Efficient overlay of molecular 3-D pharmacophores
|
|
|
- Leonard King
- 5 years ago
- Views:
Transcription
1 Efficient overlay of molecular 3D pharmacophores Gerhard Wolber*, Alois A. Dornhofer & Thierry Langer * wolber@inteligand.com Superposition of molecules 1





2 Alignment: Outline Scope, design goals & existing methods o Scope = virtual screening & pharmacophore model building Our algorithm, validation & examples o Matching and analytic o Validation Current applications & outlook o Shared and merged pharmacophores Virtual Screening Methods Descriptor/Fingerprint Filtering 3D Fitting 1DFilter 2DFilter 3DFilter Real 3DFitting 1D Filtering o Property Ranges o Fingerprints 2D Filtering o Topology, Molecular Graph o (Red. Graphs, FTrees, ) 3D Filtering o 3point pharmacophores o Distance hashing 3D Fitting o Flexible o From precomputed conformers e.g. MW Lipinsky Computationally expensive 2
3 Design Goals 1. Fast o Scalable and usable also for large small molecules or pharmacophores o Interactively usable o Applicable for virtual screening 2. Rigid Method o Save time by precomputing conformers 3. Correct geometry & correct chemistry o Produce real geometric s, not only hashing o Represent molecule in a pharmacophoric way 4. Pharmacophore applicability o Be able to align molecules to 3D pharmacophores and vice versa o Align different molecules/pharmacophores to each other Existing methods, different scopes Distancebased combinatorial approach (bruteforce) o Computationally very expensive Distancebased cliquedetection: o Feasible for small molecules, but still growing exponentially with the number of features (npcomplete) Fingerprinting: o No real 3D, only a prefilter step Reduced Graphs: o Bound to the chemical graph, problems with aggregation/fragmentation Flexible Pharmacophore Elucidation: o Expensive for virtual screening [Lemmen C, Lengauer T. 2000] Computational methods for the structural of molecules. J Comput Aided Mol Des Mar;14(3):21532 (27 methods!) 3












4 Alignment by pharmacophore points Methotrexate Dihydrofolate Wrong Correct Böhm, Klebe, Kubinyi: Wirkstoffdesign (1999) p. 320f Alignment by pharmacophore points 1RX2 1RB3 4









5 Pharmacophore Representation LigandScout [LigandScout 2005] Wolber, G.; Langer, T. 3D Pharmacophores Derived from ProteinBound Ligands and Their Use as Virtual Screening Filters J. Chem. Inf. Comput. Sci.; (Article); 2005; 45(1); DOI: /ci049885e 3D Pharmacophore: Chemical Features Hydrogen Bond Donors/Acceptors Vectors: Direction and Distance constraint Negative/Positive Ionizable Spheres Location Spheres: Distance constraint only 5






6 3D Pharmacophore: Chemical Features Hydrophobic Interactions o Location spheres by aggregation π Interactions o Center & normal Chemical features always refer to the ligand side! Chemical feature universality layers Layer 4 Layer 3 Layer 2 Layer 1 Chemical Function Subgraph Without geometry constraint Including geometry constraint Without geometry constraint Including geometry constraint Lipophilic area, positive ionizable area Hydrogen bond Donor/Acceptor Hydroxylic group, Phenol Group Phenol group facing a parallel benzene Selectivity Universality Layer 3 and Layer 4 features provide good abstraction, and still sufficient characterization 6



.")

7 Existing methods: Kabsch Geometric Alignment KABSCH: Given two pairwise defined sets of points of equal size, there is an optimal rotation to minimize RMSD, related to distances. o uses matrix algebra to find the optimal rotation of two sets of points in N dimensional space to minimize the RMSD between them o Eigenvector decomposition to derive the orthogonal matrix that describes the best rotation [Kabsch, 1976] Kabsch, W. (1976). A solution for the best rotation to relate two sets of vectors. Acta. Crystal, 32A: [Kabsch, 1978] Kabsch, W. (1978). A discussion of the solution for the best rotation to related two sets of vectors. Acta. Crystal, 34A: Kabsch Alignment For sequencealigned proteins r r r r r r r r r One single centered rotation Computationally efficient 7




8 Kabsch Superposition For molecules: atoms instead of sequence o Canonicalize atom order to create pairs Sildenafil 1TBF 1UDT Atom by Atom 8
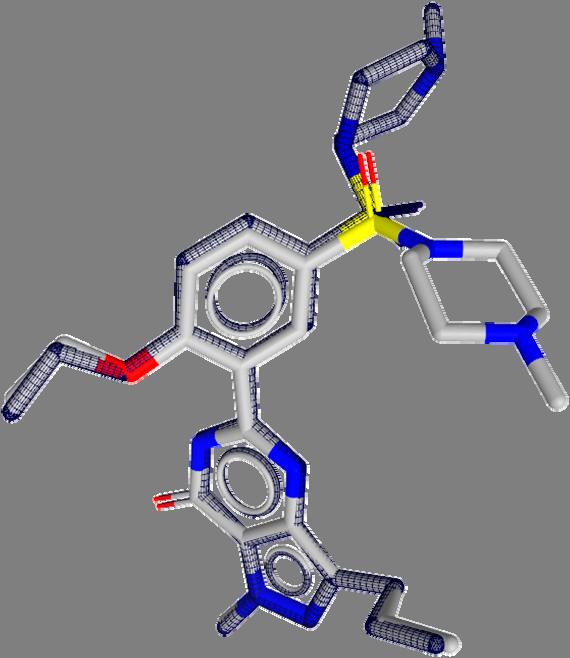

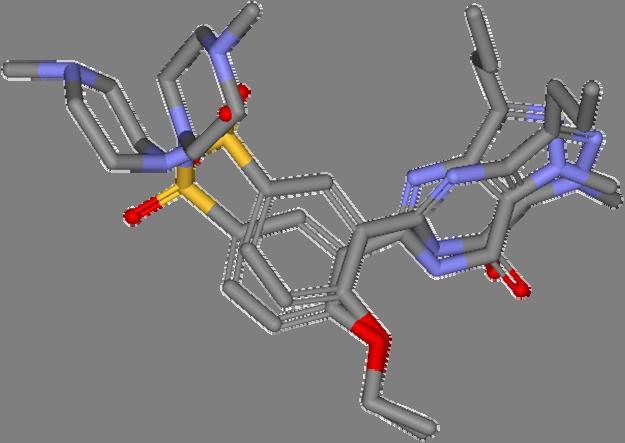





9 Pharmacophore Points Pairwise Superposition Ideal rotation using all atom pairs Ideal rotation using pharmacophore points 9
10 What Is Missing? Molecule pharmacophore Best Pairing 3D Rotation (Kabsch) Superposition Best Pairing: o Algorithm to find a maximum # of pairs with minimum matching cost o Similarity measure that describes matching cost between pharmacophore points How to get optimal pairs? Hungarian Matcher (Marrying Problem) [Edmods 1965] Matching and a Polyhedron with 01 Vertices. J. Res. NBS 69B (1965), [nonbipartite application] [Kuhn 1995] The Hungarian method for the Assignment Problem. Noval Research Quarterly, 2 (1995) [bipartite variant] [Richmond 2004] Application to chemistry: N. Richmond et al. Alignment of 3D molecules using an image recognition algorithm. J. Mol. Graph. Model. 23, 2004, pp

11 Hungarian Matching How to define pharmacophore feature matching cost (similarity)? o Few feature types o Selectivity by geometric relations => Encode geometry in each feature! Acceptor Donor Lipophilic
12 Similarity measure Similarity = Create a "typed shared shell" list for each type Donor Donor o o o Take the minimum from each typed shell. The higher the sum of the counts in each shell, the higher the potential correspondence for the respective point type. Subtract value from max. cost (normalize to minimize cost) Cost function weight( shell) ( min( nx( shell, type), ny ( shell, type ) cost ( x, y) = )) shell type n x (shell,type). # elements in shell of type for element x n y (shell,type). # elements in shell of type for element y weight( i i ) = 1 n i *2 1 n 3 shell shell shell * m. maximum element count for all shells and all types n. maximum shell index i shell shell index m 12
 Superposition Is pairing valid?")
 as reference points o Alignments of small molecules compared to alinged")
13 The Symmetry Problem o Reduced information may cause false ambiguity for some cases Molecule pharmacophore Best Pairing 3D Rotation (Kabsch) Superposition Is pairing valid? If not, remove invalid pairs and retry Validation o Bound ligands from the PDB in the same target o C α (7A) as reference points o Alignments of small molecules compared to alinged binding sites 13

 o Binding site similarity (>= 40 similar")
14 Validation Alignment within protein compared to Pure Ligandbased Success measure: RMSD between proteinbound and predicted position Test Set 4 Targets o COX2 o ABLTyrosin Kinase o PDE5 o CDK2 Prerequisites o PDB quality (resolution, reasonable) o Binding site similarity (>= 40 similar C α; RMS <1) 14


 4COX")
")



15 COX2 Pharmacophore Protein RMSD= 1CX2 4COX 6COX 1CX COX 2.8 6COX 2.7 1CX2, 6COX selective inhibitors (SC558) 4COX nonselective inhibitor (indomethacine) COX2 Pharmacophore Protein RMSD=2.4 1CX2 4COX 6COX 1CX COX 2.8 6COX 2.7 1CX2, 6COX selective inhibitors (SC558) 4COX nonselective inhibitor (indomethacine) 15






16 Abl Tyrosin Kinase/Gleevec 1FPU 1IEP 1FPU Gleevec and a variant 1IEP 0.6 Pharmacophore Protein RMSD= Phosphodiesterase 5 1UDT 1TBF 1XP0 1UHO 1UDU 1XOZ 1UDT 1TBF Sildenafil 1XP0 1UHO Tadalafil 1UDU 1XOZ Vardenafil Pharmacophore Protein RMSD= 16





17 Phosphodiesterase 5 1UDT 1TBF 1XP0 1UHO 1UDU 1XOZ 1UDT 1TBF Sildenafil 1XP0 1UHO Tadalafil 1UDU 1XOZ Vardenafil Pharmacophore Protein RMSD=4.5 Phosphodiesterase 5 Protein 17








18 Phosphodiesterase 5 1UDT 1TBF 1XP0 1UHO 1UDU 1XOZ 1UDT 1TBF Sildenafil 1XP0 1UHO Tadalafil 1UDU 1XOZ Vardenafil Protein RMSD= Pharmacophore CDK2 1KE5 1KE6 1KE7 1KE8 1KE9 1KE KE6 1KE KE8 1KE9 Protein Pharmacophore RMSD=0.2 18









19 CDK2 (all 5) Pharmacophore Protein Validation Summary o The worked perfectly in nearly all of the cases o Very Fast (up to max. 50 ms per ) o Scales polynomially with the number of features o Provides pharmacophoric view on molecule (scaffoldhopping) 19







20 Shared Feature Pharmacophore Merged Feature Pharmacophore 20

21 Summary Universal, effective and fast (2050ms) pharmacophore method Can be used for: o Comparison of molecules by their pharmacophore features o Model & compare pharmacophores (share/merge) o Fast and accurate 3D pharmacophore screening Thanks to Fabian Bendix Robert Kosara Martin Biely Eva Maria Krovat Theodora Steindl Thierry Langer Johannes Kirchmair Christian Laggner Daniela Schuster Markus Böhler 21
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
Pharmacophore Approaches In Drug Discovery
 Pharmacophore Approaches In Drug Discovery Prof. Thierry Langer Prestwick Chemical, Inc. Boulevard Gonthier d Andernach 67400 Strasbourg-Illkirch, France Contents Introduction - Non HTS Hit Recognition
Pharmacophore Approaches In Drug Discovery Prof. Thierry Langer Prestwick Chemical, Inc. Boulevard Gonthier d Andernach 67400 Strasbourg-Illkirch, France Contents Introduction - Non HTS Hit Recognition
BIOINF Drug Design 2. Jens Krüger and Philipp Thiel Summer Lecture 5: 3D Structure Comparison Part 1: Rigid Superposition, Pharmacophores
 BIOINF 472 Drug Design 2 Jens Krüger and Philipp Thiel Summer 2014 Lecture 5: D Structure Comparison Part 1: Rigid Superposition, Pharmacophores Overview Comparison of D structures Rigid superposition
BIOINF 472 Drug Design 2 Jens Krüger and Philipp Thiel Summer 2014 Lecture 5: D Structure Comparison Part 1: Rigid Superposition, Pharmacophores Overview Comparison of D structures Rigid superposition
The Conformation Search Problem
 Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Pharmacophores and Pharmacophore Searches
 Pharmacophores and Pharmacophore Searches Edited by Thierry Longer and Remy D. Hoffmann WILEY- VCH WILEY-VCH Verlag GmbH &. Co. KGaA Contents Preface XIII A Personal Foreword List of Contributors XV XVII
Pharmacophores and Pharmacophore Searches Edited by Thierry Longer and Remy D. Hoffmann WILEY- VCH WILEY-VCH Verlag GmbH &. Co. KGaA Contents Preface XIII A Personal Foreword List of Contributors XV XVII
The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score. Protein Structures: The Starting Point for New Drugs 2
 The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score Protein Structures: The Starting Point for New Drugs 2 Matthias Rarey Stefan Bietz Nadine Schneider Sascha Urbaczek University
The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score Protein Structures: The Starting Point for New Drugs 2 Matthias Rarey Stefan Bietz Nadine Schneider Sascha Urbaczek University
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
fconv Tutorial Part 2
 fconv Tutorial Part 2 Gerd Neudert Introduction to some basic fconv features based on version 1.08 Introduction Definitions Welcome to the second part of fconv tutorials! If you have not already done part
fconv Tutorial Part 2 Gerd Neudert Introduction to some basic fconv features based on version 1.08 Introduction Definitions Welcome to the second part of fconv tutorials! If you have not already done part
Author Index Volume
 Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Part 6. 3D Pharmacophore Modeling
 279 Part 6 3D Pharmacophore Modeling 281 20 3D Pharmacophore Modeling Techniques in Computer Aided Molecular Design Using LigandScout Thomas Seidel, Sharon D. Bryant, Gökhan Ibis, Giulio Poli, and Thierry
279 Part 6 3D Pharmacophore Modeling 281 20 3D Pharmacophore Modeling Techniques in Computer Aided Molecular Design Using LigandScout Thomas Seidel, Sharon D. Bryant, Gökhan Ibis, Giulio Poli, and Thierry
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Protein-Ligand Docking
 Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Journal of Pharmacology and Experimental Therapy-JPET#172536
 A NEW NON-PEPTIDIC INHIBITOR OF THE 14-3-3 DOCKING SITE INDUCES APOPTOTIC CELL DEATH IN CHRONIC MYELOID LEUKEMIA SENSITIVE OR RESISTANT TO IMATINIB Manuela Mancini, Valentina Corradi, Sara Petta, Enza
A NEW NON-PEPTIDIC INHIBITOR OF THE 14-3-3 DOCKING SITE INDUCES APOPTOTIC CELL DEATH IN CHRONIC MYELOID LEUKEMIA SENSITIVE OR RESISTANT TO IMATINIB Manuela Mancini, Valentina Corradi, Sara Petta, Enza
Training a Scoring Function for the Alignment of Small Molecules
 1724 J. Chem. Inf. Model. 2010, 50, 1724 1735 Training a Scoring Function for the Alignment of Small Molecules Shek Ling Chan* and Paul Labute Chemical Computing Group, Inc., 1010 Sherbrooke Street West,
1724 J. Chem. Inf. Model. 2010, 50, 1724 1735 Training a Scoring Function for the Alignment of Small Molecules Shek Ling Chan* and Paul Labute Chemical Computing Group, Inc., 1010 Sherbrooke Street West,
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Ligand-receptor interactions
 University of Silesia, Katowice, Poland 11 22 March 2013 Ligand-receptor interactions Dr. Pavel Polishchuk A.V. Bogatsky Physico-Chemical Institute of National Academy of Sciences of Ukraine Odessa, Ukraine
University of Silesia, Katowice, Poland 11 22 March 2013 Ligand-receptor interactions Dr. Pavel Polishchuk A.V. Bogatsky Physico-Chemical Institute of National Academy of Sciences of Ukraine Odessa, Ukraine
BioSolveIT. A Combinatorial Approach for Handling of Protonation and Tautomer Ambiguities in Docking Experiments
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Fragment based drug discovery in teams of medicinal and computational chemists. Carsten Detering
 Fragment based drug discovery in teams of medicinal and computational chemists Carsten Detering BioSolveIT Quick Facts Founded in 2001 by the developers of FlexX ~20 people Core expertise: docking, screening,
Fragment based drug discovery in teams of medicinal and computational chemists Carsten Detering BioSolveIT Quick Facts Founded in 2001 by the developers of FlexX ~20 people Core expertise: docking, screening,
New approaches to scoring function design for protein-ligand binding affinities. Richard A. Friesner Columbia University
 New approaches to scoring function design for protein-ligand binding affinities Richard A. Friesner Columbia University Overview Brief discussion of advantages of empirical scoring approaches Analysis
New approaches to scoring function design for protein-ligand binding affinities Richard A. Friesner Columbia University Overview Brief discussion of advantages of empirical scoring approaches Analysis
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
Protein-Ligand Docking Evaluations
 Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Virtual screening for drug discovery. Markus Lill Purdue University
 Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich KNIME UGM, Berlin, February 2015 The Schrödinger Extensions
The Schrödinger KNIME extensions
 The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
The Schrödinger KNIME extensions Computational Chemistry and Cheminformatics in a workflow environment Jean-Christophe Mozziconacci Volker Eyrich Topics What are the Schrödinger extensions? Workflow application
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules
 Software News and Updates tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules DANIEL SEELIGER, BERT L. DE GROOT Computational Biomolecular Dynamics Group, Max-Planck-Institute
Software News and Updates tconcoord-gui: Visually Supported Conformational Sampling of Bioactive Molecules DANIEL SEELIGER, BERT L. DE GROOT Computational Biomolecular Dynamics Group, Max-Planck-Institute
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Structure-Activity Modeling - QSAR. Uwe Koch
 Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Spatial chemical distance based on atomic property fields
 J Comput Aided Mol Des (2010) 24:173 182 DOI 10.1007/s10822-009-9316-x Spatial chemical distance based on atomic property fields A. V. Grigoryan I. Kufareva M. Totrov R. A. Abagyan Received: 20 September
J Comput Aided Mol Des (2010) 24:173 182 DOI 10.1007/s10822-009-9316-x Spatial chemical distance based on atomic property fields A. V. Grigoryan I. Kufareva M. Totrov R. A. Abagyan Received: 20 September
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
Targeting protein-protein interactions: A hot topic in drug discovery
 Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Plan. Day 2: Exercise on MHC molecules.
 Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Patrick: An Introduction to Medicinal Chemistry 5e Chapter 01
 Questions Patrick: An Introduction to Medicinal Chemistry 5e 01) Which of the following molecules is a phospholipid? a. i b. ii c. iii d. iv 02) Which of the following statements is false regarding the
Questions Patrick: An Introduction to Medicinal Chemistry 5e 01) Which of the following molecules is a phospholipid? a. i b. ii c. iii d. iv 02) Which of the following statements is false regarding the
Comparative Analysis of Pharmacophore Screening Tools
 pubs.acs.org/jcim Comparative Analysis of Pharmacophore Screening Tools Marijn P. A. Sanders,,# Armeńio J. M. Barbosa, Barbara Zarzycka, Gerry A.F. Nicolaes, Jan P.G. Klomp, Jacob de Vlieg,, and Alberto
pubs.acs.org/jcim Comparative Analysis of Pharmacophore Screening Tools Marijn P. A. Sanders,,# Armeńio J. M. Barbosa, Barbara Zarzycka, Gerry A.F. Nicolaes, Jan P.G. Klomp, Jacob de Vlieg,, and Alberto
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification
 Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Virtual Screening - The Road to Success
 Virtual Screening - The Road to Success ugo Kubinyi Germany E-Mail kubinyi@t-online.de omepage www.kubinyi.de XIXth ISMC, Istanbul, Turkey, August 29-September 02, 2006 Strategies in Design no protein
Virtual Screening - The Road to Success ugo Kubinyi Germany E-Mail kubinyi@t-online.de omepage www.kubinyi.de XIXth ISMC, Istanbul, Turkey, August 29-September 02, 2006 Strategies in Design no protein
Toward an Understanding of GPCR-ligand Interactions. Alexander Heifetz
 Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Protein Structure Prediction and Protein-Ligand Docking
 Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
11. IN SILICO DOCKING ANALYSIS
 11. IN SILICO DOCKING ANALYSIS 11.1 Molecular docking studies of major active constituents of ethanolic extract of GP The major active constituents are identified from the ethanolic extract of Glycosmis
11. IN SILICO DOCKING ANALYSIS 11.1 Molecular docking studies of major active constituents of ethanolic extract of GP The major active constituents are identified from the ethanolic extract of Glycosmis
Self-Organizing Superimposition Algorithm for Conformational Sampling
 Self-Organizing Superimposition Algorithm for Conformational Sampling FANGQIANG ZHU, DIMITRIS K. AGRAFIOTIS Johnson & Johnson Pharmaceutical Research & Development, L.L.C., 665 Stockton Drive, Exton, Pennsylvania
Self-Organizing Superimposition Algorithm for Conformational Sampling FANGQIANG ZHU, DIMITRIS K. AGRAFIOTIS Johnson & Johnson Pharmaceutical Research & Development, L.L.C., 665 Stockton Drive, Exton, Pennsylvania
Supporting Information
 Supporting Information Decoding Allosteric Networks in Biocatalysts: Rational Approach to Therapies and Biotechnologies Johannes T. Cramer 1,2, Jana I. Führing 1, Petra Baruch 2, Christian Brütting 3,
Supporting Information Decoding Allosteric Networks in Biocatalysts: Rational Approach to Therapies and Biotechnologies Johannes T. Cramer 1,2, Jana I. Führing 1, Petra Baruch 2, Christian Brütting 3,
György M. Keserű H2020 FRAGNET Network Hungarian Academy of Sciences
 Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Chemical library design
 Chemical library design Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery Today,
Chemical library design Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery Today,
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
What is Protein-Ligand Docking?
 MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Atomic and molecular interaction forces in biology
 Atomic and molecular interaction forces in biology 1 Outline Types of interactions relevant to biology Van der Waals interactions H-bond interactions Some properties of water Hydrophobic effect 2 Types
Atomic and molecular interaction forces in biology 1 Outline Types of interactions relevant to biology Van der Waals interactions H-bond interactions Some properties of water Hydrophobic effect 2 Types
Determining Flexibility of Molecules Using Resultants of Polynomial Systems
 Determining Flexibility of Molecules Using Resultants of Polynomial Systems Robert H. Lewis 1 and Evangelos A. Coutsias 2 1 Fordham University, New York, NY 10458, USA 2 University of New Mexico, Albuquerque,
Determining Flexibility of Molecules Using Resultants of Polynomial Systems Robert H. Lewis 1 and Evangelos A. Coutsias 2 1 Fordham University, New York, NY 10458, USA 2 University of New Mexico, Albuquerque,
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
CHAPTER 3. Pharmacophore modelling studies:
 CHAPTER 3 Pharmacophore modelling studies: 3.1 Introduction: According to Langer & Wolber (2004), the key goal of computer-aided molecular design methods in medicinal chemistry is to reduce the time and
CHAPTER 3 Pharmacophore modelling studies: 3.1 Introduction: According to Langer & Wolber (2004), the key goal of computer-aided molecular design methods in medicinal chemistry is to reduce the time and
Pose and affinity prediction by ICM in D3R GC3. Max Totrov Molsoft
 Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Pose and affinity prediction by ICM in D3R GC3 Max Totrov Molsoft Pose prediction method: ICM-dock ICM-dock: - pre-sampling of ligand conformers - multiple trajectory Monte-Carlo with gradient minimization
Extended examples and description of various jcompoundmapper fingerprints in string format
 Extended examples and description of various jcompoundmapper fingerprints in string format Figure 1: The geometry and topology of Oxaceprol. Pharmacophore types shown in the 3D structure are 1=[L], 3=[A
Extended examples and description of various jcompoundmapper fingerprints in string format Figure 1: The geometry and topology of Oxaceprol. Pharmacophore types shown in the 3D structure are 1=[L], 3=[A
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Different conformations of the drugs within the virtual library of FDA approved drugs will be generated.
 Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Chapter 3 Molecular Modeling 3.1. Introduction In this study pharmacophore models will be created to screen a virtual library of FDA approved drugs for compounds that may inhibit MA-A and MA-B. The virtual
Supporting Information
 Supporting Information Discovery of ovel CB Receptor Ligands by a Pharmacophore-based Virtual Screening Workflow Patrick Markt, a emens Feldmann, a Judith Maria Rollinger, b Stefan Raduner, c Daniela Schuster,
Supporting Information Discovery of ovel CB Receptor Ligands by a Pharmacophore-based Virtual Screening Workflow Patrick Markt, a emens Feldmann, a Judith Maria Rollinger, b Stefan Raduner, c Daniela Schuster,
Scoring functions for of protein-ligand docking: New routes towards old goals
 3nd Strasbourg Summer School on Chemoinformatics Strasbourg, June 25-29, 2012 Scoring functions for of protein-ligand docking: New routes towards old goals Christoph Sotriffer Institute of Pharmacy and
3nd Strasbourg Summer School on Chemoinformatics Strasbourg, June 25-29, 2012 Scoring functions for of protein-ligand docking: New routes towards old goals Christoph Sotriffer Institute of Pharmacy and
Electronic Supplementary Information Effective lead optimization targeted for displacing bridging water molecule
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Electronic Supplementary Information Effective lead optimization targeted for displacing
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Electronic Supplementary Information Effective lead optimization targeted for displacing
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites. J. Andrew Surface
 Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Analyzing Molecular Conformations Using the Cambridge Structural Database. Jason Cole Cambridge Crystallographic Data Centre
 Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Bioinformatics Workshop - NM-AIST
 Bioinformatics Workshop - NM-AIST Day 3 Introduction to Drug/Small Molecule Discovery Thomas Girke July 25, 2012 Bioinformatics Workshop - NM-AIST Slide 1/44 Introduction CMP Structure Formats Similarity
Bioinformatics Workshop - NM-AIST Day 3 Introduction to Drug/Small Molecule Discovery Thomas Girke July 25, 2012 Bioinformatics Workshop - NM-AIST Slide 1/44 Introduction CMP Structure Formats Similarity
BioSolveIT. A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
Methods for tautomer enumeration, -searching and -duplicate filtering
 Methods for tautomer enumeration, -searching and -duplicate filtering József Szegezdi, Zsolt Mohácsi, Tamás Csizmazia, Szilárd Dóránt, Ákos Papp, György Pirok, Szabolcs Csepregi, Ferenc Csizmadia Solutions
Methods for tautomer enumeration, -searching and -duplicate filtering József Szegezdi, Zsolt Mohácsi, Tamás Csizmazia, Szilárd Dóránt, Ákos Papp, György Pirok, Szabolcs Csepregi, Ferenc Csizmadia Solutions
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses
 Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
Analyzing Small Molecule Data in R
 Analyzing Small Molecule Data in R Tyler Backman and Thomas Girke December 12, 2011 Analyzing Small Molecule Data in R Slide 1/49 Introduction CMP Structure Formats Similarity Searching Background Fragment
Analyzing Small Molecule Data in R Tyler Backman and Thomas Girke December 12, 2011 Analyzing Small Molecule Data in R Slide 1/49 Introduction CMP Structure Formats Similarity Searching Background Fragment
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Protein-Ligand Docking Methods
 Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Nonlinear QSAR and 3D QSAR
 onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
Design and Synthesis of the Comprehensive Fragment Library
 YOUR INNOVATIVE CHEMISTRY PARTNER IN DRUG DISCOVERY Design and Synthesis of the Comprehensive Fragment Library A 3D Enabled Library for Medicinal Chemistry Discovery Warren S Wade 1, Kuei-Lin Chang 1,
YOUR INNOVATIVE CHEMISTRY PARTNER IN DRUG DISCOVERY Design and Synthesis of the Comprehensive Fragment Library A 3D Enabled Library for Medicinal Chemistry Discovery Warren S Wade 1, Kuei-Lin Chang 1,
Machine learning for ligand-based virtual screening and chemogenomics!
 Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
Machine learning for ligand-based virtual screening and chemogenomics! Jean-Philippe Vert Institut Curie - INSERM U900 - Mines ParisTech In silico discovery of molecular probes and drug-like compounds:
Supporting Information
 Discovery of kinase inhibitors by high-throughput docking and scoring based on a transferable linear interaction energy model Supporting Information Peter Kolb, Danzhi Huang, Fabian Dey and Amedeo Caflisch
Discovery of kinase inhibitors by high-throughput docking and scoring based on a transferable linear interaction energy model Supporting Information Peter Kolb, Danzhi Huang, Fabian Dey and Amedeo Caflisch
KNIME-based scoring functions in Muse 3.0. KNIME User Group Meeting 2013 Fabian Bös
 KIME-based scoring functions in Muse 3.0 KIME User Group Meeting 2013 Fabian Bös Certara Mission: End-to-End Model-Based Drug Development Certara was formed by acquiring and integrating Tripos, Pharsight,
KIME-based scoring functions in Muse 3.0 KIME User Group Meeting 2013 Fabian Bös Certara Mission: End-to-End Model-Based Drug Development Certara was formed by acquiring and integrating Tripos, Pharsight,
Kernel-based Machine Learning for Virtual Screening
 Kernel-based Machine Learning for Virtual Screening Dipl.-Inf. Matthias Rupp Beilstein Endowed Chair for Chemoinformatics Johann Wolfgang Goethe-University Frankfurt am Main, Germany 2008-04-11, Helmholtz
Kernel-based Machine Learning for Virtual Screening Dipl.-Inf. Matthias Rupp Beilstein Endowed Chair for Chemoinformatics Johann Wolfgang Goethe-University Frankfurt am Main, Germany 2008-04-11, Helmholtz
Computing RMSD and fitting protein structures: how I do it and how others do it
 Computing RMSD and fitting protein structures: how I do it and how others do it Bertalan Kovács, Pázmány Péter Catholic University 03/08/2016 0. Introduction All the following algorithms have been implemented
Computing RMSD and fitting protein structures: how I do it and how others do it Bertalan Kovács, Pázmány Péter Catholic University 03/08/2016 0. Introduction All the following algorithms have been implemented
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Electronic Supplementary Information. A biologically relevant fluorescent probe to assess the binding of ceramide to the CERT transfer protein
 This journal is The Royal Society of Chemistry 20 Electronic Supplementary Information A biologically relevant fluorescent probe to assess the binding of ceramide to the CERT transfer protein Stéphanie
This journal is The Royal Society of Chemistry 20 Electronic Supplementary Information A biologically relevant fluorescent probe to assess the binding of ceramide to the CERT transfer protein Stéphanie
Ranking of HIV-protease inhibitors using AutoDock
 Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Common Pharmacophore Identification Using Frequent Clique Detection Algorithm
 Common Pharmacophore Identification Using Frequent Clique Detection Algorithm Yevgeniy Podolyan and George Karypis University of Minnesota, Department of Computer Science and Computer Engineering Minneapolis,
Common Pharmacophore Identification Using Frequent Clique Detection Algorithm Yevgeniy Podolyan and George Karypis University of Minnesota, Department of Computer Science and Computer Engineering Minneapolis,
Pharmacophore-Based Molecular Docking to Account for Ligand Flexibility
 PROTEINS: Structure, Function, and Genetics 51:172 188 (2003) Pharmacophore-Based Molecular Docking to Account for Ligand Flexibility Diane Joseph-McCarthy,* Bert E. Thomas IV, Michael Belmarsh, Demetri
PROTEINS: Structure, Function, and Genetics 51:172 188 (2003) Pharmacophore-Based Molecular Docking to Account for Ligand Flexibility Diane Joseph-McCarthy,* Bert E. Thomas IV, Michael Belmarsh, Demetri