Small Molecule & Protein Docking CHEM 430
|
|
|
- Laurence Payne
- 5 years ago
- Views:
Transcription
1 Small Molecule & Protein Docking CHEM 430

2 Molecular Docking Models Over the years biochemists have developed numerous models to capture the key elements of the molecular recognition process. Although very simpli%ed, these models have proven highly useful to the scienti%c community.

3 The Lock and Key Theory As far back as 1890 Emil Fischer proposed a model called the "lock-and-key model" that explained how biological systems function. A substrate %ts into the active site of a macromolecule, just like a key %ts into a lock. Biological 'locks' have unique stereochemical features that are necessary to their function.


4 The Induced-Fit Theory In 1958 Daniel Koshland introduced the "induced-%t theory". The basic idea is that in the recognition process, both ligand and target mutually adapt to each other through small conformational changes, until an optimal %t is achieved.

5 The Conformation Ensemble Model In addition to small induced-%t adaptation, it has been observed that proteins can undergo much larger conformational changes. A recent model describes proteins as a pre-existing ensemble of conformational states. The plasticity of the protein allows it to switch from one state to another.

6 From the Lock and Key to the Ensemble Model Lock-and-key, induced-%t and the conformation ensemble model are not contradictory. Each one focuses on a particular aspect of the recognition process. The lock-and-key model introduces the principle of 3D complementarity, the induced-%t model explains how complementarity is achieved, and the ensemble model depicts the conformational complexity of proteins.

7 Number of Citations for Docking Programs ISI Web of Science (2005) Sousa, S.F., Fernandes, P.A. & Ramos, M.J. (2006) Protein-Ligand Docking: Current Status and Future Challenges Proteins, 65:15-26
8 Introduction A significant portion of biology is built on the paradigm sequence structure function As we sequence more genomes and get more structural information, the next challenge will be to predict interactions and binding for two or more biomolecules (nucleic acids, proteins, peptides, drugs or other small molecules)
9 Introduction The questions we are interested in are: Do two biomolecules bind each other? If so, how do they bind? What is the binding free energy or affinity? The goals we have are: Searching for lead compounds Estimating effect of modifications General understanding of binding
10 Rationale The ability to predict the binding site and binding affinity of a drug or compound is immensely valuable in the area or pharmaceutical design Most (if not all) drug companies use computational methods as one of the first methods of screening or development Computer-aided drug design is a more daunting task, but there are several examples of drugs developed with a significant contribution from computational methods
11 Examples Tacrine inhibits acetylcholinesterase and boost acetylcholine levels (for treating Alzheimer s disease) Relenza targets influenza Invirase, Norvir, Crixivan Various HIV protease inhibitors Celebrex inhibits Cox-2 enzyme which causes inflammation (not our fault)
12 Docking Docking refers to a computational scheme that tries to find the best binding orientation between two biomolecules where the starting point is the atomic coordinates of the two molecules Additional data may be provided (biochemical, mutational, conservation, etc.) and this can significantly improve the performance, however this extra information is not required
13 What is Docking? Given the 3D structures of two molecules, determine the best binding modes.


14 Molecular Complementarity in Computational Docking Computational docking exploit the concept of molecular complementarity. The structures interact like a hand in a glove, where both the shape and the physico-chemical properties of the structures contribute to the %t. Shape complementarity is the primary criterion for evaluating the %t in the computational docking of two candidate structures In addition to shape compatibility, chemical and physicochemical complementarity are also important criteria in the docking between candidate structures


15 Energy Dictates Molecular Associations The process of "self-assembly" is dictated by forces that are energy based: the complex has a lower potential energy than its constituent parts, and this keeps the parts together The goal of computational docking is to %nd the 3D con%guration of the complex that minimizes the energy
16 Defining a Docking Position x, y, z y Orientation qx, qy, qz, qw Torsions τ 1 τ 1, τ 2, τ n x z

17 De(nition of the "Pose" A "pose" is a term widely adopted for describing the geometry of a particular complex (also called "binding mode") It refers to a precise con%guration which is characterized not only by the relative orientation of the docked molecules but also their respective conformations
18 Key aspects of docking Scoring Functions Predicting the energy of a particular pose Often a trade-off between speed and accuracy Search Methods Finding an optimal pose Which search method should I use? Dimensionality Can we trust the answer?
19 Bound vs. Unbound Docking The simplest problem is the bound docking problem. The goal in this case is to reproduce a known complex where the starting point is atomic structures from a co-crystal The unbound docking problem is significantly more difficult task. Here the starting point is structure in their unbound conformation, perhaps the native structure, perhaps a modeled structure, etc.


20 Bound vs. Unbound Receptor surface 10 highly penetrating residues Ligand Kallikrein A/trypsin inhibitor complex (PDB codes 2KAI,6PTI)
21 Molecular Flexibility in Protein-Ligand Docking The mutual adaptation of a ligand with its receptor is crucial to understanding ligand binding and protein function One of the major challenges in molecular docking is how to account for this adaptation in docking calculations The docking problem can be classi%ed according to the way Dexibility is modeled. In ascending order of complexity: 1. Rigid body docking ignores the Dexibility of the molecules and treats them like rigid objects 2. Rigid receptor Dexible ligand docking: only the ligand is treated as Dexible, receptor is rigid 3. Flexible receptor Dexible ligand docking: both protein and ligand are treated as Dexible.

22 Molecular Flexibility in Protein-Ligand Docking
23 Approach to the Problem One of the first suggestions on how to tackle this problem was given by Crick who postulated that knobs could be fitted into holes on the protein surface Lee & Richards had already described how to treat the van der Waals surface of a protein, however the van der Waals surface was not appropriate for docking purposes (too many crevices) Connolly showed how to calculate the molecular surface (and also the solvent accessible surface) which allowed for many docking programs to start being developed in the mid 1980 s
24 Docking Methodology All small molecule docking programs have three main components Representation of system The search algorithm The scoring or energy evaluation routine In order to do a good job of docking, you need to search efficiently and evaluate the energy accurately
25 A Matter of Size For two proteins, the docking problem is very difficult since the search space (all possible relative conformations) is extremely large In the case of a small molecule (drug, peptide or ligand) binding to a protein, we have a chance of exploring the conformational space, at least for the small molecule Now we want to consider the case where we have limited or no a priori knowledge and the mode of binding
26 Protein-Protein Docking Methods The approaches to protein-protein docking have a lot in common with small molecule docking The methods are still based on the combination of search algorithm coupled to a scoring function The scoring functions are essentially the same (since we are still dealing with atomic interactions), however the major problem is that the conformational space we now need to search is massive
27 FFT Methods A clever method developed by Katchalski-Katzir et al. uses FFT methods to search the translational and rotational degrees of freedom H. A. Gabb, R. M Jackson and M. Sternberg Modelling protein docking using shape complementarity, electrostatics and biochemical information J. Mol Biol. (1997) 272:
28 Katzir et al - ummary Exhaustive enumeration of the rotational and translational degrees of freedom. Since the scoring function is represented as a convolution, the enumeration of the translational degrees of freedom is made efficient exploiting FFT - O(N 3 logn). Allows introduction of electrostatics as the complex number component of the convolution. Time consuming, yet conceptually simple and appealing. Volumetric does not use MS representation.
29 Representation of the System or Site Characterization
30 Representation of Phase Space The type or choice of representation for a system is really a reflection of the type of energy evaluation or scoring function that will be used If we would choose the most straightforward or logical representation of the atomic coordinates of the of the two biomolecules, we would simply use a molecular mechanics force field such as Amber, CHARMM or OPLS However, this may not be the best choice in terms of computational efficiency or practicality

31 Protein Preparation The preparation of the protein calls for great care. Important decisions include the choice of the tautomeric forms of histidine residues, the protonation states of amino-acids and conformations of some residues; their incorrect assignments may lead to docking errors.
32 Small Molecule Preparation Before generating and docking the 3D structures of a library of ligands, it is important to "clean up" the 2D structures being used by removing any counter ions, salts, or water molecules that might be part of the registered structure All reactive or otherwise undesirable compounds must also be removed Possibly generate all optical isomers (enantiomers), cis/trans isomers, tautomers, and protonation states of the structures For most docking programs the tautomeric and protonation state of the ligands to be docked is de%ned by the user; in general the structure considered to be dominant at a neutral ph is generated; here also, incorrect assignments may lead to docking errors
 binding site as a collection of overlapping")
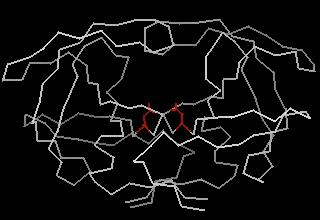
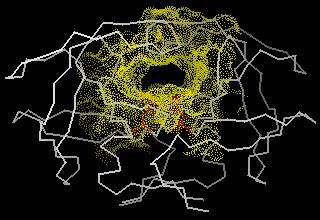
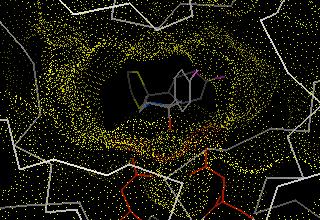
33 DOCK The DOCK program is from the Kuntz group at UCSF It was the first docking program developed in 1982 It represents the (negative image of the) binding site as a collection of overlapping spheres
34 DOCK This method of a negative image is targeted at finding complexes with a high degree of shape complementarity The ligand is fit into the image by a least squares fitting of the atomic positions to the sphere centers Creating the negative image is obviously not a problem with a unique solution. Hence, factors such as the sphere radius and center-to-center distance of the spheres must be carefully controlled.
 Sparse surface (Shuo Lin et al.")
35 Surface Representation Dense MS surface (Connolly) Sparse surface (Shuo Lin et al.)
36 The DOCK algorithm Rigid docking The DOCK algorithm developed by Kuntz and co-workers is generally considered one of the major advances in protein ligand docking [Kuntz et al., JMB, 1982, 161, 269] The earliest version of the DOCK algorithm only considered rigid body docking and was designed to identify molecules with a high degree of shape complementarity to the protein binding site. The first stage of the DOCK method involves the construction of a negative image of the binding site consisting of a series of overlapping spheres of varying radii, derived from the molecular surface of the protein AR Leach, VJ Gillet, An Introduction to Cheminformatics
37 The DOCK algorithm Rigid docking Ligand atoms are then matched to the sphere centres so that the distances between the atoms equal the distances between the corresponding sphere centres, within some tolerance. The ligand conformation is then oriented into the binding site. After checking to ensure that there are no unacceptable steric interactions, it is then scored. New orientations are produced by generating new sets of matching ligand atoms and sphere centres. The procedure continues until all possible matches have been considered. AR Leach, VJ Gillet, An Introduction to Cheminformatics

38 DOCK
39 DOCK Extensions Dock 4.0 has recently been extended to include: Several different scoring schemes Ligand flexibility (via incremental construction and fragment joining) Chemical properties of receptor (each sphere assume a chemical characteristic)
40 CLIX CLIX uses a chemical description of the receptor and distance constraints on the atoms Lawrence et al., Proteins: Struct. Func. And Gen. 12:31 (1992)

41 Clique-Detection Nodes comprise of matches between protein and ligand Edges connect distance compatible pairs of nodes In a clique all pair of nodes are connected
42 ESCHER ESCHER uses the solvent accessible surface from a Connolly algorithm This surface is cut into 1.5 Å slabs that are transformed into polygons and used for rigid docking (again image matching) G. Ausiello, G. Cesareni, M. Helmer Citterich, "ESCHER: a new docking procedure applied to the reconstruction of protein tertiary structure", Proteins, 28: (1997)
43 Search Methods
44 Search Basics One of the more difficult tasks in computational docking is simply enumerating the number of ways two molecules can be put together (3 translational plus 3 rotational degrees of freedom) The size of this search space grows exponentially as we increase the size of the molecules, however this is still only for rigid structures
45 Sampling Hyperspace Say we are searching in D-dimensional hyperspace We want to evaluate each of the D dimensions N times. The number of evals needed, n, is: n = N D N = n 1/D For example, if n = 10 6 and D=6, N = (10 6 ) 1/6 = 10 evaluations per dimension D=36, N = (10 6 ) 1/36 = ~1.5 evaluations per dimension Clearly, the more dimensions, the tougher it gets.

46 SAMPLING HYPERSPACE! Very CPU time consuming... N=T 360/i Methotrexate N: number of conformations T: number of rotable bonds I: incremental degrees 10 rotable bonds 30º increments (discrete) plausible conformations! Dihydrofolate reductase with a methotrexate (4dfr.pdb)
47 Search Difficulties If we allow flexibility of one or both of the binding partners, this quickly becomes an intractable problem If we manage to solve/circumvent the problem of flexibility, we then want to be able to screen large databases of structures or drugs, so our troubles are again compounded
48 Spectrum of Search: Breadth and Level-of-Detail Search Breadth Local Molecular Mechanics (MM) Intermediate Monte Carlo Simulated Annealing (MC SA) Brownian Dynamics Molecular Dynamics (MD) Global Docking Level-of-Detail Atom types Bond stretching Bond-angle bending Rotational barrier potentials Implicit solvation Polarizability What s rigid and what s flexible?

49 Two Kinds of Search Systematic Exhaustive, deterministic Outcome is dependent on granularity of sampling Feasible only for lowdimensional problems Stochastic Random, outcome varies Must repeat the search or perform more steps to improve chances of success Feasible for larger problems
50 Stochastic Search Methods Simulated Annealing (SA)* Evolutionary Algorithms (EA) Genetic Algorithm (GA)* Others Tabu Search (TS) Particle Swarm Optimisation (PSO) Hybrid Global-Local Search Methods Lamarckian GA (LGA)* *Supported in AutoDock
51 Monte Carlo Simulated Annealing Also known as Metropolis Monte Carlo The basic steps are 1. The ligand performs a random walk around the protein 2. At each step, a random displacement, rotation, etc. is applied and the energy is evaluated and compared to the previous energy 3. If the new energy is lower, the step is accepted 4. If the energy is higher, the step is accepted with a probability exp( E/kT)
52 Simulated Annealing If we were to perform this at a constant temperature, it would be basic Monte Carlo In this case, after a specified number of steps, the temperature is lowered and the search repeated As the temperature continues to go down, steps which increase the energy become less likely and the system moves to the minimum (or minima)
53 Simulated Annealing Raising the temperature allows the system to cross large barriers and explore the full phase space Lowering the temperature drives the system into a minimum, and hopefully after repeated cycles, the global minimum
54 Genetic Algorithms Genetic algorithms belong to a class of stochastic search methods, but rather than operating on a single solution, they function on a population As implied by the name, you must encode your solution, structure or problem as a genome or chromosome The translation, orientation and conformation of the ligand is encoded (the state variables)
55 Genetic Algorithms The algorithm starts by generating a population of genomes and then applies crossover and mutation to the individuals to create a new population The fitness of each structure has to be evaluated, in our case by our estimate of the binding free energy The best member or members survive to the next generation This procedure is repeated for some number of generations or energy evaluations
56 Crossover and Mutation There are several possible methods of crossover Single Point Crossover Parent A Parent B Offspring + = Two Point Crossover Parent A Parent B Offspring + =
57 Crossover and Mutation Uniform Crossover Parent A Parent B Offspring + = For mutation, selected bits are simply inverted Before mutation After mutation
58 Structure Representation in a Genetic Algorithm Use of a Genetic Algorithm as a sampling method Each conformation is described as a set of rotational angles. 64 possible angles are allowed to each of the bond in the ligand. Each plausible dihedral angle is codified in a set of binary bits (2 6 =64) Each conformation is codified by a so called chromosome with 4 6 bits (0 or 1) Φ 1 Φ Φ 1 = = 58
59 Population of Structures Population (ie, set of chromosomes or configurations) Chromosome Gene
60 Discrete Mutation Operator Single Mutation
61 Crossover Operator Recombination (Crossover)
62 Migration between Populations Migration
63 Efficiency Random mutations are also possible (perhaps the orientation or position is shifted by some random amount) Binary crossover and mutation can introduce inefficiencies into the algorithm since they can easily drive the system away from the region of interest For this reason, genetic searches are often combined with local searches (producing a Lamarckian Genetic Algorithm)
64
65 Local Searches In a Lamarckian GA, the genetic representations of the ligands starting point is replaced with the results of the local search The mapping of the local search back on to the genetic representation is a biologically unrealistic task, but it works well and makes the algorithm more efficient One of the more common local search methods is the Solis and Wets algorithm
66 Solis and Wets Algorithm Starting point x Set bias vector b to 0 Initialize While max iterations not exceeded: Add deviate d to each dimension (from distribution with width ) If new solution is better: else success++ b = 0.4 d b failures++ b = b d If too much success: increase If too many failures: decrease Adaptive step size which biases the search in the direction of success
67 Start LGA Scheme Generate Population Local Search Mutation and Crossover Pick Top Member(s) Final Structure
68 Approaches to Flexibility A relatively simple molecule with 10 rotatable bonds has more than 10 9 possible conformation if we only consider 6 possible positions for each bond Monte Carlo, Simulated Annealing and Genetic Algorithm can help navigate this vast space Other methods have been developed to again circumvent this problem

69 Side Chain Rotamer Libraries Exploring the conformational space of protein side chains is a complex optimization problem that leads to a combinatorial explosion of conformers Protein side chains can be represented adequately by a small set of discrete rotamers Analysis of side-chain Ramachadran plots of pdb structures show that 17 of the 20 amino acids can be represented adequately by 67 side-chain rotamers This approach greatly simpli%ed the problem and enabled sidechain Dexiblity to be tackled
70 Flexibility Some algorithms (call Place & Join algorithms) break the ligand up into pieces, dock the individual pieces, and try and reconnect the bound conformations FlexX uses a library of precomputed, minimized geometries from the Cambridge database with up to 12 minima per bond. Sets of alternative fragments are selected by choosing single or multiple pieces in combination Flexible docking via molecular dynamics with minimization can handle arbitrary flexibility, however it is extremely slow

71 Place-and-Join Algorithm The place-and-join method splits the molecule into rigid subparts Each subpart is docked independently Assembly of the fragments is then done by looking at their relative location and assessing the possibility of re-connecting them with the connectivity of the initial molecule, in geometrically correct conformations

72 Incremental-Based Methods The incremental based approach starts with an initial core docked in the active site, and new fragments are progressively added and minimized; the treatment is terminated when the entire molecule is formed
73 Hybrid Methods Some of the newer methods use a hybrid scheme of a quick docking using a GA or other scheme followed by molecular dynamics to refine the prediction These methods are likely to be some of the best once they are correctly parameterized, etc.
, molecular and mechanics term for internal energy.")
74 GOLD (Genetic Optimisation for Ligand Docking) Performs automated docking with full acyclic ligand flexibility, partial cyclic ligand flexibility and partial protein flexibility in and around active site. Scoring: includes H-bonding term, pairwise dispersion potential (hydrophobic interactions), molecular and mechanics term for internal energy. Analysis shows algorithm more likely to fail if ligand is large or highly flexible, and more likely to succeed if ligand is polar The GA is encoded to search for H-bonding networks first; Fitness function contains a term for dispersive interactions but takes no account of desolvation, thus underestimates The Hydrophobic Effect
75 Scoring Functions
76 Scoring We covered some of the most common representations of the system as well as the most commonly used search methods All of these search methods involve evaluating the fitness or energy of a given binding conformation In order to do this effectively, we must have a good scoring function that can give an accurate estimate of the binding free energy (or relative free energy)
77 Practical Considerations If we have developed an efficient search algorithm, it may produce 10 9 or more potential solutions Many solutions can be immediately eliminated due to atomic clashes or other obvious problems, but we still must evaluate the fitness of a large number of structures For this reason, our scoring function must not only accurate, but it must be fast and efficient
78 Scoring Accuracy If we were scoring a single ligand-protein complex, we could adopt much more sophisticated methods to arrive at an accurate value for the binding free energy Requiring true accuracy in a scoring function is not a realistic expectation, however there are two features that a good scoring function should possess When docking a database of compounds, a good scoring function should Give the best rank to the true bound structure Give the correct relative rank of each ligand in the database And again, it must be able to do these things relatively quickly
79 Types of Scoring Functions There are several types of scoring functions that we have discussed previously First Principles Methods Semiempirical methods Empirical methods Knowledge based potentials All of these are used by various docking programs
80 Clustering No docking algorithm can produce a single, trustworthy structure for the bound complex, but instead they produce an ensemble of predictions Each predicted structure has an associated energy, but we need to consider both the binding free energy (or enthalpy as the case may be) as well as the relative population By clustering our data based on some distance criteria, we can gain some sense of similarity between predictions The distance can be any of a number of similarity measures, but for 3D structures, RMSD is the standard choice
81 Clustering One of the more commonly used methods is hierarchical clustering
82 Agglomerative Clustering In the agglomerative hierarchical clustering method, all structures begin as individual clusters, and the closest clusters are subsequently (iteratively) merged together There are again several different methods of measuring the distance between clusters Simple linkage Complete linkage Group average
83 Linkage Criterion Simple linkage selects the distance as the gives the minimum distance between any two members Complete linkage selects the maximum distance between any two members Group average approaches vary in effectiveness depending on the nature of the data
84 Example: Autodock Autodock uses pre-calculated affinity maps for each atom type in the substrate molecule, usually C, N, O and H, plus an electrostatic map These grids include energetic contributions from all the usual sources ( G = G 1 i,j + G 3 i,j A ij r 12 ij B ij r 6 ij ) ( + G 2 i,j C ij r 12 ij q i q j + G 4 G tor + G 5 ϵ(r)r ij i,j D ij r 10 ij S i V j e r2 ij /2σ2 + E hbond )
85 Each type of atom is placed at each individual grid point and the change in free energy is calculated Grid Maps

86 Grid Maps Precompute interactions for each type of atom 100X faster than pairwise methods Drawbacks: receptor is conformationally rigid, limits the search space
. Number of grid points in each dimension: only give even numbers (from 2 2 2 to 126 126 126). AutoGrid adds one point to each dimension.")
87 Setting up the AutoGrid Box Macromolecule atoms in the rigid part Center: center of ligand; center of macromolecule; a picked atom; or typed-in x-, y- and z-coordinates. Grid point spacing: default is 0.375Å (from 0.2Å to 1.0Å: ). Number of grid points in each dimension: only give even numbers (from to ). AutoGrid adds one point to each dimension. Grid Maps depend on the orientation of the macromolecule.

88 Scoring Functions G binding = G vdw + G elec + G hbond + G desolv + G tors

89 Dispersion/Repulsion G binding = G vdw + G elec + G hbond + G desolv + G tors
 into a softer curve (such as the yellow one) enables them to be less dominant and to simulate the plasticity of the receptor without changing its geometry Fig.")
90 Soft Van der Waals Repulsion Functions When calculated with force %elds from molecular mechanics, steric clashes correspond to high energies Modifying the normal Van der Waals repulsion function (in blue) into a softer curve (such as the yellow one) enables them to be less dominant and to simulate the plasticity of the receptor without changing its geometry Fig. 2: Another approach implemented in AutoDock software

91 Electrostatics and Hydrogen Bonds G binding = G vdw + G elec + G hbond + G desolv + G tors
 Letts. Drug Des. & Disc.")
92 Improved H-bond Directionality Hydrogen affinity AutoGrid 3 Oxygen affinity Guanine Cytosine AutoGrid 4 Guanine Cytosine Huey, Goodsell, Morris, and Olson (2004) Letts. Drug Des. & Disc., 1:

93 Desolvation G binding = G vdw + G elec + G hbond + G desolv + G tors
94 Torsional Entropy G binding = G vdw + G elec + G hbond + G desolv + G tors
95 AutoDock4 Scoring Function Terms ΔG binding = ΔG vdw + ΔG elec + ΔG hbond + ΔG desolv + Δg tors ΔG vdw = ΔG vdw 12-6 Lennard-Jones potential (with 0.5 Å smoothing) ΔG elec with Solmajer & Mehler distance-dependent dielectric ΔG hbond H-bonding Potential with Goodford Directionality ΔG desolv Charge-dependent variant of Stouten Pairwise Atomic Solvation Parameters ΔG tors Number of rotatable bonds
96 AutoDock Empirical Free Energy Force Field W vdw W hbond A ij 12 r B ij 6 ij r + i, j ij E(t) C ij 12 r D ij 10 ij r + i, j ij Physics-based approach from molecular mechanics Calibrated with 188 complexes from LPDB, K i s from PDB-Bind Standard error = 2.52 kcal/mol W elec W sol q i q j ε(r ij )r + ij i, j i, j W tor N tor ( S i V j + S j V i ) e ( r ij 2 /2σ 2 ) +
97 Scoring Function in AutoDock 4 A V = W ij vdw 12 r B ij +W 6 hbond E(t) C ij 12 i, j ij r ij r D ij +W q i q j 10 elec i, j ij r ij i, j ε(r ij )r +W S V + S V sol i j j i ij i, j ( ) e ( r ij 2 /2σ 2 ) Desolvation includes terms for all atom types Favorable term for C, A (aliphatic and aromatic carbons) Unfavorable term for O, N Proportional to the absolute value of the charge on the atom Computes the intramolecular desolvation energy for moving atoms Calibrated with 188 complexes from LPDB, K i s from PDB-Bind Standard error (in Kcal/mol): 2.62 (extended) 2.72 (compact) 2.52 (bound) 2.63 (AutoDock 3, bound) Improved H-bond directionality
98 AutoDock Vina Scoring Function Combination of knowledge-based and empirical approach G binding = G gauss + G repulsion + G hbond + G hydrophobic + G tors G gauss Attractive term for dispersion, two gaussian functions G repulsion Square of the distance if closer than a threshold value G hbond Ramp function - also used for interactions with metal ions G hydrophobic Ramp function G tors Proportional to the number of rotatable bonds Calibrated with 1,300 complexes from PDB-Bind Standard error = 2.85 kcal/mol
99 Other Energetic Notes Also includes torsional energy (if the ligand is flexible) Entropic contributions due to rotatable bonds Desolvation is calculated, but only for heavy atoms on the marcromolecule
100 Search Methods Autodock has three search methods Monte Carlo simulated annealing A global genetic algorithm search A local Solis and Wets search Combining the last two search methods gives the Lamarckian Genetic Algorithm which is what we typically use
101 AutoDock and Vina Search Methods Global search algorithms: Simulated Annealing (Goodsell et al. 1990) Genetic Algorithm (Morris et al. 1998) Local search algorithm: Solis & Wets (Morris et al. 1998) Hybrid global-local search algorithm: Lamarckian GA (Morris et al. 1998) Iterated Local Search: Genetic Algorithm with Local Gradient Optimization (Trott and Olson 2010)
102 Methodology Require structures for both ligand and macromolecule Add charges to both structures (create a pdbq file) For proteins or polypeptides we use the Kollman united atom model For drugs and other molecule, we use Gasteiger charges
103 Methodology For the macromolecule, solvation needs to be calculated (producing a pdbqs file) Based on the atom types in the ligand, the appropriate maps need to be calculated for the macromolecule Flexible bonds (if any) need to be assigned to the ligand
104 Practical Considerations What problems are feasible? Depends on the search method: Vina > LGA > GA >> SA >> LS AutoDock SA : can output trajectories, D < 8 torsions. AutoDock LGA : D < 8-16 torsions. Vina : good for torsions. When are AutoDock and Vina not suitable? Modeled structure of poor quality; Too many torsions (32 max); Target protein too flexible. Redocking studies are used to validate the method
105 Using AutoDock: Step-by-Step Set up ligand PDBQT using ADT s Ligand menu OPTIONAL: Set up flexible receptor PDBQT using ADT s Flexible Residues menu Set up macromolecule & grid maps using ADT s Grid menu Pre-compute AutoGrid maps for all atom types in your set of ligands using autogrid4 Perform dockings of ligand to target using autodock4, and in parallel if possible. Visualize AutoDock results using ADT s Analyze menu Cluster dockings using analysis DPF command in autodock4 or ADT s Analyze menu for parallel docking results.
106 AutoDock 4 File Formats Prepare the Following Input Files Ligand PDBQT file Rigid Macromolecule PDBQT file Flexible Macromolecule PDBQT file ( Flexres ) AutoGrid Parameter File (GPF) GPF depends on atom types in: Ligand PDBQT file Optional flexible residue PDBQT files) AutoDock Parameter File (DPF) Run AutoGrid 4 Macromolecule PDBQT + GPF Grid Maps, GLG Run AutoDock 4 Grid Maps + Ligand PDBQT [+ Flexres PDBQT] + DPF DLG (dockings & clustering)
107 Things you need to do before using Ligand: AutoDock 4 Add all hydrogens, compute Gasteiger charges, and merge non-polar H; also assign AutoDock 4 atom types Ensure total charge corresponds to tautomeric state Choose torsion tree root & rotatable bonds Macromolecule: Add all hydrogens (PDB2PQR flips Asn, Gln, His), compute Gasteiger charges, and merge non-polar H; also assign AutoDock 4 atom types Assign Stouten atomic solvation parameters Optionally, create a flexible residues PDBQT in addition to the rigid PDBQT file Compute AutoGrid maps
108 Preparing Ligands and Receptors AutoDock uses United Atom model Reduces number of atoms, speeds up docking Need to: Add polar Hs. Remove non-polar Hs. Both Ligand & Macromolecule Replace missing atoms (disorder). Fix hydrogens at chain breaks. Need to consider ph: Acidic & Basic residues, Histidines. Other molecules in receptor: Waters; Cofactors; Metal ions. Molecular Modelling Using AutoDock elsewhere. 4 with ADT
109 Atom Types in AutoDock 4 One-letter or two-letter atom type codes More atom types than AD3: 22 Same atom types in both ligand and receptor how-do-i-add-new-atom-types-to-autodock-4 where-do-i-set-the-autodock-4-force-field-parameters
110 Partial Atomic Charges are required for both Ligand and Receptor Partial Atomic Charges: Peptides & Proteins; DNA & RNA Gasteiger (PEOE) - AD4 Force Field Organic compounds; Cofactors Gasteiger (PEOE) - AD4 Force Field; MOPAC (MNDO, AM1, PM3); Gaussian (6-31G*). Integer total charge per residue. Non-polar hydrogens: Always merge
111 Carbon Atoms can be either Aliphatic or Aromatic Atom Types Solvation Free Energy Based on a partial-charge-dependent variant of Stouten method. Treats aliphatic ( C ) and aromatic ( A ) carbons differently. Need to rename ligand aromatic C to A. ADT determines if ligand is a peptide: If so, uses a look-up dictionary. If not, inspects geometry of C s in rings. Renames C to A if flat enough. Can adjust planarity criterion (15 detects more rings than default 7.5 ).
112 Defining Ligand Flexibility Set Root of Torsion Tree: By interactively picking, or Automatically. Smallest largest sub-tree. Interactively Pick Rotatable Bonds: No leaves ; No bonds in rings; Can freeze: Peptide/amide/selected/all; Can set the number of active torsions that move either the most or the fewest atoms
113 Setting Up Your Environment At TSRI: Modify.cshrc Change PATH & stacksize: setenv PATH (/mgl/prog/$archosv/bin:/tsri/python:$path) % limit stacksize unlimited ADT Tutorial, every time you open a Shell or Terminal, type: % source /tsri/python/share/bin/initadtcsh To start AutoDockTools (ADT), type: Web % cd tutorial % adt1
114 Choose the Docking Algorithm SA.dpf Simulated Annealing GA.dpf Genetic Algorithm LS.dpf Local Search Solis-Wets (SW) Pseudo Solis-Wets (psw) GALS.dpf Genetic Algorithm with Local Search, i.e. Lamarckian GA
115 Run AutoGrid Check: Enough disk space? Maps are ASCII, but can be ~2-8MB! Start AutoGrid from the Shell: % autogrid4 p mol.gpf l mol.glg & % autogrid4 -p mol.gpf -l mol.glg ; autodock4 -p mol.dpf -l mol.dlg Follow the log file using: % tail -f mol.glg Type <Ctrl>-C to break out of the tail -f command Wait for Successful Completion before starting AutoDock
116 Run AutoDock Do a test docking, ~ 25,000 evals Do a full docking, if test is OK, ~ 250,000 to 50,000,000 evals From the Shell: % autodock4 p yourfile.dpf l yourfile.dlg & Expected time? Size of docking log? Distributed computation At TSRI, Linux Clusters % submit.py stem 20 % recluster.py stem 20 during 3.5
117 Analyzing AutoDock Results In ADT, you can: Read & view a single DLG, or Read & view many DLG results files in a single directory Re-cluster docking results by conformation & view these Outside ADT, you can re-cluster several DLGs Useful in distributed docking % recluster.py stem 20 [during end] 3.5
118 Viewing Conformational Clusters by RMSD List the RMSD tolerances Separated by spaces Histogram of conformational clusters Number in cluster versus lowest energy in that cluster Picking a cluster makes a list of the conformations in that cluster; set these to be the current sequence for states player.
Using AutoDock With AutoDockTools: A Tutorial
 Using AutoDock With AutoDockTools: A Tutorial Dr. Ruth Huey & Dr. Garrett M. Morris 6/6/06 AutoDock & ADT Tutorial 1 What is Docking? Best ways to put two molecules together. Three steps: (1) Obtain the
Using AutoDock With AutoDockTools: A Tutorial Dr. Ruth Huey & Dr. Garrett M. Morris 6/6/06 AutoDock & ADT Tutorial 1 What is Docking? Best ways to put two molecules together. Three steps: (1) Obtain the
Using AutoDock 4 with ADT: A Tutorial
 Using AutoDock 4 with ADT: A Tutorial Ruth Huey Sargis Dallakyan Alex Perryman David S. Goodsell (Garrett Morris) 9/2/08 Using AutoDock 4 with ADT 1 What is Docking? Predicting the best ways two molecules
Using AutoDock 4 with ADT: A Tutorial Ruth Huey Sargis Dallakyan Alex Perryman David S. Goodsell (Garrett Morris) 9/2/08 Using AutoDock 4 with ADT 1 What is Docking? Predicting the best ways two molecules
Using AutoDock 4 with ADT: A Tutorial
 Using AutoDock 4 with ADT: A Tutorial Dr. Garrett M. Morris 1 What is Docking? A computational procedure for predicting the best way(s) two molecules will interact in 3D. (1) Obtain the 3D structures of
Using AutoDock 4 with ADT: A Tutorial Dr. Garrett M. Morris 1 What is Docking? A computational procedure for predicting the best way(s) two molecules will interact in 3D. (1) Obtain the 3D structures of
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Softwares for Molecular Docking. Lokesh P. Tripathi NCBS 17 December 2007
 Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Softwares for Molecular Docking Lokesh P. Tripathi NCBS 17 December 2007 Molecular Docking Attempt to predict structures of an intermolecular complex between two or more molecules Receptor-ligand (or drug)
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
BCB410 Protein-Ligand Docking Exercise Set Shirin Shahsavand December 11, 2011
 BCB410 Protein-Ligand Docking Exercise Set Shirin Shahsavand December 11, 2011 1. Describe the search algorithm(s) AutoDock uses for solving protein-ligand docking problems. AutoDock uses 3 different approaches
BCB410 Protein-Ligand Docking Exercise Set Shirin Shahsavand December 11, 2011 1. Describe the search algorithm(s) AutoDock uses for solving protein-ligand docking problems. AutoDock uses 3 different approaches
Protein-Ligand Docking Methods
 Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Kd = koff/kon = [R][L]/[RL]
![Kd = koff/kon = [R][L]/[RL]](/thumbs/96/127564193.jpg "Kd = koff/kon = [R][L]/[RL]") Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Taller de docking y cribado virtual: Uso de herramientas computacionales en el diseño de fármacos Docking program GLIDE El programa de docking GLIDE Sonsoles Martín-Santamaría Shrödinger is a scientific
Ranking of HIV-protease inhibitors using AutoDock
 Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Ranking of HIV-protease inhibitors using AutoDock 1. Task Calculate possible binding modes and estimate the binding free energies for 1 3 inhibitors of HIV-protease. You will learn: Some of the theory
Protein-Ligand Docking
 Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
Protein-Ligand Docking Matthias Rarey GMD - German National Research Center for Information Technology Institute for Algorithms and Scientific Computing (SCAI) 53754Sankt Augustin, Germany rarey@gmd.de
What is Protein-Ligand Docking?
 MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
MOLECULAR DOCKING Definition: What is Protein-Ligand Docking? Computationally predict the structures of protein-ligand complexes from their conformations and orientations. The orientation that maximizes
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Protein-Ligand Docking Methods
 Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
Review Goal: Given a protein structure, predict its ligand bindings Protein-Ligand Docking Methods Applications: Function prediction Drug discovery etc. Thomas Funkhouser Princeton University S597A, Fall
MOLECULAR RECOGNITION DOCKING ALGORITHMS. Natasja Brooijmans 1 and Irwin D. Kuntz 2
 Annu. Rev. Biophys. Biomol. Struct. 2003. 32:335 73 doi: 10.1146/annurev.biophys.32.110601.142532 Copyright c 2003 by Annual Reviews. All rights reserved First published online as a Review in Advance on
Annu. Rev. Biophys. Biomol. Struct. 2003. 32:335 73 doi: 10.1146/annurev.biophys.32.110601.142532 Copyright c 2003 by Annual Reviews. All rights reserved First published online as a Review in Advance on
Protein Structure Prediction and Protein-Ligand Docking
 Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Dr. Sander B. Nabuurs. Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre
 Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Dr. Sander B. Nabuurs Computational Drug Discovery group Center for Molecular and Biomolecular Informatics Radboud University Medical Centre The road to new drugs. How to find new hits? High Throughput
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes Introduction The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes
 Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Introduction Chemical properties that affect binding of enzyme-inhibiting drugs to enzymes The production of new drugs requires time for development and testing, and can result in large prohibitive costs
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
User Guide for LeDock
 User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
User Guide for LeDock Hongtao Zhao, PhD Email: htzhao@lephar.com Website: www.lephar.com Copyright 2017 Hongtao Zhao. All rights reserved. Introduction LeDock is flexible small-molecule docking software,
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
DOCKING TUTORIAL. A. The docking Workflow
 2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
2 nd Strasbourg Summer School on Chemoinformatics VVF Obernai, France, 20-24 June 2010 E. Kellenberger DOCKING TUTORIAL A. The docking Workflow 1. Ligand preparation It consists in the standardization
Introduction to AutoDock and AutoDock Tools
 Introduction to AutoDock and AutoDock Tools Alexander B. Pacheco User Services Consultant LSU HPC & LONI sys-help@loni.org HPC Training Series Louisiana State University Baton Rouge Mar. 28, 2012 HPC@LSU
Introduction to AutoDock and AutoDock Tools Alexander B. Pacheco User Services Consultant LSU HPC & LONI sys-help@loni.org HPC Training Series Louisiana State University Baton Rouge Mar. 28, 2012 HPC@LSU
Conformational Searching using MacroModel and ConfGen. John Shelley Schrödinger Fellow
 Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
Conformational Searching using MacroModel and ConfGen John Shelley Schrödinger Fellow Overview Types of conformational searching applications MacroModel s conformation generation procedure General features
PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS
 TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation
 OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation Gareth Price Computational MiniProject OpenDiscovery Aims + Achievements Produce a high-throughput protocol to screen a
OpenDiscovery: Automated Docking of Ligands to Proteins and Molecular Simulation Gareth Price Computational MiniProject OpenDiscovery Aims + Achievements Produce a high-throughput protocol to screen a
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Why Proteins Fold. How Proteins Fold? e - ΔG/kT. Protein Folding, Nonbonding Forces, and Free Energy
 Why Proteins Fold Proteins are the action superheroes of the body. As enzymes, they make reactions go a million times faster. As versatile transport vehicles, they carry oxygen and antibodies to fight
Why Proteins Fold Proteins are the action superheroes of the body. As enzymes, they make reactions go a million times faster. As versatile transport vehicles, they carry oxygen and antibodies to fight
Computational protein design
 Computational protein design There are astronomically large number of amino acid sequences that needs to be considered for a protein of moderate size e.g. if mutating 10 residues, 20^10 = 10 trillion sequences
Computational protein design There are astronomically large number of amino acid sequences that needs to be considered for a protein of moderate size e.g. if mutating 10 residues, 20^10 = 10 trillion sequences
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation
 Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
CS 273 Prof. Serafim Batzoglou Prof. Jean-Claude Latombe Spring Lecture 12 : Energy maintenance (1) Lecturer: Prof. J.C.
 Lecturer: Prof. J.C.") CS 273 Prof. Serafim Batzoglou Prof. Jean-Claude Latombe Spring 2006 Lecture 12 : Energy maintenance (1) Lecturer: Prof. J.C. Latombe Scribe: Neda Nategh How do you update the energy function during the
CS 273 Prof. Serafim Batzoglou Prof. Jean-Claude Latombe Spring 2006 Lecture 12 : Energy maintenance (1) Lecturer: Prof. J.C. Latombe Scribe: Neda Nategh How do you update the energy function during the
Flexibility and Constraints in GOLD
 Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Flexibility and Constraints in GOLD Version 2.1 August 2018 GOLD v5.6.3 Table of Contents Purpose of Docking... 3 GOLD s Evolutionary Algorithm... 4 GOLD and Hermes... 4 Handling Flexibility and Constraints
Autodock tutorial VINA with UCSF Chimera
 Autodock tutorial VINA with UCSF Chimera José R. Valverde CNB/CSIC jrvalverde@cnb.csic.es José R. Valverde, 2014 CC-BY-NC-SA Loading the receptor Open UCSF Chimera and then load the protein: File Open
Autodock tutorial VINA with UCSF Chimera José R. Valverde CNB/CSIC jrvalverde@cnb.csic.es José R. Valverde, 2014 CC-BY-NC-SA Loading the receptor Open UCSF Chimera and then load the protein: File Open
Receptor Based Drug Design (1)
") Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Induced Fit Model For more than 100 years, the behaviour of enzymes had been explained by the "lock-and-key" mechanism developed by pioneering German chemist Emil Fischer. Fischer thought that the chemicals
Crystal Structure Prediction using CRYSTALG program
 Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Structural Bioinformatics (C3210) Molecular Mechanics
 Molecular Mechanics") Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Structural Bioinformatics (C3210) Molecular Mechanics How to Calculate Energies Calculation of molecular energies is of key importance in protein folding, molecular modelling etc. There are two main computational
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
Protein-Ligand Docking Evaluations
 Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
Introduction Protein-Ligand Docking Evaluations Protein-ligand docking: Given a protein and a ligand, determine the pose(s) and conformation(s) minimizing the total energy of the protein-ligand complex
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015,
 Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Abstract. Introduction
 In silico protein design: the implementation of Dead-End Elimination algorithm CS 273 Spring 2005: Project Report Tyrone Anderson 2, Yu Bai1 3, and Caroline E. Moore-Kochlacs 2 1 Biophysics program, 2
In silico protein design: the implementation of Dead-End Elimination algorithm CS 273 Spring 2005: Project Report Tyrone Anderson 2, Yu Bai1 3, and Caroline E. Moore-Kochlacs 2 1 Biophysics program, 2
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
Introduction to Structure Preparation and Visualization
 Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
ONETEP PB/SA: Application to G-Quadruplex DNA Stability. Danny Cole
 ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
ONETEP PB/SA: Application to G-Quadruplex DNA Stability Danny Cole Introduction Historical overview of structure and free energy calculation of complex molecules using molecular mechanics and continuum
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Towards Physics-based Models for ADME/Tox. Tyler Day
 Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Towards Physics-based Models for ADME/Tox Tyler Day Overview Motivation Application: P450 Site of Metabolism Application: Membrane Permeability Future Directions and Applications Motivation Advantages
Generating Small Molecule Conformations from Structural Data
 Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Generating Small Molecule Conformations from Structural Data Jason Cole cole@ccdc.cam.ac.uk Cambridge Crystallographic Data Centre 1 The Cambridge Crystallographic Data Centre About us A not-for-profit,
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Docking with Water in the Binding Site using GOLD
 Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
Docking with Water in the Binding Site using GOLD Version 2.0 November 2017 GOLD v5.6 Table of Contents Docking with Water in the Binding Site... 2 Case Study... 3 Introduction... 3 Provided Input Files...
CHAPTER-5 MOLECULAR DOCKING STUDIES
 CHAPTER-5 MOLECULAR DOCKING STUDIES 156 CHAPTER -5 MOLECULAR DOCKING STUDIES 5. Molecular docking studies This chapter discusses about the molecular docking studies of the synthesized compounds with different
CHAPTER-5 MOLECULAR DOCKING STUDIES 156 CHAPTER -5 MOLECULAR DOCKING STUDIES 5. Molecular docking studies This chapter discusses about the molecular docking studies of the synthesized compounds with different
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
Virtual screening for drug discovery. Markus Lill Purdue University
 Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Virtual screening for drug discovery Markus Lill Purdue University mlill@purdue.edu Lecture material http://people.pharmacy.purdue.edu/~mlill/teaching/eidelberg/ I.1 Drug discovery Cl N Disease I.1 Drug
Using AutoDock for Virtual Screening
 Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
Using AutoDock for Virtual Screening CUHK Croucher ASI Workshop 2011 Stefano Forli, PhD Prof. Arthur J. Olson, Ph.D Molecular Graphics Lab Screening and Virtual Screening The ultimate tool for identifying
NIH Public Access Author Manuscript J Comput Chem. Author manuscript; available in PMC 2011 February 18.
 NIH Public Access Author Manuscript Published in final edited form as: J Comput Chem. 2010 January 30; 31(2): 455 461. doi:10.1002/jcc.21334. AutoDock Vina: improving the speed and accuracy of docking
NIH Public Access Author Manuscript Published in final edited form as: J Comput Chem. 2010 January 30; 31(2): 455 461. doi:10.1002/jcc.21334. AutoDock Vina: improving the speed and accuracy of docking
Computational modeling of G-Protein Coupled Receptors (GPCRs) has recently become
 has recently become") Homology Modeling and Docking of Melatonin Receptors Andrew Kohlway, UMBC Jeffry D. Madura, Duquesne University 6/18/04 INTRODUCTION Computational modeling of G-Protein Coupled Receptors (GPCRs) has recently
Homology Modeling and Docking of Melatonin Receptors Andrew Kohlway, UMBC Jeffry D. Madura, Duquesne University 6/18/04 INTRODUCTION Computational modeling of G-Protein Coupled Receptors (GPCRs) has recently
Other Cells. Hormones. Viruses. Toxins. Cell. Bacteria
 Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Other Cells Hormones Viruses Toxins Cell Bacteria ΔH < 0 reaction is exothermic, tells us nothing about the spontaneity of the reaction Δ H > 0 reaction is endothermic, tells us nothing about the spontaneity
Protein Docking by Exploiting Multi-dimensional Energy Funnels
 Protein Docking by Exploiting Multi-dimensional Energy Funnels Ioannis Ch. Paschalidis 1 Yang Shen 1 Pirooz Vakili 1 Sandor Vajda 2 1 Center for Information and Systems Engineering and Department of Manufacturing
Protein Docking by Exploiting Multi-dimensional Energy Funnels Ioannis Ch. Paschalidis 1 Yang Shen 1 Pirooz Vakili 1 Sandor Vajda 2 1 Center for Information and Systems Engineering and Department of Manufacturing
Assignment 2 Atomic-Level Molecular Modeling
 Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
Alchemical free energy calculations in OpenMM
 Alchemical free energy calculations in OpenMM Lee-Ping Wang Stanford Department of Chemistry OpenMM Workshop, Stanford University September 7, 2012 Special thanks to: John Chodera, Morgan Lawrenz Outline
Alchemical free energy calculations in OpenMM Lee-Ping Wang Stanford Department of Chemistry OpenMM Workshop, Stanford University September 7, 2012 Special thanks to: John Chodera, Morgan Lawrenz Outline
schematic diagram; EGF binding, dimerization, phosphorylation, Grb2 binding, etc.
 Lecture 1: Noncovalent Biomolecular Interactions Bioengineering and Modeling of biological processes -e.g. tissue engineering, cancer, autoimmune disease Example: RTK signaling, e.g. EGFR Growth responses
Lecture 1: Noncovalent Biomolecular Interactions Bioengineering and Modeling of biological processes -e.g. tissue engineering, cancer, autoimmune disease Example: RTK signaling, e.g. EGFR Growth responses
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Analyzing Molecular Conformations Using the Cambridge Structural Database. Jason Cole Cambridge Crystallographic Data Centre
 Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Analyzing Molecular Conformations Using the Cambridge Structural Database Jason Cole Cambridge Crystallographic Data Centre 1 The Cambridge Structural Database (CSD) 905,284* USOPEZ a natural product intermediate,
Homework Problem Set 4 Solutions
 Chemistry 380.37 Dr. Jean M. Standard omework Problem Set 4 Solutions 1. A conformation search is carried out on a system and four low energy stable conformers are obtained. Using the MMFF force field,
Chemistry 380.37 Dr. Jean M. Standard omework Problem Set 4 Solutions 1. A conformation search is carried out on a system and four low energy stable conformers are obtained. Using the MMFF force field,
Build_model v User Guide
 Build_model v.2.0.1 User Guide MolTech Build_model User Guide 2008-2011 Molecular Technologies Ltd. www.moltech.ru Please send your comments and suggestions to contact@moltech.ru. Table of Contents Input
Build_model v.2.0.1 User Guide MolTech Build_model User Guide 2008-2011 Molecular Technologies Ltd. www.moltech.ru Please send your comments and suggestions to contact@moltech.ru. Table of Contents Input
Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions Van der Waals Interactions
Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions Van der Waals Interactions
1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?!
 for structure refinement from X-ray diffraction data?!") 1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?! Visual analysis: Identification of ligand density from
1.b What are current best practices for selecting an initial target ligand atomic model(s) for structure refinement from X-ray diffraction data?! Visual analysis: Identification of ligand density from
Using AutoDock with AutoDockTools: A Tutorial
 Using AutoDock with AutoDockTools: A Tutorial Written by Ruth Huey and Garrett M. Morris The Scripps Resea rch Institute Molecula r Graphics Labora tory 10550 N. Torrey Pines Rd. La Jolla, California 92037-1000
Using AutoDock with AutoDockTools: A Tutorial Written by Ruth Huey and Garrett M. Morris The Scripps Resea rch Institute Molecula r Graphics Labora tory 10550 N. Torrey Pines Rd. La Jolla, California 92037-1000
Tutorial: Structural Analysis of a Protein-Protein Complex
 Molecular Modeling Section (MMS) Department of Pharmaceutical and Pharmacological Sciences University of Padova Via Marzolo 5-35131 Padova (IT) @contact: stefano.moro@unipd.it Tutorial: Structural Analysis
Molecular Modeling Section (MMS) Department of Pharmaceutical and Pharmacological Sciences University of Padova Via Marzolo 5-35131 Padova (IT) @contact: stefano.moro@unipd.it Tutorial: Structural Analysis
Computational Molecular Modeling
 Computational Molecular Modeling Lecture 1: Structure Models, Properties Chandrajit Bajaj Today s Outline Intro to atoms, bonds, structure, biomolecules, Geometry of Proteins, Nucleic Acids, Ribosomes,
Computational Molecular Modeling Lecture 1: Structure Models, Properties Chandrajit Bajaj Today s Outline Intro to atoms, bonds, structure, biomolecules, Geometry of Proteins, Nucleic Acids, Ribosomes,
Fondamenti di Chimica Farmaceutica. Computer Chemistry in Drug Research: Introduction
 Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Fondamenti di Chimica Farmaceutica Computer Chemistry in Drug Research: Introduction Introduction Introduction Introduction Computer Chemistry in Drug Design Drug Discovery: Target identification Lead
Cheminformatics platform for drug discovery application
 EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
EGI-InSPIRE Cheminformatics platform for drug discovery application Hsi-Kai, Wang Academic Sinica Grid Computing EGI User Forum, 13, April, 2011 1 Introduction to drug discovery Computing requirement of
Exercise 2: Solvating the Structure Before you continue, follow these steps: Setting up Periodic Boundary Conditions
 Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Exercise 2: Solvating the Structure HyperChem lets you place a molecular system in a periodic box of water molecules to simulate behavior in aqueous solution, as in a biological system. In this exercise,
Portal. User Guide Version 1.0. Contributors
 Portal www.dockthor.lncc.br User Guide Version 1.0 Contributors Diogo A. Marinho, Isabella A. Guedes, Eduardo Krempser, Camila S. de Magalhães, Hélio J. C. Barbosa and Laurent E. Dardenne www.gmmsb.lncc.br
Portal www.dockthor.lncc.br User Guide Version 1.0 Contributors Diogo A. Marinho, Isabella A. Guedes, Eduardo Krempser, Camila S. de Magalhães, Hélio J. C. Barbosa and Laurent E. Dardenne www.gmmsb.lncc.br
Atomic and molecular interaction forces in biology
 Atomic and molecular interaction forces in biology 1 Outline Types of interactions relevant to biology Van der Waals interactions H-bond interactions Some properties of water Hydrophobic effect 2 Types
Atomic and molecular interaction forces in biology 1 Outline Types of interactions relevant to biology Van der Waals interactions H-bond interactions Some properties of water Hydrophobic effect 2 Types
High Throughput In-Silico Screening Against Flexible Protein Receptors
 John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
John von Neumann Institute for Computing High Throughput In-Silico Screening Against Flexible Protein Receptors H. Sánchez, B. Fischer, H. Merlitz, W. Wenzel published in From Computational Biophysics
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Length Scales (c) ICAMS: http://www.icams.de/cms/upload/01_home/01_research_at_icams/length_scales_1024x780.png Goals for today: Background
André Schleife Department of Materials Science and Engineering Length Scales (c) ICAMS: http://www.icams.de/cms/upload/01_home/01_research_at_icams/length_scales_1024x780.png Goals for today: Background
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
5.1. Hardwares, Softwares and Web server used in Molecular modeling
 5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
5. EXPERIMENTAL The tools, techniques and procedures/methods used for carrying out research work reported in this thesis have been described as follows: 5.1. Hardwares, Softwares and Web server used in
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Schrodinger ebootcamp #3, Summer EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist
 Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Schrodinger ebootcamp #3, Summer 2016 EXPLORING METHODS FOR CONFORMER SEARCHING Jas Bhachoo, Senior Applications Scientist Numerous applications Generating conformations MM Agenda http://www.schrodinger.com/macromodel
Context of the project...3. What is protein design?...3. I The algorithms...3 A Dead-end elimination procedure...4. B Monte-Carlo simulation...
 Laidebeure Stéphane Context of the project...3 What is protein design?...3 I The algorithms...3 A Dead-end elimination procedure...4 B Monte-Carlo simulation...5 II The model...6 A The molecular model...6
Laidebeure Stéphane Context of the project...3 What is protein design?...3 I The algorithms...3 A Dead-end elimination procedure...4 B Monte-Carlo simulation...5 II The model...6 A The molecular model...6
Biophysics II. Hydrophobic Bio-molecules. Key points to be covered. Molecular Interactions in Bio-molecular Structures - van der Waals Interaction
 Biophysics II Key points to be covered By A/Prof. Xiang Yang Liu Biophysics & Micro/nanostructures Lab Department of Physics, NUS 1. van der Waals Interaction 2. Hydrogen bond 3. Hydrophilic vs hydrophobic
Biophysics II Key points to be covered By A/Prof. Xiang Yang Liu Biophysics & Micro/nanostructures Lab Department of Physics, NUS 1. van der Waals Interaction 2. Hydrogen bond 3. Hydrophilic vs hydrophobic
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
The PhilOEsophy. There are only two fundamental molecular descriptors
 The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
The PhilOEsophy There are only two fundamental molecular descriptors Where can we use shape? Virtual screening More effective than 2D Lead-hopping Shape analogues are not graph analogues Molecular alignment
Preparing a PDB File
 Figure 1: Schematic view of the ligand-binding domain from the vitamin D receptor (PDB file 1IE9). The crystallographic waters are shown as small spheres and the bound ligand is shown as a CPK model. HO
Figure 1: Schematic view of the ligand-binding domain from the vitamin D receptor (PDB file 1IE9). The crystallographic waters are shown as small spheres and the bound ligand is shown as a CPK model. HO
Using AutoDock 4 and Vina with AutoDockTools: A Tutorial
 Using AutoDock 4 and Vina with AutoDockTools: A Tutorial Written by Ruth Huey, Garrett M. Morris and Stefano Forli The Scripps Research Institute Molecular Graphics Laboratory 10550 N. Torrey Pines Rd.
Using AutoDock 4 and Vina with AutoDockTools: A Tutorial Written by Ruth Huey, Garrett M. Morris and Stefano Forli The Scripps Research Institute Molecular Graphics Laboratory 10550 N. Torrey Pines Rd.
Protein Structure. W. M. Grogan, Ph.D. OBJECTIVES
 Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Molecular Mechanics. Yohann Moreau. November 26, 2015
 Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
Molecular Mechanics Yohann Moreau yohann.moreau@ujf-grenoble.fr November 26, 2015 Yohann Moreau (UJF) Molecular Mechanics, Label RFCT 2015 November 26, 2015 1 / 29 Introduction A so-called Force-Field
BUDE. A General Purpose Molecular Docking Program Using OpenCL. Richard B Sessions
 BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
BUDE A General Purpose Molecular Docking Program Using OpenCL Richard B Sessions 1 The molecular docking problem receptor ligand Proteins typically O(1000) atoms Ligands typically O(100) atoms predicted
April, The energy functions include:
 REDUX A collection of Python scripts for torsion angle Monte Carlo protein molecular simulations and analysis The program is based on unified residue peptide model and is designed for more efficient exploration
REDUX A collection of Python scripts for torsion angle Monte Carlo protein molecular simulations and analysis The program is based on unified residue peptide model and is designed for more efficient exploration
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University
 Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites. J. Andrew Surface
 Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Using Bayesian Statistics to Predict Water Affinity and Behavior in Protein Binding Sites Introduction J. Andrew Surface Hampden-Sydney College / Virginia Commonwealth University In the past several decades
Self-Organizing Superimposition Algorithm for Conformational Sampling
 Self-Organizing Superimposition Algorithm for Conformational Sampling FANGQIANG ZHU, DIMITRIS K. AGRAFIOTIS Johnson & Johnson Pharmaceutical Research & Development, L.L.C., 665 Stockton Drive, Exton, Pennsylvania
Self-Organizing Superimposition Algorithm for Conformational Sampling FANGQIANG ZHU, DIMITRIS K. AGRAFIOTIS Johnson & Johnson Pharmaceutical Research & Development, L.L.C., 665 Stockton Drive, Exton, Pennsylvania
Protein Folding & Stability. Lecture 11: Margaret A. Daugherty. Fall How do we go from an unfolded polypeptide chain to a
 Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2004 How do we go from an unfolded polypeptide chain to a compact folded protein? (Folding of thioredoxin, F. Richards) Structure - Function
Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2004 How do we go from an unfolded polypeptide chain to a compact folded protein? (Folding of thioredoxin, F. Richards) Structure - Function
Performing a Pharmacophore Search using CSD-CrossMiner
 Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Table of Contents Introduction... 2 CSD-CrossMiner Terminology... 2 Overview of CSD-CrossMiner... 3 Searching with a Pharmacophore... 4 Performing a Pharmacophore Search using CSD-CrossMiner Version 2.0
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
The protein folding problem consists of two parts:
 Energetics and kinetics of protein folding The protein folding problem consists of two parts: 1)Creating a stable, well-defined structure that is significantly more stable than all other possible structures.
Energetics and kinetics of protein folding The protein folding problem consists of two parts: 1)Creating a stable, well-defined structure that is significantly more stable than all other possible structures.