michne James R. Damewood American Chemical Society ational Meeting August, 2004 Philadelphia, PA
Plan. Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics.
 and its used in Chemoinformatics.") Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Studying the effect of noise on Laplacian-modified Bayesian Analysis and Tanimoto Similarity
 Studying the effect of noise on Laplacian-modified Bayesian nalysis and Tanimoto Similarity David Rogers, Ph.D. SciTegic, Inc. (Division of ccelrys, Inc.) drogers@scitegic.com Description of: nalysis methods
Studying the effect of noise on Laplacian-modified Bayesian nalysis and Tanimoto Similarity David Rogers, Ph.D. SciTegic, Inc. (Division of ccelrys, Inc.) drogers@scitegic.com Description of: nalysis methods
Binding Response: A Descriptor for Selecting Ligand Binding Site on Protein Surfaces
 J. Chem. Inf. Model. 2007, 47, 2303-2315 2303 Binding Response: A Descriptor for Selecting Ligand Binding Site on Protein Surfaces Shijun Zhong and Alexander D. MacKerell, Jr.* Computer-Aided Drug Design
J. Chem. Inf. Model. 2007, 47, 2303-2315 2303 Binding Response: A Descriptor for Selecting Ligand Binding Site on Protein Surfaces Shijun Zhong and Alexander D. MacKerell, Jr.* Computer-Aided Drug Design
Improving protein-ligand binding site prediction accuracy by classification of inner pocket points using local features
 Krivák and Hoksza Journal of Cheminformatics (2015) 7:12 DOI 10.1186/s13321-015-0059-5 RESEARCH ARTICLE Open Access Improving protein-ligand binding site prediction accuracy by classification of inner
Krivák and Hoksza Journal of Cheminformatics (2015) 7:12 DOI 10.1186/s13321-015-0059-5 RESEARCH ARTICLE Open Access Improving protein-ligand binding site prediction accuracy by classification of inner
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Machine-learning scoring functions for docking
 Machine-learning scoring functions for docking Dr Pedro J Ballester MRC Methodology Research Fellow EMBL-EBI, Cambridge, United Kingdom EBI is an Outstation of the European Molecular Biology Laboratory.
Machine-learning scoring functions for docking Dr Pedro J Ballester MRC Methodology Research Fellow EMBL-EBI, Cambridge, United Kingdom EBI is an Outstation of the European Molecular Biology Laboratory.
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
Bridging the Dimensions:
 Bridging the Dimensions: Seamless Integration of 3D Structure-based Design and 2D Structure-activity Relationships to Guide Medicinal Chemistry ACS Spring National Meeting. COMP, March 13 th 2016 Marcus
Bridging the Dimensions: Seamless Integration of 3D Structure-based Design and 2D Structure-activity Relationships to Guide Medicinal Chemistry ACS Spring National Meeting. COMP, March 13 th 2016 Marcus
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Identifying Interaction Hot Spots with SuperStar
 Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Comparative Analysis of Pharmacophore Screening Tools
 pubs.acs.org/jcim Comparative Analysis of Pharmacophore Screening Tools Marijn P. A. Sanders,,# Armeńio J. M. Barbosa, Barbara Zarzycka, Gerry A.F. Nicolaes, Jan P.G. Klomp, Jacob de Vlieg,, and Alberto
pubs.acs.org/jcim Comparative Analysis of Pharmacophore Screening Tools Marijn P. A. Sanders,,# Armeńio J. M. Barbosa, Barbara Zarzycka, Gerry A.F. Nicolaes, Jan P.G. Klomp, Jacob de Vlieg,, and Alberto
Ultra High Throughput Screening using THINK on the Internet
 Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
Ultra High Throughput Screening using THINK on the Internet Keith Davies Central Chemistry Laboratory, Oxford University Cathy Davies Treweren Consultants, UK Blue Sky Objectives Reduce Development Failures
What is Protein-Ligand Docking?
 MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
Structure-Activity Modeling - QSAR. Uwe Koch
 Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
proteins Comparison of structure-based and threading-based approaches to protein functional annotation Michal Brylinski, and Jeffrey Skolnick*
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS Comparison of structure-based and threading-based approaches to protein functional annotation Michal Brylinski, and Jeffrey Skolnick* Center for the Study
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Comparison of structure-based and threading-based approaches to protein functional annotation Michal Brylinski, and Jeffrey Skolnick* Center for the Study
Supplementary Material
 upplementary Material Molecular docking and ligand specificity in fragmentbased inhibitor discovery Chen & hoichet 26 27 (a) 2 1 2 3 4 5 6 7 8 9 10 11 12 15 16 13 14 17 18 19 (b) (c) igure 1 Inhibitors
upplementary Material Molecular docking and ligand specificity in fragmentbased inhibitor discovery Chen & hoichet 26 27 (a) 2 1 2 3 4 5 6 7 8 9 10 11 12 15 16 13 14 17 18 19 (b) (c) igure 1 Inhibitors
Performance Evaluation
 Performance Evaluation Confusion Matrix: Detected Positive Negative Actual Positive A: True Positive B: False Negative Negative C: False Positive D: True Negative Recall or Sensitivity or True Positive
Performance Evaluation Confusion Matrix: Detected Positive Negative Actual Positive A: True Positive B: False Negative Negative C: False Positive D: True Negative Recall or Sensitivity or True Positive
New approaches to scoring function design for protein-ligand binding affinities. Richard A. Friesner Columbia University
 New approaches to scoring function design for protein-ligand binding affinities Richard A. Friesner Columbia University Overview Brief discussion of advantages of empirical scoring approaches Analysis
New approaches to scoring function design for protein-ligand binding affinities Richard A. Friesner Columbia University Overview Brief discussion of advantages of empirical scoring approaches Analysis
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Molecular Complexity Effects and Fingerprint-Based Similarity Search Strategies
 Molecular Complexity Effects and Fingerprint-Based Similarity Search Strategies Dissertation zur Erlangung des Doktorgrades (Dr. rer. nat.) der Mathematisch-aturwissenschaftlichen Fakultät der Rheinischen
Molecular Complexity Effects and Fingerprint-Based Similarity Search Strategies Dissertation zur Erlangung des Doktorgrades (Dr. rer. nat.) der Mathematisch-aturwissenschaftlichen Fakultät der Rheinischen
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
How IJC is Adding Value to a Molecular Design Business
 How IJC is Adding Value to a Molecular Design Business James Mills Sexis LLP ChemAxon TechTalk Stevenage, ov 2012 james.mills@sexis.co.uk Overview Introduction to Sexis Sexis IJC use cases Data visualisation
How IJC is Adding Value to a Molecular Design Business James Mills Sexis LLP ChemAxon TechTalk Stevenage, ov 2012 james.mills@sexis.co.uk Overview Introduction to Sexis Sexis IJC use cases Data visualisation
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
An Integrated Approach to in-silico
 An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
Overview of IslandPick pipeline and the generation of GI datasets
 Overview of IslandPick pipeline and the generation of GI datasets Predicting GIs using comparative genomics By using whole genome alignments we can identify regions that are present in one genome but not
Overview of IslandPick pipeline and the generation of GI datasets Predicting GIs using comparative genomics By using whole genome alignments we can identify regions that are present in one genome but not
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors
 MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
MM-GBSA for Calculating Binding Affinity A rank-ordering study for the lead optimization of Fxa and COX-2 inhibitors Thomas Steinbrecher Senior Application Scientist Typical Docking Workflow Databases
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Dock Ligands from a 2D Molecule Sketch
 Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
Dock Ligands from a 2D Molecule Sketch March 31, 2016 Sample to Insight CLC bio, a QIAGEN Company Silkeborgvej 2 Prismet 8000 Aarhus C Denmark Telephone: +45 70 22 32 44 www.clcbio.com support-clcbio@qiagen.com
CSCE555 Bioinformatics. Protein Function Annotation
 CSCE555 Bioinformatics Protein Function Annotation Why we need to do function annotation? Fig from: Network-based prediction of protein function. Molecular Systems Biology 3:88. 2007 What s function? The
CSCE555 Bioinformatics Protein Function Annotation Why we need to do function annotation? Fig from: Network-based prediction of protein function. Molecular Systems Biology 3:88. 2007 What s function? The
CS4445 Data Mining and Knowledge Discovery in Databases. B Term 2014 Solutions Exam 2 - December 15, 2014
 CS4445 Data Mining and Knowledge Discovery in Databases. B Term 2014 Solutions Exam 2 - December 15, 2014 Prof. Carolina Ruiz Department of Computer Science Worcester Polytechnic Institute NAME: Prof.
CS4445 Data Mining and Knowledge Discovery in Databases. B Term 2014 Solutions Exam 2 - December 15, 2014 Prof. Carolina Ruiz Department of Computer Science Worcester Polytechnic Institute NAME: Prof.
Hit Finding and Optimization Using BLAZE & FORGE
 Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Hit Finding and Optimization Using BLAZE & FORGE Kevin Cusack,* Maria Argiriadi, Eric Breinlinger, Jeremy Edmunds, Michael Hoemann, Michael Friedman, Sami Osman, Raymond Huntley, Thomas Vargo AbbVie, Immunology
Structure based drug design and LIE models for GPCRs
 Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
A.I. in health informatics lecture 2 clinical reasoning & probabilistic inference, I. kevin small & byron wallace
 A.I. in health informatics lecture 2 clinical reasoning & probabilistic inference, I kevin small & byron wallace today a review of probability random variables, maximum likelihood, etc. crucial for clinical
A.I. in health informatics lecture 2 clinical reasoning & probabilistic inference, I kevin small & byron wallace today a review of probability random variables, maximum likelihood, etc. crucial for clinical
A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery
 AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
Cross Discipline Analysis made possible with Data Pipelining. J.R. Tozer SciTegic
 Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
Cross Discipline Analysis made possible with Data Pipelining J.R. Tozer SciTegic System Genesis Pipelining tool created to automate data processing in cheminformatics Modular system built with generic
CHAPTER 3. Pharmacophore modelling studies:
 CHAPTER 3 Pharmacophore modelling studies: 3.1 Introduction: According to Langer & Wolber (2004), the key goal of computer-aided molecular design methods in medicinal chemistry is to reduce the time and
CHAPTER 3 Pharmacophore modelling studies: 3.1 Introduction: According to Langer & Wolber (2004), the key goal of computer-aided molecular design methods in medicinal chemistry is to reduce the time and
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
Introduction to Structure Preparation and Visualization
 Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining
![Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining](/thumbs/91/107031261.jpg "Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining") Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
An Enhancement of Bayesian Inference Network for Ligand-Based Virtual Screening using Features Selection
 American Journal of Applied Sciences 8 (4): 368-373, 2011 ISSN 1546-9239 2010 Science Publications An Enhancement of Bayesian Inference Network for Ligand-Based Virtual Screening using Features Selection
American Journal of Applied Sciences 8 (4): 368-373, 2011 ISSN 1546-9239 2010 Science Publications An Enhancement of Bayesian Inference Network for Ligand-Based Virtual Screening using Features Selection
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Chemical library design
 Chemical library design Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery Today,
Chemical library design Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery Today,
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Cheminformatics platform for drug discovery application
 EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
Virtual screening for drug discovery. Markus Lill Purdue University
 Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Structure-based maximal affinity model predicts small-molecule druggability
 Structure-based maximal affinity model predicts small-molecule druggability Alan Cheng alan.cheng@amgen.com IMA Workshop (Jan 17, 2008) Druggability prediction Introduction Affinity model Some results
Structure-based maximal affinity model predicts small-molecule druggability Alan Cheng alan.cheng@amgen.com IMA Workshop (Jan 17, 2008) Druggability prediction Introduction Affinity model Some results
Overview. Descriptors. Definition. Descriptors. Overview 2D-QSAR. Number Vector Function. Physicochemical property (log P) Atom
 Atom") verview D-QSAR Definition Examples Features counts Topological indices D fingerprints and fragment counts R-group descriptors ow good are D descriptors in practice? Summary Peter Gedeck ovartis Institutes
verview D-QSAR Definition Examples Features counts Topological indices D fingerprints and fragment counts R-group descriptors ow good are D descriptors in practice? Summary Peter Gedeck ovartis Institutes
PerspectiVe. Recent Developments in Fragment-Based Drug Discovery. 1. Introduction. 2. Trends and Developments
 J. Med. Chem. XXXX, xxx, 000 A PerspectiVe Recent Developments in Fragment-Based Drug Discovery Miles Congreve,* Gianni Chessari, Dominic Tisi, and Andrew J. Woodhead Astex Therapeutics Ltd., 436 Cambridge
J. Med. Chem. XXXX, xxx, 000 A PerspectiVe Recent Developments in Fragment-Based Drug Discovery Miles Congreve,* Gianni Chessari, Dominic Tisi, and Andrew J. Woodhead Astex Therapeutics Ltd., 436 Cambridge
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Expanded Interaction Fingerprint Method for Analyzing Ligand Binding Modes in Docking and Structure-Based Drug Design
 1942 J. Chem. Inf. Comput. Sci. 2004, 44, 1942-1951 Expanded Interaction Fingerprint Method for Analyzing Ligand Binding Modes in Docking and Structure-Based Drug Design Matthew D. Kelly and Ricardo L.
1942 J. Chem. Inf. Comput. Sci. 2004, 44, 1942-1951 Expanded Interaction Fingerprint Method for Analyzing Ligand Binding Modes in Docking and Structure-Based Drug Design Matthew D. Kelly and Ricardo L.
KNIME-based scoring functions in Muse 3.0. KNIME User Group Meeting 2013 Fabian Bös
 KIME-based scoring functions in Muse 3.0 KIME User Group Meeting 2013 Fabian Bös Certara Mission: End-to-End Model-Based Drug Development Certara was formed by acquiring and integrating Tripos, Pharsight,
KIME-based scoring functions in Muse 3.0 KIME User Group Meeting 2013 Fabian Bös Certara Mission: End-to-End Model-Based Drug Development Certara was formed by acquiring and integrating Tripos, Pharsight,
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Development of a Structure Generator to Explore Target Areas on Chemical Space
 Development of a Structure Generator to Explore Target Areas on Chemical Space Kimito Funatsu Department of Chemical System Engineering, This materials will be published on Molecular Informatics Drug Development
Development of a Structure Generator to Explore Target Areas on Chemical Space Kimito Funatsu Department of Chemical System Engineering, This materials will be published on Molecular Informatics Drug Development
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
11. IN SILICO DOCKING ANALYSIS
 11. IN SILICO DOCKING ANALYSIS 11.1 Molecular docking studies of major active constituents of ethanolic extract of GP The major active constituents are identified from the ethanolic extract of Glycosmis
11. IN SILICO DOCKING ANALYSIS 11.1 Molecular docking studies of major active constituents of ethanolic extract of GP The major active constituents are identified from the ethanolic extract of Glycosmis
Rationalizing Tight Ligand Binding through Cooperative Interaction Networks
 pubs.acs.org/jcim Rationalizing Tight Ligand Binding through Cooperative Interaction Networks Bernd Kuhn,, * Julian E. Fuchs,, Michael Reutlinger,, Martin Stahl, and Neil R. Taylor, * Discovery Chemistry,
pubs.acs.org/jcim Rationalizing Tight Ligand Binding through Cooperative Interaction Networks Bernd Kuhn,, * Julian E. Fuchs,, Michael Reutlinger,, Martin Stahl, and Neil R. Taylor, * Discovery Chemistry,
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Scoring functions for of protein-ligand docking: New routes towards old goals
 3nd Strasbourg Summer School on Chemoinformatics Strasbourg, June 25-29, 2012 Scoring functions for of protein-ligand docking: New routes towards old goals Christoph Sotriffer Institute of Pharmacy and
3nd Strasbourg Summer School on Chemoinformatics Strasbourg, June 25-29, 2012 Scoring functions for of protein-ligand docking: New routes towards old goals Christoph Sotriffer Institute of Pharmacy and
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors
 A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
An optimized energy potential can predict SH2 domainpeptide
 An optimized energy potential can predict SH2 domainpeptide interactions Running Title Predicting SH2 interactions Authors Zeba Wunderlich 1, Leonid A. Mirny 2 Affiliations 1. Biophysics Program, Harvard
An optimized energy potential can predict SH2 domainpeptide interactions Running Title Predicting SH2 interactions Authors Zeba Wunderlich 1, Leonid A. Mirny 2 Affiliations 1. Biophysics Program, Harvard
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Drug Informatics for Chemical Genomics...
 Drug Informatics for Chemical Genomics... An Overview First Annual ChemGen IGERT Retreat Sept 2005 Drug Informatics for Chemical Genomics... p. Topics ChemGen Informatics The ChemMine Project Library Comparison
Drug Informatics for Chemical Genomics... An Overview First Annual ChemGen IGERT Retreat Sept 2005 Drug Informatics for Chemical Genomics... p. Topics ChemGen Informatics The ChemMine Project Library Comparison
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Benchmarking of protein descriptor sets in proteochemometric modeling (part 2): modeling performance of 13 amino acid descriptor sets
: modeling performance of 13 amino acid descriptor sets") van Westen et al. Journal of Cheminformatics 2013, 5:42 RESEARCH ARTICLE Open Access Benchmarking of protein descriptor sets in proteochemometric modeling (part 2): modeling performance of 13 amino acid
van Westen et al. Journal of Cheminformatics 2013, 5:42 RESEARCH ARTICLE Open Access Benchmarking of protein descriptor sets in proteochemometric modeling (part 2): modeling performance of 13 amino acid
PL-PatchSurfer: A Novel Molecular Local Surface-Based Method for Exploring Protein-Ligand Interactions
 Int. J. Mol. Sci. 2014, 15, 15122-15145; doi:10.3390/ijms150915122 Article OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms PL-PatchSurfer: A Novel Molecular
Int. J. Mol. Sci. 2014, 15, 15122-15145; doi:10.3390/ijms150915122 Article OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms PL-PatchSurfer: A Novel Molecular
bcl::cheminfo Suite Enables Machine Learning-Based Drug Discovery Using GPUs Edward W. Lowe, Jr. Nils Woetzel May 17, 2012
 bcl::cheminfo Suite Enables Machine Learning-Based Drug Discovery Using GPUs Edward W. Lowe, Jr. Nils Woetzel May 17, 2012 Outline Machine Learning Cheminformatics Framework QSPR logp QSAR mglur 5 CYP
bcl::cheminfo Suite Enables Machine Learning-Based Drug Discovery Using GPUs Edward W. Lowe, Jr. Nils Woetzel May 17, 2012 Outline Machine Learning Cheminformatics Framework QSPR logp QSAR mglur 5 CYP
Mechanistic insight into inhibition of two-component system signaling
 Supporting Information Mechanistic insight into inhibition of two-component system signaling Samson Francis, a Kaelyn E. Wilke, a Douglas E. Brown a and Erin E. Carlson a,b* a Department of Chemistry,
Supporting Information Mechanistic insight into inhibition of two-component system signaling Samson Francis, a Kaelyn E. Wilke, a Douglas E. Brown a and Erin E. Carlson a,b* a Department of Chemistry,
Computational Methods and Drug-Likeness. Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004
 Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score. Protein Structures: The Starting Point for New Drugs 2
 The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score Protein Structures: The Starting Point for New Drugs 2 Matthias Rarey Stefan Bietz Nadine Schneider Sascha Urbaczek University
The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score Protein Structures: The Starting Point for New Drugs 2 Matthias Rarey Stefan Bietz Nadine Schneider Sascha Urbaczek University
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today
 est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
est Drive K20 GPUs! Experience The Acceleration Run Computational Chemistry Codes on Tesla K20 GPU today Sign up for FREE GPU Test Drive on remotely hosted clusters www.nvidia.com/gputestd rive Shape Searching
How Diverse Are Diversity Assessment Methods? A Comparative Analysis and Benchmarking of Molecular Descriptor Space
 pubs.acs.org/jcim How Diverse Are Diversity Assessment Methods? A Comparative Analysis and Benchmarking of Molecular Descriptor Space Alexios Koutsoukas,, Shardul Paricharak,,, Warren R. J. D. Galloway,
pubs.acs.org/jcim How Diverse Are Diversity Assessment Methods? A Comparative Analysis and Benchmarking of Molecular Descriptor Space Alexios Koutsoukas,, Shardul Paricharak,,, Warren R. J. D. Galloway,
Model Accuracy Measures
 Model Accuracy Measures Master in Bioinformatics UPF 2017-2018 Eduardo Eyras Computational Genomics Pompeu Fabra University - ICREA Barcelona, Spain Variables What we can measure (attributes) Hypotheses
Model Accuracy Measures Master in Bioinformatics UPF 2017-2018 Eduardo Eyras Computational Genomics Pompeu Fabra University - ICREA Barcelona, Spain Variables What we can measure (attributes) Hypotheses