Virtual Screening - The Road to Success
|
|
|
- Shanon Osborne
- 5 years ago
- Views:
Transcription


1 Virtual Screening - The Road to Success ugo Kubinyi Germany kubinyi@t-online.de omepage XIXth ISMC, Istanbul, Turkey, August 29-September 02, 2006
2 Strategies in Design no protein 3D structure, no ligands combichem, TS, virtual screening no protein 3D structure, ligands protein 3D structure, no ligands de novo design (protein flexibility!) protein 3D structure, ligands pharmacophores, (3D) similarity, (3D) QSAR structure-based design LBLD SBLD

3 Drug Research is... the Search for a eedle in a aystack
4 Virtual Screening Reduces the Size of the aystack by Selecting: Compounds or libraries that are either lead-like, or drug-like, or have the potential of oral bioavailability, or are similar to a lead, or fit the binding site of a certain protein by rules (e.g. Lipinski bioavailability rules), neural nets (e.g. drug-like character), similarity analyses, pharmacophore analyses, scaffold hopping, or docking and scoring

5 Chris Rescues CombiChem and Screening Collections
6 Drug-like Character % Compounds ACD 100 Top-Selling Drugs, 1997 Training sets WDI Test set predictions Drug Score
7 Filters for Virtual Screening remaining Garbage filter Druglike / on-druglike Bioavailability Cytotoxicity herg channel inhibiton Antitargets α1a (orthostatic hypotension) D2 (extrapyramidal syndrome) 5-T2c (obesity) musc. M1 (hallucinations, memory) CYP inhibition (3A4, 2C9, 2D6) 90% 60% 40% : : : : : : : 0%?
8 Pharmacophore Generation and Searches Catalyst (Accelrys) established tool for hypothesis generation and 3D searches CATS topological pharmacophores (Roche) no 3D structures required FTree (feature trees; BioSolveIT) no 3D searches required, ultrafast searches LigandScout (inte:ligand) automated generation of bioactive pharmacophores from protein 3D structures
9 Problems in Pharmacophore Generation Isomers, enantiomers, diastereomers Superposition of flexible molecules Ionisation and Dissoziation (Sadowski rules, ACS Boston, 2002) Tautomeric and protomeric forms (program AGET, ET Zurich; ChemoSoft tautomer recognition, ChemDiv) Acceptor properties of oxygen and sulfur atoms (esters, aromatic ethers, oxazoles, isoxazoles, thiazoles, etc.)
10 Pharmacophore ypotheses - istamine D P3 A P3 P P3 A Pharmacophores D P A Pharmacophores D P D P3 A P3 P P3

11
12 The Discovery of the DA Double elix Summer 1952: Erwin Chargaff critisizes that Francis Crick and James Watson are ignorant about the structures of the bases adenine guanine cytosine thymine J.. Davidson, The Biochemistry of ucleic Acids, London, 1950 early 1953: Pauling publishes a DA model with a phosphate core February 27, 1953: Jerry Donohue corrects the formulas of the bases February 28, 1953: Watson and Crick derive the correct DA model April 02, 1953: Manuscript sent to ature; published April 25, 1953 cited from: J. Watson and A. Berry, DA. The Secret of Life, 2003 Me
13 Tautomeric Forms of an MMP-8 Inhibitor (1jj9) R1 Glu 198 Zn 2+ Ro "enantiomer I" prochiral "enantiomer II" form * * * R2 Ala161 - R1 Leu160 R2 Glu 198 Zn 2+ R1 Ala161 - R2 Batimastat. Brandstetter et al., J. Biol. Chem. 276, (2001) R
14 Donor and Acceptor Properties of and aliph aliph arom aliph arom arom +
15 FTree (feature tree) Generation for Fast Similarity Searches 2 + C Vol.: Ring cl: 1 Profile: Vol.: 33.9 Ring cl: 0 Profile: yd Aro Amd Acc Don yd Aro Amd Acc Don M. Rarey and J. S. Dixon, J. Comput.- Aided Mol. Design 12, (1998); M. Rarey and M. Stahl, J. Comput.- Aided Mol. Design 15, (2001)
16 FTree Query Results for 1 Antagonists and Antidepressants Dimethindene R R = CEt Cl Mianserin I known actives plausible hits known actives related to the plausible hit M. Rarey and M. Stahl, J. Comput.-Aided Mol. Design 15, (2001)
17 LigandScout (inte:ligand) 1dhf G. Wolber and T. Langer, J. Chem. Inf. Model. 45, (2005)
")
18 LigandScout (inte:ligand) 1dhf G. Wolber and T. Langer, J. Chem. Inf. Model. 45, (2005)
")
19 LigandScout (inte:ligand) 1dhf G. Wolber and T. Langer, J. Chem. Inf. Model. 45, (2005)

20 LigandScout Superposition: Zanamivir vs. GS nnc 2qwk

21 LigandScout Superposition: Zanamivir vs. GS nnc 2qwk

22 LigandScout Superposition: Zanamivir vs. GS nnc 2qwk



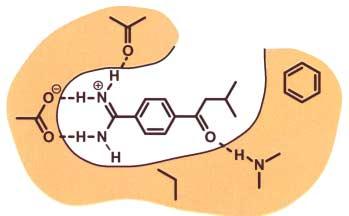
23 Computer-Aided Drug Design LUDI.-J. Böhm, 1992
24 Problems in Docking and Scoring Pre-processing of the protein lacking hydrogens, hydrogen bonds network, protonation states of his, lys, asp, glu Pre-processing of the ligands protonation states, tautomers Flexibility of the ligand (no serious problem) Flexibility of the protein / binding site (the real problem) Fuzzy scoring functions (the biggest problem)


25 IV Protease, without a ligand IV Protease, with a ligand

26 Consideration of Water, Flexibility and Mobility Ligand Receptor in aqueous environment S int
27 Factors to be Considered in Scoring Functions Desolvation enthalpy and entropy (ligand and protein) Protonation state of the ligand and the binding site Distortion energy of the ligand and its binding site Loss of translational and rotational degrees of freedom of the ligand MEP + dielectric constant at the binding site Dipole moment of the ligand and local dipole moment at the binding site Binding enthalpy of the ligand-protein complex Repulsive effects (e.g ) Inserted water molecules Solvation enthalpy and entropy of the complex
28 Virtual Screening vs. igh-throughput Screening Comparison of the performance of high-throughput screening and virtual screening of potential leads of protein tyrosine phosphatase 1B (PTP1B): a) igh throughput screening of 400,000 compounds from a corporate collection! 300 hits < 300 µm, 85 validated hits with IC 50 < 100 µm = % hit rate (many violate Lipinski rules) b) Virtual screening of 235,000 commercially available compounds, using DCK, version 3.5! 365 high-scoring molecules are tested 127 with IC 50 < 100 µm = 34.8% hit rate (hits are more drug-like?) T.. Doman et al., J. Med. Chem. 45, (2002)
29 Stepwise Virtual Screening 560,000 compounds (subsection of AstraZeneca repository) MW, rot-bond filter, presence of hinge region 199,000 hits binding motif FlexX-Pharm docking into ATP binding site 250 highest-scoring hits visual inspection for unrealistic conformations 103 compounds tested, 36 hits in the range 110 nm to 68 µm C Br Me Checkpoint kinase 1 (Chk-1) inhibitor IC 50 = 450 nm P. D. Lyne et al., J. Med. Chem. 47, (2004)
30 Stepwise Virtual Screening Aventis in-house compound repository MW, rot-bond filter, 3D pharmacophore search 22,950 compounds docking into an α 1A receptor model (GLD, PMF) 300 top-scoring compounds clustering, diversity selection 80 compounds tested, 37 hits with K i < 10 µm α 1A adrenergic receptor antagonist, K i S F = 1.4 nm A. Evers and T. Klabunde, J. Med. Chem. 48, (2005)
31 Stepwise Virtual Screening 250,251 CI compounds (3D database) 3D pharmacophore search 6,727 hits docking into four conformational clusters of a D 3 receptor homology model 2,478 potential ligands elimination of known chemotypes by similarity 20 compounds tested, 8 hits with K i < 0.5 µm F dopamine D 3 receptor antagonist, K i = 11 nm J. Varady et al., J. Med. Chem. 46, (2003)
32 2 Stepwise Virtual Screening 259,747 ACD compounds Ro5 filter with MW < 350 and rot-bond < 9, 12,545 candidates presence of -C - or equivalent 3D pharmacophore search (derived from binding site analysis) 1,261 hits FlexX docking into 0.66 Å aldose reductase 3D structure 216 highest-scoring compounds, after clustering and visual inspection: 9 hits for biological testing C aldose reductase inhibitor, IC 50 = 2.4 µm. Krämer et al., Proteins Struct. Funct. Genet. 55, (2004)
33 Virtual Screening of Carbonic Anhydrase Inhibitors 98,850 compounds (LeadQuest and Maybridge libraries) filter for Zn 2+ -binding anchor groups 5,904 hits 2D and 3D pharmacophore searches (derived from binding site analysis) 3,314 hits FlexS superposition with dorzolamide, followed by FlexX docking of 100 hits 13 hits into carbonic anhydrase binding site S X S 2 S X = S K i = 0.9 nm X = S 2 K i = 0.8 nm 2 S 2 K i = 0.6 nm S. Grüneberg et al., Angew. Chem., Int. Ed. Engl. 40, (2001); S. Grüneberg et al., J. Med. Chem. 45, (2002).
34 Combinatorial Design of Carbonic Anhydrase Inhibitors start structure 2 S K d = 120 nm 2 optimized structure 2 S C 3 R enantiomer, K d = 30 pm (S enantiomer: K d = 230 pm) Program CombiSMoG, best -substituents from 100,000 candidates (20 scored by knowledge-based potentials) B. A. Grzybowski et al., Acc. Chem. Res. 35, (2002); B. A. Grzybowski et al., Proc. atl. Acad. Sci. USA 99, (2002)

35 The Future: Combinatorial Drug Design FlexX docking of MTX vs. X-ray structure
36 Summary and Conclusions Virtual screening is a powerful tool to enrich libraries and compound collections A proper preprocessing of the compound database is of utmost importance Further experimental data and theoretical investigations are needed for better pk a estimations and better scoring functions Stepwise procedures (filters, pharmacophore searches, docking and scoring, visual inspection) are most efficient Fragment-based approaches are a promising new strategy in lead structure search and optimization Further progess needed in the understanding and scoring of ligand-receptor interactions
37 References Böhm,.-J., and Schneider, G., Eds., Virtual Screening for Bioactive Molecules (Volume 10 of Methods and Principles in Medicinal Chemistry, Mannhold, R., Kubinyi,., and Timmerman,., Eds.), Wiley-VC, Weinheim, Klebe, G., Ed., Virtual Screening: An Alternative or Complement to igh Throughput Screening, Kluwer Academic Publ, Dordrecht, 2000; also published in Persp. Drug Discov. Design 20, (2000). Böhm,.-J., and Schneider, G., Eds., Protein-Ligand Interactions. From Molecular Recognition to Drug Design, (Volume 19 of Methods and Principles in Medicinal Chemistry,, Mannhold, R., Kubinyi,., and Folkers, G., Eds.), Wiley-VC, Weinheim, Alvarez, J., and Shoichet, B., Eds., Virtual Screening in Drug Discovery, CRC Press, Taylor & Francis Group, Boca Raton, FL, USA, T. Langer and R. offmann, Pharmacophores and Pharmacophore Searches (Volume 32 of Methods and principles in medicinal chemistry, R. Mannhold,. Kubinyi and G. Folkers, Eds), Wiley-VC, Weinheim, Kubinyi, Success Stories of Computer-Aided Design, in: Computer Applications in Pharmaceutical Research and Development, S. Ekins, Ed. (Wiley Series in Drug Discovery and Development, B. Wang, Ed.), Wiley- Interscience, ew York, 2006, pp G. Klebe, Virtual ligand screening: strategies, perspectives and limitations, Drug Discov. Today 11, (2006).
Structural biology and drug design: An overview
 Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Structural biology and drug design: An overview livier Taboureau Assitant professor Chemoinformatics group-cbs-dtu otab@cbs.dtu.dk Drug discovery Drug and drug design A drug is a key molecule involved
Drug Discovery Technologies - A Closer Look
 Drug Discovery Technologies - A Closer Look Hugo Kubinyi Weisenheim am Sand E-mail kubinyi@t-online.de URL www.kubinyi.de Mastering Medicinal Chemistry RSC, Cambridge, April 27, 2009 Yesterday s Drug Discovery
Drug Discovery Technologies - A Closer Look Hugo Kubinyi Weisenheim am Sand E-mail kubinyi@t-online.de URL www.kubinyi.de Mastering Medicinal Chemistry RSC, Cambridge, April 27, 2009 Yesterday s Drug Discovery
Virtual Screening: How Are We Doing?
 Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
Virtual Screening: How Are We Doing? Mark E. Snow, James Dunbar, Lakshmi Narasimhan, Jack A. Bikker, Dan Ortwine, Christopher Whitehead, Yiannis Kaznessis, Dave Moreland, Christine Humblet Pfizer Global
BioSolveIT. A Combinatorial Approach for Handling of Protonation and Tautomer Ambiguities in Docking Experiments
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Approach for andling of Protonation and Tautomer Ambiguities in Docking Experiments Ingo Dramburg BioSolve IT Gmb An der
Introduction. OntoChem
 Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Introduction ntochem Providing drug discovery knowledge & small molecules... Supporting the task of medicinal chemistry Allows selecting best possible small molecule starting point From target to leads
Retrieving hits through in silico screening and expert assessment M. N. Drwal a,b and R. Griffith a
 Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Retrieving hits through in silico screening and expert assessment M.. Drwal a,b and R. Griffith a a: School of Medical Sciences/Pharmacology, USW, Sydney, Australia b: Charité Berlin, Germany Abstract:
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Computational Chemistry in Drug Design. Xavier Fradera Barcelona, 17/4/2007
 Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Computational Chemistry in Drug Design Xavier Fradera Barcelona, 17/4/2007 verview Introduction and background Drug Design Cycle Computational methods Chemoinformatics Ligand Based Methods Structure Based
Author Index Volume
 Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Perspectives in Drug Discovery and Design, 20: 289, 2000. KLUWER/ESCOM Author Index Volume 20 2000 Bradshaw,J., 1 Knegtel,R.M.A., 191 Rose,P.W., 209 Briem, H., 231 Kostka, T., 245 Kuhn, L.A., 171 Sadowski,
Fragment based drug discovery in teams of medicinal and computational chemists. Carsten Detering
 Fragment based drug discovery in teams of medicinal and computational chemists Carsten Detering BioSolveIT Quick Facts Founded in 2001 by the developers of FlexX ~20 people Core expertise: docking, screening,
Fragment based drug discovery in teams of medicinal and computational chemists Carsten Detering BioSolveIT Quick Facts Founded in 2001 by the developers of FlexX ~20 people Core expertise: docking, screening,
LigandScout. Automated Structure-Based Pharmacophore Model Generation. Gerhard Wolber* and Thierry Langer
 LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
LigandScout Automated Structure-Based Pharmacophore Model Generation Gerhard Wolber* and Thierry Langer * E-Mail: wolber@inteligand.com Pharmacophores from LigandScout Pharmacophores & the Protein Data
György M. Keserű H2020 FRAGNET Network Hungarian Academy of Sciences
 Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Fragment based lead discovery - introduction György M. Keserű H2020 FRAGET etwork Hungarian Academy of Sciences www.fragnet.eu Hit discovery from screening Druglike library Fragment library Large molecules
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME
 Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
Virtual Libraries and Virtual Screening in Drug Discovery Processes using KNIME Iván Solt Solutions for Cheminformatics Drug Discovery Strategies for known targets High-Throughput Screening (HTS) Cells
BioSolveIT. A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities
 BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
BioSolveIT Biology Problems Solved using Information Technology A Combinatorial Docking Approach for Dealing with Protonation and Tautomer Ambiguities Ingo Dramburg BioSolve IT Gmb An der Ziegelei 75 53757
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Virtual screening in drug discovery
 Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Virtual screening in drug discovery Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery
Plan. Day 2: Exercise on MHC molecules.
 Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Plan Day 1: What is Chemoinformatics and Drug Design? Methods and Algorithms used in Chemoinformatics including SVM. Cross validation and sequence encoding Example and exercise with herg potassium channel:
Computational chemical biology to address non-traditional drug targets. John Karanicolas
 Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Computational chemical biology to address non-traditional drug targets John Karanicolas Our computational toolbox Structure-based approaches Ligand-based approaches Detailed MD simulations 2D fingerprints
Introduction to Chemoinformatics and Drug Discovery
 Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Introduction to Chemoinformatics and Drug Discovery Irene Kouskoumvekaki Associate Professor February 15 th, 2013 The Chemical Space There are atoms and space. Everything else is opinion. Democritus (ca.
Hydrogen Bonding & Molecular Design Peter
 Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
Hydrogen Bonding & Molecular Design Peter Kenny(pwk.pub.2008@gmail.com) Hydrogen Bonding in Drug Discovery & Development Interactions between drug and water molecules (Solubility, distribution, permeability,
Ligand-receptor interactions
 University of Silesia, Katowice, Poland 11 22 March 2013 Ligand-receptor interactions Dr. Pavel Polishchuk A.V. Bogatsky Physico-Chemical Institute of National Academy of Sciences of Ukraine Odessa, Ukraine
University of Silesia, Katowice, Poland 11 22 March 2013 Ligand-receptor interactions Dr. Pavel Polishchuk A.V. Bogatsky Physico-Chemical Institute of National Academy of Sciences of Ukraine Odessa, Ukraine
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Nonlinear QSAR and 3D QSAR
 onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
onlinear QSAR and 3D QSAR Hugo Kubinyi Germany E-Mail kubinyi@t-online.de HomePage www.kubinyi.de onlinear Lipophilicity-Activity Relationships drug receptor Possible Reasons for onlinear Lipophilicity-Activity
Advanced Medicinal Chemistry SLIDES B
 Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Advanced Medicinal Chemistry Filippo Minutolo CFU 3 (21 hours) SLIDES B Drug likeness - ADME two contradictory physico-chemical parameters to balance: 1) aqueous solubility 2) lipid membrane permeability
Chemical library design
 Chemical library design Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery Today,
Chemical library design Pavel Polishchuk Institute of Molecular and Translational Medicine Palacky University pavlo.polishchuk@upol.cz Drug development workflow Vistoli G., et al., Drug Discovery Today,
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Design and Synthesis of the Comprehensive Fragment Library
 YOUR INNOVATIVE CHEMISTRY PARTNER IN DRUG DISCOVERY Design and Synthesis of the Comprehensive Fragment Library A 3D Enabled Library for Medicinal Chemistry Discovery Warren S Wade 1, Kuei-Lin Chang 1,
YOUR INNOVATIVE CHEMISTRY PARTNER IN DRUG DISCOVERY Design and Synthesis of the Comprehensive Fragment Library A 3D Enabled Library for Medicinal Chemistry Discovery Warren S Wade 1, Kuei-Lin Chang 1,
Protein-Ligand Docking Evaluations
 Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design
 Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Medicinal Chemistry/ CHEM 458/658 Chapter 4- Computer-Aided Drug Design Bela Torok Department of Chemistry University of Massachusetts Boston Boston, MA 1 Computer Aided Drug Design - Introduction Development
Efficient overlay of molecular 3-D pharmacophores
 Efficient overlay of molecular 3D pharmacophores Gerhard Wolber*, Alois A. Dornhofer & Thierry Langer * EMail: wolber@inteligand.com Superposition of molecules 1 Alignment: Outline Scope, design goals
Efficient overlay of molecular 3D pharmacophores Gerhard Wolber*, Alois A. Dornhofer & Thierry Langer * EMail: wolber@inteligand.com Superposition of molecules 1 Alignment: Outline Scope, design goals
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses
 Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
Dispensing Processes Profoundly Impact Biological, Computational and Statistical Analyses Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc,
An Integrated Approach to in-silico
 An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
An Integrated Approach to in-silico Screening Joseph L. Durant Jr., Douglas. R. Henry, Maurizio Bronzetti, and David. A. Evans MDL Information Systems, Inc. 14600 Catalina St., San Leandro, CA 94577 Goals
Ignasi Belda, PhD CEO. HPC Advisory Council Spain Conference 2015
 Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Ignasi Belda, PhD CEO HPC Advisory Council Spain Conference 2015 Business lines Molecular Modeling Services We carry out computational chemistry projects using our selfdeveloped and third party technologies
Fragment-based de novo Design
 ragment-based de novo Design Gisbert Schneider & Uli echner gisbert.schneider@modlab.de www.modlab.de Beilstein Endowed Chair for Cheminformatics Institute of rganic Chemistry & Chemical Biology Johann
ragment-based de novo Design Gisbert Schneider & Uli echner gisbert.schneider@modlab.de www.modlab.de Beilstein Endowed Chair for Cheminformatics Institute of rganic Chemistry & Chemical Biology Johann
Targeting protein-protein interactions: A hot topic in drug discovery
 Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Michal Kamenicky; Maria Bräuer; Katrin Volk; Kamil Ödner; Christian Klein; Norbert Müller Targeting protein-protein interactions: A hot topic in drug discovery 104 Biomedizin Innovativ patientinnenfokussierte,
Structure based drug design and LIE models for GPCRs
 Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
Structure based drug design and LIE models for GPCRs Peter Kolb kolb@docking.org Shoichet Lab ACS 237 th National Meeting, March 24, 2009 p.1/26 [Acknowledgements] Brian Shoichet John Irwin Mike Keiser
Using Phase for Pharmacophore Modelling. 5th European Life Science Bootcamp March, 2017
 Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Using Phase for Pharmacophore Modelling 5th European Life Science Bootcamp March, 2017 Phase: Our Pharmacohore generation tool Significant improvements to Phase methods in 2016 New highly interactive interface
Biologically Relevant Molecular Comparisons. Mark Mackey
 Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Biologically Relevant Molecular Comparisons Mark Mackey Agenda > Cresset Technology > Cresset Products > FieldStere > FieldScreen > FieldAlign > FieldTemplater > Cresset and Knime About Cresset > Specialist
Building innovative drug discovery alliances. Just in KNIME: Successful Process Driven Drug Discovery
 Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
Building innovative drug discovery alliances Just in KIME: Successful Process Driven Drug Discovery Berlin KIME Spring Summit, Feb 2016 Research Informatics @ Evotec Evotec s worldwide operations 2 Pharmaceuticals
In silico pharmacology for drug discovery
 In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
In silico pharmacology for drug discovery In silico drug design In silico methods can contribute to drug targets identification through application of bionformatics tools. Currently, the application of
Medicinal Chemist s Relationship with Additivity: Are we Taking the Fundamentals for Granted?
 Medicinal Chemist s Relationship with Additivity: Are we Taking the Fundamentals for Granted? J. Guy Breitenbucher Streamlining Drug Discovery Conference San Francisco, CA ct. 25, 2018 Additivity as the
Medicinal Chemist s Relationship with Additivity: Are we Taking the Fundamentals for Granted? J. Guy Breitenbucher Streamlining Drug Discovery Conference San Francisco, CA ct. 25, 2018 Additivity as the
1. (18) Multiple choice questions. Please place your answer on the line preceding each question.
 Multiple choice questions. Please place your answer on the line preceding each question.") CEM 5720 ame KEY Exam 2 ctober 21, 2015 Read all questions carefully and attempt those questions you are sure of first. Remember to proof your work; art work carries as much importance as written responses.
CEM 5720 ame KEY Exam 2 ctober 21, 2015 Read all questions carefully and attempt those questions you are sure of first. Remember to proof your work; art work carries as much importance as written responses.
The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score. Protein Structures: The Starting Point for New Drugs 2
 The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score Protein Structures: The Starting Point for New Drugs 2 Matthias Rarey Stefan Bietz Nadine Schneider Sascha Urbaczek University
The Long and Rocky Road from a PDB File to a Protein Ligand Docking Score Protein Structures: The Starting Point for New Drugs 2 Matthias Rarey Stefan Bietz Nadine Schneider Sascha Urbaczek University
Structure Investigation of Fam20C, a Golgi Casein Kinase
 Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Protein-Ligand Docking
 Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification
 Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Fragment Hotspot Maps: A CSD-derived Method for Hotspot identification Chris Radoux www.ccdc.cam.ac.uk radoux@ccdc.cam.ac.uk 1 Introduction Hotspots Strongly attractive to organic molecules Organic molecules
Structure-Activity Modeling - QSAR. Uwe Koch
 Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Structure-Activity Modeling - QSAR Uwe Koch QSAR Assumption: QSAR attempts to quantify the relationship between activity and molecular strcucture by correlating descriptors with properties Biological activity
Data Quality Issues That Can Impact Drug Discovery
 Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Data Quality Issues That Can Impact Drug Discovery Sean Ekins 1, Joe Olechno 2 Antony J. Williams 3 1 Collaborations in Chemistry, Fuquay Varina, NC. 2 Labcyte Inc, Sunnyvale, CA. 3 Royal Society of Chemistry,
Navigation in Chemical Space Towards Biological Activity. Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland
 Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Navigation in Chemical Space Towards Biological Activity Peter Ertl Novartis Institutes for BioMedical Research Basel, Switzerland Data Explosion in Chemistry CAS 65 million molecules CCDC 600 000 structures
Protein-Ligand Docking Methods
 Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Synthetic organic compounds
 Synthetic organic compounds for research and drug discovery Compounds for TS Fragment libraries Target-focused libraries Chemical building blocks Custom synthesis Drug discovery services Contract research
Synthetic organic compounds for research and drug discovery Compounds for TS Fragment libraries Target-focused libraries Chemical building blocks Custom synthesis Drug discovery services Contract research
Supporting Information
 Discovery of kinase inhibitors by high-throughput docking and scoring based on a transferable linear interaction energy model Supporting Information Peter Kolb, Danzhi Huang, Fabian Dey and Amedeo Caflisch
Discovery of kinase inhibitors by high-throughput docking and scoring based on a transferable linear interaction energy model Supporting Information Peter Kolb, Danzhi Huang, Fabian Dey and Amedeo Caflisch
Plan. Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics.
 and its used in Chemoinformatics.") Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Plan Lecture: What is Chemoinformatics and Drug Design? Description of Support Vector Machine (SVM) and its used in Chemoinformatics. Exercise: Example and exercise with herg potassium channel: Use of
Drug Design 2. Oliver Kohlbacher. Winter 2009/ QSAR Part 4: Selected Chapters
 Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Drug Design 2 Oliver Kohlbacher Winter 2009/2010 11. QSAR Part 4: Selected Chapters Abt. Simulation biologischer Systeme WSI/ZBIT, Eberhard-Karls-Universität Tübingen Overview GRIND GRid-INDependent Descriptors
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining
![Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining](/thumbs/91/107031261.jpg "Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining") Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining Samer Haidar 1, Zouhair Bouaziz 2, Christelle Marminon 2, Tiomo Laitinen 3, Anti Poso
ChemDiv Beyond the Flatland 3D-Fragment Library for Fragment-Based Drug Discovery (FBDD)
") ChemDiv Beyond the Flatland 3D-Fragment Library for Fragment-Based Drug Discovery (FBDD) CEMDIV BEYD TE FLATLAD 3D-FRAGMET LIBRARY BACKGRUD Fragment-based drug discovery (FBDD) has become an efficient
ChemDiv Beyond the Flatland 3D-Fragment Library for Fragment-Based Drug Discovery (FBDD) CEMDIV BEYD TE FLATLAD 3D-FRAGMET LIBRARY BACKGRUD Fragment-based drug discovery (FBDD) has become an efficient
Bridging the Dimensions:
 Bridging the Dimensions: Seamless Integration of 3D Structure-based Design and 2D Structure-activity Relationships to Guide Medicinal Chemistry ACS Spring National Meeting. COMP, March 13 th 2016 Marcus
Bridging the Dimensions: Seamless Integration of 3D Structure-based Design and 2D Structure-activity Relationships to Guide Medicinal Chemistry ACS Spring National Meeting. COMP, March 13 th 2016 Marcus
Pharmacophores and Pharmacophore Searches
 Pharmacophores and Pharmacophore Searches Edited by Thierry Longer and Remy D. Hoffmann WILEY- VCH WILEY-VCH Verlag GmbH &. Co. KGaA Contents Preface XIII A Personal Foreword List of Contributors XV XVII
Pharmacophores and Pharmacophore Searches Edited by Thierry Longer and Remy D. Hoffmann WILEY- VCH WILEY-VCH Verlag GmbH &. Co. KGaA Contents Preface XIII A Personal Foreword List of Contributors XV XVII
Similarity Search. Uwe Koch
 Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Similarity Search Uwe Koch Similarity Search The similar property principle: strurally similar molecules tend to have similar properties. However, structure property discontinuities occur frequently. Relevance
Chemogenomic: Approaches to Rational Drug Design. Jonas Skjødt Møller
 Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
Chemogenomic: Approaches to Rational Drug Design Jonas Skjødt Møller Chemogenomic Chemistry Biology Chemical biology Medical chemistry Chemical genetics Chemoinformatics Bioinformatics Chemoproteomics
The Conformation Search Problem
 Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
Jon Sutter Senior Manager Life Sciences R&D jms@accelrys.com Jiabo Li Senior Scientist Life Sciences R&D jli@accelrys.com CAESAR: Conformer Algorithm based on Energy Screening and Recursive Buildup The
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors
 A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
A Tiered Screen Protocol for the Discovery of Structurally Diverse HIV Integrase Inhibitors Rajarshi Guha, Debojyoti Dutta, Ting Chen and David J. Wild School of Informatics Indiana University and Dept.
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
ChemBioNet: Chemical Biology supported by Networks of Chemists and Biologists. Affinity Proteomics Meeting Alpbach. Michael Lisurek 15.3.
 ChemBioet: Chemical Biology supported by etworks of Chemists and Biologists Affinity Proteomics Meeting Alpbach Michael Lisurek 15.3.2007 - Introduction - Equipment Screening Unit - Screening Compound
ChemBioet: Chemical Biology supported by etworks of Chemists and Biologists Affinity Proteomics Meeting Alpbach Michael Lisurek 15.3.2007 - Introduction - Equipment Screening Unit - Screening Compound
Applying Bioisosteric Transformations to Predict Novel, High Quality Compounds
 Applying Bioisosteric Transformations to Predict Novel, High Quality Compounds Dr James Chisholm,* Dr John Barnard, Dr Julian Hayward, Dr Matthew Segall*, Mr Edmund Champness*, Dr Chris Leeding,* Mr Hector
Applying Bioisosteric Transformations to Predict Novel, High Quality Compounds Dr James Chisholm,* Dr John Barnard, Dr Julian Hayward, Dr Matthew Segall*, Mr Edmund Champness*, Dr Chris Leeding,* Mr Hector
Towards Physics-based Models for ADME/Tox. Tyler Day
 Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
PATTERN RECOGNITION AND DISTRIBUTED COMPUTING
 49 Molecular Informatics: Confronting Complexity, May 13 th - 16 th 2002, Bozen, Italy PATTERN RECOGNITION AND DISTRIBUTED COMPUTING IN DRUG DESIGN W. GRAHAM RICHARDS Department of Chemistry, University
49 Molecular Informatics: Confronting Complexity, May 13 th - 16 th 2002, Bozen, Italy PATTERN RECOGNITION AND DISTRIBUTED COMPUTING IN DRUG DESIGN W. GRAHAM RICHARDS Department of Chemistry, University
Virtual screening for drug discovery. Markus Lill Purdue University
 Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Lecture for Molekylär bioinformatik X3 Feb Computational Chemistry in Drug Discovery. Mats Kihlén. Head of Research Informatics.
 Lecture for Molekylär bioinformatik X3 Feb 24 2004 Computational Chemistry in Drug Discovery Mats Kihlén Head of Research Informatics Biovitrum AB verview» The role of computational chemistry» Basic concepts
Lecture for Molekylär bioinformatik X3 Feb 24 2004 Computational Chemistry in Drug Discovery Mats Kihlén Head of Research Informatics Biovitrum AB verview» The role of computational chemistry» Basic concepts
Computational Methods and Drug-Likeness. Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004
 Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
Computational Methods and Drug-Likeness Benjamin Georgi und Philip Groth Pharmakokinetik WS 2003/2004 The Problem Drug development in pharmaceutical industry: >8-12 years time ~$800m costs >90% failure
CHAPTER-2. Drug discovery is a comprehensive approach wherein several disciplines
 36 CAPTER-2 Molecular Modeling Analog Based Studies Drug discovery is a comprehensive approach wherein several disciplines are used to design or discover the drugs. The R&D expenditure incurred to bring
36 CAPTER-2 Molecular Modeling Analog Based Studies Drug discovery is a comprehensive approach wherein several disciplines are used to design or discover the drugs. The R&D expenditure incurred to bring
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Principles of Drug Design
 (16:663:502) Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu For more current information, please check WebCT at https://webct.rutgers.edu
(16:663:502) Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu For more current information, please check WebCT at https://webct.rutgers.edu
Integrated Cheminformatics to Guide Drug Discovery
 Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Integrated Cheminformatics to Guide Drug Discovery Matthew Segall, Ed Champness, Peter Hunt, Tamsin Mansley CINF Drug Discovery Cheminformatics Approaches August 23 rd 2017 Optibrium, StarDrop, Auto-Modeller,
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites. J. Andrew Surface
 Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Practical QSAR and Library Design: Advanced tools for research teams
 DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
DS QSAR and Library Design Webinar Practical QSAR and Library Design: Advanced tools for research teams Reservationless-Plus Dial-In Number (US): (866) 519-8942 Reservationless-Plus International Dial-In
JCICS Major Research Areas
 JCICS Major Research Areas Chemical Information Text Searching Structure and Substructure Searching Databases Patents George W.A. Milne C571 Lecture Fall 2002 1 JCICS Major Research Areas Chemical Computation
JCICS Major Research Areas Chemical Information Text Searching Structure and Substructure Searching Databases Patents George W.A. Milne C571 Lecture Fall 2002 1 JCICS Major Research Areas Chemical Computation
Toward an Understanding of GPCR-ligand Interactions. Alexander Heifetz
 Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Toward an Understanding of GPCR-ligand Interactions Alexander Heifetz UK QSAR and ChemoInformatics Group Conference, Cambridge, UK October 6 th, 2015 Agenda Fragment Molecular Orbitals (FMO) for GPCR exploration
Identifying Interaction Hot Spots with SuperStar
 Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Identifying Interaction Hot Spots with SuperStar Version 1.0 November 2017 Table of Contents Identifying Interaction Hot Spots with SuperStar... 2 Case Study... 3 Introduction... 3 Generate SuperStar Maps
Synthetic organic compounds
 Synthetic organic compounds for research and drug discovery chemicals Compounds for TS Fragment libraries Target-focused libraries Chemical building blocks Custom synthesis Drug discovery services Contract
Synthetic organic compounds for research and drug discovery chemicals Compounds for TS Fragment libraries Target-focused libraries Chemical building blocks Custom synthesis Drug discovery services Contract
Comparative Analysis of Pharmacophore Screening Tools
 pubs.acs.org/jcim Comparative Analysis of Pharmacophore Screening Tools Marijn P. A. Sanders,,# Armeńio J. M. Barbosa, Barbara Zarzycka, Gerry A.F. Nicolaes, Jan P.G. Klomp, Jacob de Vlieg,, and Alberto
pubs.acs.org/jcim Comparative Analysis of Pharmacophore Screening Tools Marijn P. A. Sanders,,# Armeńio J. M. Barbosa, Barbara Zarzycka, Gerry A.F. Nicolaes, Jan P.G. Klomp, Jacob de Vlieg,, and Alberto
Journal of Pharmacology and Experimental Therapy-JPET#172536
 A NEW NON-PEPTIDIC INHIBITOR OF THE 14-3-3 DOCKING SITE INDUCES APOPTOTIC CELL DEATH IN CHRONIC MYELOID LEUKEMIA SENSITIVE OR RESISTANT TO IMATINIB Manuela Mancini, Valentina Corradi, Sara Petta, Enza
A NEW NON-PEPTIDIC INHIBITOR OF THE 14-3-3 DOCKING SITE INDUCES APOPTOTIC CELL DEATH IN CHRONIC MYELOID LEUKEMIA SENSITIVE OR RESISTANT TO IMATINIB Manuela Mancini, Valentina Corradi, Sara Petta, Enza
Relative Drug Likelihood: Going beyond Drug-Likeness
 Relative Drug Likelihood: Going beyond Drug-Likeness ACS Fall National Meeting, August 23rd 2012 Matthew Segall, Iskander Yusof Optibrium, StarDrop, Auto-Modeller and Glowing Molecule are trademarks of
Relative Drug Likelihood: Going beyond Drug-Likeness ACS Fall National Meeting, August 23rd 2012 Matthew Segall, Iskander Yusof Optibrium, StarDrop, Auto-Modeller and Glowing Molecule are trademarks of
CSD. CSD-Enterprise. Access the CSD and ALL CCDC application software
 CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
CSD CSD-Enterprise Access the CSD and ALL CCDC application software CSD-Enterprise brings it all: access to the Cambridge Structural Database (CSD), the world s comprehensive and up-to-date database of
Keywords: anti-coagulants, factor Xa, QSAR, Thrombosis. Introduction
 PostDoc Journal Vol. 2, No. 3, March 2014 Journal of Postdoctoral Research www.postdocjournal.com QSAR Study of Thiophene-Anthranilamides Based Factor Xa Direct Inhibitors Preetpal S. Sidhu Department
PostDoc Journal Vol. 2, No. 3, March 2014 Journal of Postdoctoral Research www.postdocjournal.com QSAR Study of Thiophene-Anthranilamides Based Factor Xa Direct Inhibitors Preetpal S. Sidhu Department
Hugo Kubinyi. Strategies in Lead Optimization
 University of eidelberg From Leads to Drugs ugo Kubinyi University of eidelberg Germany E-Mail kubinyi@t-online.de omepage www.kubinyi.de Joint eting on dicinal Chemistry, Vienna, June 2005 University
University of eidelberg From Leads to Drugs ugo Kubinyi University of eidelberg Germany E-Mail kubinyi@t-online.de omepage www.kubinyi.de Joint eting on dicinal Chemistry, Vienna, June 2005 University
Life Science Webinar Series
 Life Science Webinar Series Elegant protein- protein docking in Discovery Studio Francisco Hernandez-Guzman, Ph.D. November 20, 2007 Sr. Solutions Scientist fhernandez@accelrys.com Agenda In silico protein-protein
Life Science Webinar Series Elegant protein- protein docking in Discovery Studio Francisco Hernandez-Guzman, Ph.D. November 20, 2007 Sr. Solutions Scientist fhernandez@accelrys.com Agenda In silico protein-protein
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs
 Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Next Generation Computational Chemistry Tools to Predict Toxicity of CWAs William (Bill) Welsh welshwj@umdnj.edu Prospective Funding by DTRA/JSTO-CBD CBIS Conference 1 A State-wide, Regional and National
Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery
 21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
21 th /June/2018@CUGM Progress of Compound Library Design Using In-silico Approach for Collaborative Drug Discovery Kaz Ikeda, Ph.D. Keio University Self Introduction Keio University, Tokyo, Japan (Established
What is Protein-Ligand Docking?
 MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
Electronic Supplementary Information Effective lead optimization targeted for displacing bridging water molecule
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Electronic Supplementary Information Effective lead optimization targeted for displacing
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2018 Electronic Supplementary Information Effective lead optimization targeted for displacing
Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner
 Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
Table of Contents Creating a Pharmacophore Query from a Reference Molecule & Scaffold Hopping in CSD-CrossMiner Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Features
A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery
 AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
AtomNet A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery Izhar Wallach, Michael Dzamba, Abraham Heifets Victor Storchan, Institute for Computational and
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics
 Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Contents 1 Open-Source Tools, Techniques, and Data in Chemoinformatics... 1 1.1 Chemoinformatics... 2 1.1.1 Open-Source Tools... 2 1.1.2 Introduction to Programming Languages... 3 1.2 Chemical Structure
Principles of Drug Design
 Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction
Advanced Medicinal Chemistry II Principles of Drug Design Tentative Course Outline Instructors: Longqin Hu and John Kerrigan Direct questions and enquiries to the Course Coordinator: Longqin Hu I. Introduction