Charge equilibration
|
|
|
- Esther Burns
- 5 years ago
- Views:
Transcription
1 Charge equilibration Taylor expansion of energy of atom E A (Q) =E A0 + Q A + A 0 2 Q2 2 2 A The corresponding energy of cation/anion and neutral atom E A (+1) = E E A ( 1) = + 1 A 0 2 A A 0 A 0 E A (0) = E A0
2 Charge equilibration Ionization potential IP = E A (+1) E A (0) = Electron + A 0 1 EA = E A (0) E A ( 1) A 0 2 (IP + 2 E IP EA 2 =? A 0 A 0 = A 0 A 0 electronegativity: ability of atom to gain or loose an electron
3 Charge equilibration How about the 2nd derivative? IP EA 2 2 A 0 =? Consider simple scheme of a single molecular orbital -1 Neutral +1 E HF =2h A + J AA E HF = h A E HF =0
4 Charge equilibration HF energies -1 Neutral +1 E HF =2h A + J AA E HF = h A E HF =0 IP & EA IP = h A EA = h A J 2 E Coulomb repulsion between IP EA 2 = J AA 2 electrons in orbital A 0
5 Charge equilibration Back to the energy of atom A E A (Q) =E A0 + 0 AQ A J 0 AAQ 2 A Total electrostatic energy E(Q 1,Q 2,...Q N )= X A E A + X A<B Q A Q B J AB Chemical potential A = 0 A + Q A J 0 AA + X A<B Q B J AB
6 Charge equilibration Chemical potential Equilibrium A = 0 A + Q A J 0 AA + X A<B Q B J AB A = B =... N-1 equations Total charge Q tot = NX A Q A
7 Charge equilibration Chemical potential A = Three parameters: 0 A + Q A J 0 AA + X A<B Mulliken-Pauling electronegativity JAA 0 1 RA 0 Atom size 0 A = 1 2 J 0 AA Parr and Pearson hardness 0, 0 and 0 Q B J AB Inverse separation between atom A-B shielding constant for JAB
8 Charge equilibration Calculation of partial charge from atomic structure and chemical parameters SCF: expensive in computer time Not long range because need a cutoff most of the time ~ 10A ACKS2: Atom-Condensed Kohn-Sham DFT approximated to second order From Adri van Duin CH121
9 Long-range Coulomb Computation is very time consuming (N 2 ) Even more complicated by PBC (infinite replica) (NboxN 2 ) E 1/r 6 1/r Poor convergence due to its long-range nature 1. Ewald summation (N 3/2 ): E Coul = C distance q i q j hr 3 ij + 1 3i 1/3 split long-range/short-range real-space/reciprocal-space 2. Particle-particle particle-mesh (PPPM) method (NlogN) de Leeuw et al. Proc. Roy. Soc. Lond. A 373, 27 (1980) de Leeuw et al. Proc. Roy. Soc. Lond. A 373, 57 (1980)
10 BO 0 ij = exp +exp +exp Example reaxff: parametrization apple p bo,1 rij r 0 apple p bo,3 rij r 0 apple p bo,5 rij r 0 pbo,2 pbo,4 pbo,6 E bond = D e BO ij f(bo ij ) De BOij De BOij E over = f(bo ij ) E angle = 1 exp 3BO 3 a i 1 1+exp( i) 1 exp 3BO 3 b ka k a exp k b ( 0) 2 etc.
11 Example ReaxFF: parametrization Bond dissociation curve Angle bending
12 Example ReaxFF: parametrization Angle bending Reaction path Dihedral angles


13 Example ReaxFF: parametrization Dihedral angles ΔE = kcal/mol Echair= kcal/mol Eboat= kcal/mol ΔE = kcal/mol Eplanar= kcal/mol

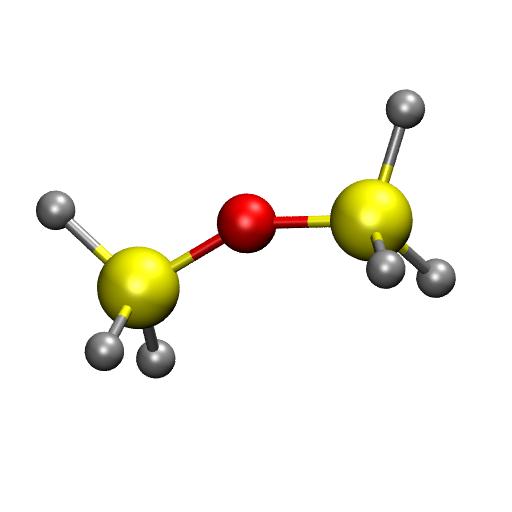
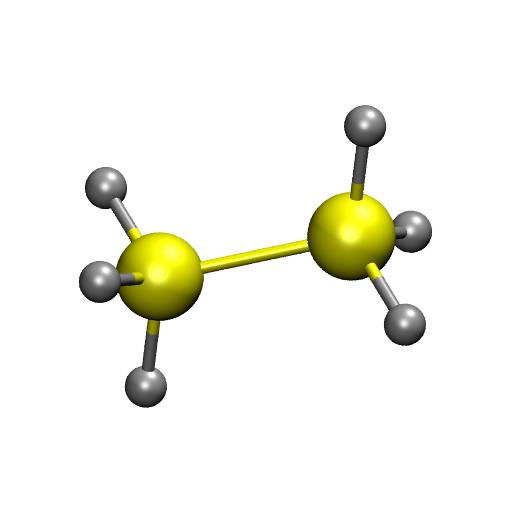

14 Example ReaxFF: parametrization Over/Under-coordination +
15 Example ReaxFF: parametrization Monte-Carlo Initial guess of parameters Loop over parameters Pnew(i) = P(i) + rand Error: If err < olderr: P(i) = Pnew(i) err = X i (E MD E DFT ) 2 E 0

16 Example ReaxFF: Development Wide range of element parametrized + some interaction between different elements


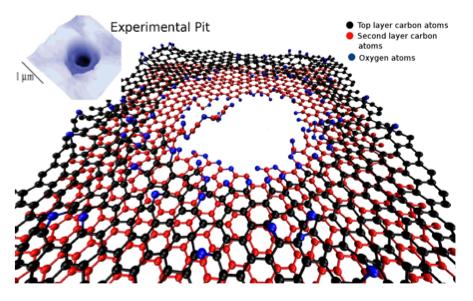
17 Example ReaxFF: chemistry and more
18 Reactive force field ReaxFF: van Duin, Adri C. T.; Dasgupta, Siddharth; Lorant, Francois; Goddard, William A. (2001). "ReaxFF: A Reactive Force Field for Hydrocarbons". The Journal of Physical Chemistry A 105 (41): Adri C. T. van Duin, Alejandro Strachan, Shannon Stewman, Qingsong Zhang, Xin Xu and William A. Goddard, III ReaxFFSiO Reactive Force Field for Silicon and Silicon Oxide Systems Anthony K. Rap and William A. Goddard III Charge equilibration for molecular dynamics Charge-Optimized Many-Body (COMB) potential: T.-R. Shan, B. D. Devine, T. W. Kemper, S. B. Sinnott, and S. R. Phillpot, Phys. Rev. B 81, (2010) The reactive empirical bond-order (REBO): Stuart, Tutein, Harrison, J Chem Phys, 112, (2000)
19 Conclusion: interatomic potentials Empirical potential type Usage Feature Names Pair-potential Noble gaz E Z LJ, Morse, Buckingham Pair-functional Metals E Z EAM Many-body potentials Semiconductors Angles Tersoff, SW Pair-potential /Reactive Organic materials Many-body/ BO DREIDING/ ReaxFF
20 Saving CPU time The force calculation is most expensive part in MD Interaction particle i with all its neighbors ~ N 2 Verlet list Cell list Combination Parallelism
21 Verlet list or neighbor list δ rc rs For each atom keep a list of the neighbor within rs = rc + δ Calculate the force if rij < rc Update the neighbor list every few timestep
22 Verlet list Construction Later Too late Good choice for δ Temperature, diffusion speed, density, etc. usually ~ timestep but depend of system studied
23 Verlet list Nneigh i = 1 j = 1 j = 2 Nneigh i = 2 j = 1 j = 2 Nneigh i = 3 j = 1 Number of neighbor of atom i = 1 Index of neighbors of i = 1 Index of neighbors Number of of i = 2 neighbor of atom i = 2 Loop over i = 1 to N Loop over j neighbor of i However building the list still N 2

24 Linked cell method Bin the atoms into 3D cells of size d rc Only check the atoms in the 26 surrounding cells Binning requires N operations
25 Combined neighbor-list / linked-cell Bin the atoms in 3D cells every few timestep: build the neighbor-list Compute the force using the neighbor-list: Less atoms in a sphere of volume 3/4πrs 3 than cube 27rc 3 Speed up with Newton s 3rd law: half-list, etc
26 Parallelism: Spacial decomposition The physical simulation domain is divided in small 3D boxes, one for each processor Each processor compute the force and update positions within the box Atom are reassign to other processor as they move through the physical domain In order to compute the force, the processor need to know the positions of atoms nearby boxes only (26) proc 1 proc 2 etc. Empty space will slow the scaling down
27 Periodic boundary conditions ghost cells containing ghost atoms
28 Parallelism: other algorithm Atom decomposition: Each processor receive a subset of atom N/P Each processor keep updating positions all along the simulation no matter the displacement of the atoms (local integration) Each processor need to know the whole vector position Force decomposition Each processor receive a subset of the force matrix Each processor need to know only part of the position vector J. Comput. Phys. 117, 1-19 (1995)
29 Macroscopic property from MD Macroscopic property from microscopic simulation Diffusion: Fick s law of diffusion Ex. study diffusion of copper in silica Z r 2 = Dr 2 c(r, t) <r2 (t) >= 2dD Microscopic (average displacement) Concentration of diffusive specie (defect, etc.) Laplacian Macroscopic (diffusion constant)
30 Mean square displacement <r 2 (t) >= 1 N NX r i (t) 2 = MSD i=1 Solid (crystal) vibrate around equilibrium Liquid diffusion
31 Copper diffusion in a-sio Log(MSD) (Å 2 ) K K K K Log(t) (ns) 1600K Log(t) (ns) 2400K Temp%(K)% D%(cm 2 /s)% Temp%(K)% D%(cm 2 /s)% Temp%(K)% D%(cm 2 /s)% 800# E*07# 1400# e*05# 2000# e*05# 1000# E*06# 1600# e*05# 2200# e*05# 1200# e*06# 1800# e*05# 2400# e*04#
32 Copper diffusion in a-sio Ln(D) vs 1/T Y= E+03X E A =0.566 ev 11 Ln(D) E A = kcal / mol = 0.566eV /T (K 1 ) x 10 4
33 Velocity <r2 (t) >= 2dD x = D = Z 1 With MSD be careful at PBC 0 Z t 0 vdt d < v x ( )v x (0) > Ensemble average velocity-velocity auto-correlation function No need with velocity Limits of velocity auto-correlation function: Small tau Large tau


34 Velocity <r2 (t) >= 2dD x = D = Z 1 If period of tau close to the oscillation of your system: peaks in auto-correlation function 0 Z t 0 vdt d < v x ( )v x (0) > Ensemble average velocity-velocity auto-correlation function correlation Fourier transform of auto-correlation function Integral: D no more correlation
 =4 r 2")
35 Radial distribution function How density varies as a function of distance from a reference particle Probability finding particle at r away from a given reference particle g(r) =4 r 2 dr

36 Nudged elastic band Calculate the barrier between two configurations: activation energy 1. Extrapolate the path between the two configurations 2. Relax the structures by constraining successive configurations

37 Nudged elastic band Dehydrogenation of ethene C 2 H 6 C 2 H 4 + H 2

38 Copper diffusion in silica Nudged elastic band
Reactive Empirical Force Fields
 Reactive Empirical Force Fields Jason Quenneville jasonq@lanl.gov X-1: Solid Mechanics, EOS and Materials Properties Applied Physics Division Los Alamos National Laboratory Timothy C. Germann, Los Alamos
Reactive Empirical Force Fields Jason Quenneville jasonq@lanl.gov X-1: Solid Mechanics, EOS and Materials Properties Applied Physics Division Los Alamos National Laboratory Timothy C. Germann, Los Alamos
Reactive potentials and applications
 1.021, 3.021, 10.333, 22.00 Introduction to Modeling and Simulation Spring 2011 Part I Continuum and particle methods Reactive potentials and applications Lecture 8 Markus J. Buehler Laboratory for Atomistic
1.021, 3.021, 10.333, 22.00 Introduction to Modeling and Simulation Spring 2011 Part I Continuum and particle methods Reactive potentials and applications Lecture 8 Markus J. Buehler Laboratory for Atomistic
Material Surfaces, Grain Boundaries and Interfaces: Structure-Property Relationship Predictions
 Material Surfaces, Grain Boundaries and Interfaces: Structure-Property Relationship Predictions Susan B. Sinnott Department of Materials Science and Engineering Penn State University September 16, 2016
Material Surfaces, Grain Boundaries and Interfaces: Structure-Property Relationship Predictions Susan B. Sinnott Department of Materials Science and Engineering Penn State University September 16, 2016
Reactive Force Field & Molecular Dynamics Simulations (Theory & Applications)
") Reactive Force Field & Molecular Dynamics Simulations (Theory & Applications) Ying Li Collaboratory for Advanced Computing & Simulations Department of Chemical Engineering & Materials Science Department
Reactive Force Field & Molecular Dynamics Simulations (Theory & Applications) Ying Li Collaboratory for Advanced Computing & Simulations Department of Chemical Engineering & Materials Science Department
Potentials, periodicity
 Potentials, periodicity Lecture 2 1/23/18 1 Survey responses 2 Topic requests DFT (10), Molecular dynamics (7), Monte Carlo (5) Machine Learning (4), High-throughput, Databases (4) NEB, phonons, Non-equilibrium
Potentials, periodicity Lecture 2 1/23/18 1 Survey responses 2 Topic requests DFT (10), Molecular dynamics (7), Monte Carlo (5) Machine Learning (4), High-throughput, Databases (4) NEB, phonons, Non-equilibrium
Computer simulation methods (2) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Introduction to ReaxFF: Reactive Molecular Dynamics
 Introduction to ReaxFF: Reactive Molecular Dynamics Ole Carstensen carstensen@scm.com TCCM ADF Tutorial April 21 Amsterdam Outline ReaxFF - general aspects Molecular Dynamics Intro 200 DFT 100 ReaxFF Harmonic
Introduction to ReaxFF: Reactive Molecular Dynamics Ole Carstensen carstensen@scm.com TCCM ADF Tutorial April 21 Amsterdam Outline ReaxFF - general aspects Molecular Dynamics Intro 200 DFT 100 ReaxFF Harmonic
Electronegativity is a very useful concept for the explanation or understanding of chemical reactivity throughout the periodic table.
 1.6. Review of Electronegativity (χ) CONCEPT: Electronegativity is a very useful concept for the explanation or understanding of chemical reactivity throughout the periodic table. There are many definitions
1.6. Review of Electronegativity (χ) CONCEPT: Electronegativity is a very useful concept for the explanation or understanding of chemical reactivity throughout the periodic table. There are many definitions
Introduction to DFTB. Marcus Elstner. July 28, 2006
 Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Introduction to DFTB Marcus Elstner July 28, 2006 I. Non-selfconsistent solution of the KS equations DFT can treat up to 100 atoms in routine applications, sometimes even more and about several ps in MD
Advanced Molecular Molecular Dynamics
 Advanced Molecular Molecular Dynamics Technical details May 12, 2014 Integration of harmonic oscillator r m period = 2 k k and the temperature T determine the sampling of x (here T is related with v 0
Advanced Molecular Molecular Dynamics Technical details May 12, 2014 Integration of harmonic oscillator r m period = 2 k k and the temperature T determine the sampling of x (here T is related with v 0
Parameterization of a reactive force field using a Monte Carlo algorithm
 Parameterization of a reactive force field using a Monte Carlo algorithm Eldhose Iype (e.iype@tue.nl) November 19, 2015 Where innovation starts Thermochemical energy storage 2/1 MgSO 4.xH 2 O+Q MgSO 4
Parameterization of a reactive force field using a Monte Carlo algorithm Eldhose Iype (e.iype@tue.nl) November 19, 2015 Where innovation starts Thermochemical energy storage 2/1 MgSO 4.xH 2 O+Q MgSO 4
Additional Comparison of QM and ReaxFF Data Included in the ReaxFF Training Set
 Additional omparison of and Data Included in the Training Set Supporting information for the manuscript A Reactive Force Field for Molecular Dynamics Simulations of ydrocarbon Oxidation by Kimberly henoweth,
Additional omparison of and Data Included in the Training Set Supporting information for the manuscript A Reactive Force Field for Molecular Dynamics Simulations of ydrocarbon Oxidation by Kimberly henoweth,
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
Atomistics of the Lithiation of Oxidized Silicon. Dynamics Simulations
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2016 Electronic Supplementary Information (ESI) Atomistics of the Lithiation of Oxidized
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2016 Electronic Supplementary Information (ESI) Atomistics of the Lithiation of Oxidized
Using Molecular Dynamics to Compute Properties CHEM 430
 Using Molecular Dynamics to Compute Properties CHEM 43 Heat Capacity and Energy Fluctuations Running an MD Simulation Equilibration Phase Before data-collection and results can be analyzed the system
Using Molecular Dynamics to Compute Properties CHEM 43 Heat Capacity and Energy Fluctuations Running an MD Simulation Equilibration Phase Before data-collection and results can be analyzed the system
Report on Atomistic Modeling of Bonding in Carbon-Based Nanostructures
 Report on Atomistic Modeling of Bonding in Carbon-Based Nanostructures Timothy Stillings Department of Physics, Astronomy and Materials Science Missouri State University Advisor: Ridwan Sakidja Abstract
Report on Atomistic Modeling of Bonding in Carbon-Based Nanostructures Timothy Stillings Department of Physics, Astronomy and Materials Science Missouri State University Advisor: Ridwan Sakidja Abstract
Formation of water at a Pt(111) surface: A study using reactive force fields (ReaxFF)
 surface: A study using reactive force fields (ReaxFF)") Formation of water at a Pt(111) surface: A study using reactive force fields (ReaxFF) Markus J. Buehler 1, Adri C.T. van Duin 2, Timo Jacob 3, Yunhee Jang 2, Boris Berinov 2, William A. Goddard III 2 1
Formation of water at a Pt(111) surface: A study using reactive force fields (ReaxFF) Markus J. Buehler 1, Adri C.T. van Duin 2, Timo Jacob 3, Yunhee Jang 2, Boris Berinov 2, William A. Goddard III 2 1
Supplementary Figure 1. Schematic of rapid thermal annealing process: (a) indicates schematics and SEM cross-section of the initial layer-by-layer
 indicates schematics and SEM cross-section of the initial layer-by-layer") Supplementary Figure 1. Schematic of rapid thermal annealing process: (a) indicates schematics and SEM cross-section of the initial layer-by-layer film configuration, (b) demonstrates schematic and cross-section
Supplementary Figure 1. Schematic of rapid thermal annealing process: (a) indicates schematics and SEM cross-section of the initial layer-by-layer film configuration, (b) demonstrates schematic and cross-section
Molecular Mechanics / ReaxFF
 Molecular Dynamics simulations Lecture 09: Molecular Mechanics / ReaxFF Dr. Olli Pakarinen University of Helsinki Fall 2012 Lecture notes based on notes by Dr. Jani Kotakoski, 2010 CONTENTS Molecular mechanics
Molecular Dynamics simulations Lecture 09: Molecular Mechanics / ReaxFF Dr. Olli Pakarinen University of Helsinki Fall 2012 Lecture notes based on notes by Dr. Jani Kotakoski, 2010 CONTENTS Molecular mechanics
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Molecular Dynamics Simulations
 Molecular Dynamics Simulations Dr. Kasra Momeni www.knanosys.com Outline Long-range Interactions Ewald Sum Fast Multipole Method Spherically Truncated Coulombic Potential Speeding up Calculations SPaSM
Molecular Dynamics Simulations Dr. Kasra Momeni www.knanosys.com Outline Long-range Interactions Ewald Sum Fast Multipole Method Spherically Truncated Coulombic Potential Speeding up Calculations SPaSM
Interatomic Potentials. The electronic-structure problem
 Interatomic Potentials Before we can start a simulation, we need the model! Interaction between atoms and molecules is determined by quantum mechanics: Schrödinger Equation + Born-Oppenheimer approximation
Interatomic Potentials Before we can start a simulation, we need the model! Interaction between atoms and molecules is determined by quantum mechanics: Schrödinger Equation + Born-Oppenheimer approximation
A Modified Embedded-Atom Method Interatomic Potential for Ionic Systems: 2NNMEAM+Qeq
 A Modified Embedded-Atom Method Interatomic Potential for Ionic Systems: 2NNMEAM+Qeq Eunkoo Lee, 1 Kwang-Ryeol Lee, 2 M. I. Baskes, 3 and Byeong-Joo Lee 1,* 1 Department of Materials Science and Engineering,
A Modified Embedded-Atom Method Interatomic Potential for Ionic Systems: 2NNMEAM+Qeq Eunkoo Lee, 1 Kwang-Ryeol Lee, 2 M. I. Baskes, 3 and Byeong-Joo Lee 1,* 1 Department of Materials Science and Engineering,
Coupling ReaxFF and DREIDING to Model Enzymatic Reactions. Li Tao, Markus J. Buehler and William A. Goddard
 Coupling ReaxFF and DREIDING to Model Enzymatic Reactions Li Tao, Markus J. Buehler and William A. Goddard Motivation Find efficient computational method to model reactivity in large biological systems
Coupling ReaxFF and DREIDING to Model Enzymatic Reactions Li Tao, Markus J. Buehler and William A. Goddard Motivation Find efficient computational method to model reactivity in large biological systems
Periodic Properties (Booklet Solution)
") Periodic Properties (Booklet Solution) Foundation Builders (Objective). (B) Law of triads states that in the set of three elements arranged in increasing order of atomic weight, having similar properties,
Periodic Properties (Booklet Solution) Foundation Builders (Objective). (B) Law of triads states that in the set of three elements arranged in increasing order of atomic weight, having similar properties,
Multiple time step Monte Carlo
 JOURNAL OF CHEMICAL PHYSICS VOLUME 117, NUMBER 18 8 NOVEMBER 2002 Multiple time step Monte Carlo Balázs Hetényi a) Department of Chemistry, Princeton University, Princeton, NJ 08544 and Department of Chemistry
JOURNAL OF CHEMICAL PHYSICS VOLUME 117, NUMBER 18 8 NOVEMBER 2002 Multiple time step Monte Carlo Balázs Hetényi a) Department of Chemistry, Princeton University, Princeton, NJ 08544 and Department of Chemistry
Application to modeling brittle materials
 1.01, 3.01, 10.333,.00 Introduction to Modeling and Simulation Spring 011 Part I Continuum and particle methods Application to modeling brittle materials Lecture 7 Markus J. Buehler Laboratory for Atomistic
1.01, 3.01, 10.333,.00 Introduction to Modeling and Simulation Spring 011 Part I Continuum and particle methods Application to modeling brittle materials Lecture 7 Markus J. Buehler Laboratory for Atomistic
A COMPARATIVE STUDY OF OXYGEN VACANCY MIGRATION PATHWAYS IN CRYSTALLINE POLYMORPHS OF SILICA
 A COMPARATIVE STUDY OF OXYGEN VACANCY MIGRATION PATHWAYS IN CRYSTALLINE POLYMORPHS OF SILICA L. René Corrales Pacific Northwest National Laboratory Richland, WA 99352 Corresponding author: rene.corrales@pnl.gov
A COMPARATIVE STUDY OF OXYGEN VACANCY MIGRATION PATHWAYS IN CRYSTALLINE POLYMORPHS OF SILICA L. René Corrales Pacific Northwest National Laboratory Richland, WA 99352 Corresponding author: rene.corrales@pnl.gov
Introduction to Simulation - Lectures 17, 18. Molecular Dynamics. Nicolas Hadjiconstantinou
 Introduction to Simulation - Lectures 17, 18 Molecular Dynamics Nicolas Hadjiconstantinou Molecular Dynamics Molecular dynamics is a technique for computing the equilibrium and non-equilibrium properties
Introduction to Simulation - Lectures 17, 18 Molecular Dynamics Nicolas Hadjiconstantinou Molecular Dynamics Molecular dynamics is a technique for computing the equilibrium and non-equilibrium properties
Periodic Properties (Booklet Solution)
") Periodic Properties (Booklet Solution) Foundation Builders (Objective). (B) Law of triads states that in the set of three elements arranged in increasing order of atomic weight, having similar properties,
Periodic Properties (Booklet Solution) Foundation Builders (Objective). (B) Law of triads states that in the set of three elements arranged in increasing order of atomic weight, having similar properties,
Analysis of the simulation
 Analysis of the simulation Marcus Elstner and Tomáš Kubař January 7, 2014 Thermodynamic properties time averages of thermodynamic quantites correspond to ensemble averages (ergodic theorem) some quantities
Analysis of the simulation Marcus Elstner and Tomáš Kubař January 7, 2014 Thermodynamic properties time averages of thermodynamic quantites correspond to ensemble averages (ergodic theorem) some quantities
MD Thermodynamics. Lecture 12 3/26/18. Harvard SEAS AP 275 Atomistic Modeling of Materials Boris Kozinsky
 MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
MD Thermodynamics Lecture 1 3/6/18 1 Molecular dynamics The force depends on positions only (not velocities) Total energy is conserved (micro canonical evolution) Newton s equations of motion (second order
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
From Atoms to Materials: Predictive Theory and Simulations
 From Atoms to Materials: Predictive Theory and Simulations Week 3 Lecture 4 Potentials for metals and semiconductors Ale Strachan strachan@purdue.edu School of Materials Engineering & Birck anotechnology
From Atoms to Materials: Predictive Theory and Simulations Week 3 Lecture 4 Potentials for metals and semiconductors Ale Strachan strachan@purdue.edu School of Materials Engineering & Birck anotechnology
VIRTUAL LATTICE TECHNIQUE AND THE INTERATOMIC POTENTIALS OF ZINC-BLEND-TYPE BINARY COMPOUNDS
 Modern Physics Letters B, Vol. 16, Nos. 5 & 6 (2002) 187 194 c World Scientific Publishing Company VIRTUAL LATTICE TECHNIQUE AND THE INTERATOMIC POTENTIALS OF ZINC-BLEND-TYPE BINARY COMPOUNDS LIU YING,
Modern Physics Letters B, Vol. 16, Nos. 5 & 6 (2002) 187 194 c World Scientific Publishing Company VIRTUAL LATTICE TECHNIQUE AND THE INTERATOMIC POTENTIALS OF ZINC-BLEND-TYPE BINARY COMPOUNDS LIU YING,
Studying the properties of substances and the reactions that transform substances into other substances.
 3 CHEMISTRY Studying the properties of substances and the reactions that transform substances into other substances. Improving agricultural production, curing many diseases, increasing the efficiency of
3 CHEMISTRY Studying the properties of substances and the reactions that transform substances into other substances. Improving agricultural production, curing many diseases, increasing the efficiency of
Introduction to model potential Molecular Dynamics A3hourcourseatICTP
 Introduction to model potential Molecular Dynamics A3hourcourseatICTP Alessandro Mattoni 1 1 Istituto Officina dei Materiali CNR-IOM Unità di Cagliari SLACS ICTP School on numerical methods for energy,
Introduction to model potential Molecular Dynamics A3hourcourseatICTP Alessandro Mattoni 1 1 Istituto Officina dei Materiali CNR-IOM Unità di Cagliari SLACS ICTP School on numerical methods for energy,
Nanotube AFM Probe Resolution
 Influence of Elastic Deformation on Single-Wall Carbon Nanotube AFM Probe Resolution Ian R. Shapiro, Santiago D. Solares, Maria J. Esplandiu, Lawrence A. Wade, William A. Goddard,* and C. Patrick Collier*
Influence of Elastic Deformation on Single-Wall Carbon Nanotube AFM Probe Resolution Ian R. Shapiro, Santiago D. Solares, Maria J. Esplandiu, Lawrence A. Wade, William A. Goddard,* and C. Patrick Collier*
References in the Supporting Information:
 Identification of the Selective Sites for Electrochemical Reduction of CO to C2+ Products on Copper Nanoparticles by Combining Reactive Force Fields, Density Functional Theory, and Machine Learning Supporting
Identification of the Selective Sites for Electrochemical Reduction of CO to C2+ Products on Copper Nanoparticles by Combining Reactive Force Fields, Density Functional Theory, and Machine Learning Supporting
LAMMPS Performance Benchmark on VSC-1 and VSC-2
 LAMMPS Performance Benchmark on VSC-1 and VSC-2 Daniel Tunega and Roland Šolc Institute of Soil Research, University of Natural Resources and Life Sciences VSC meeting, Neusiedl am See, February 27-28,
LAMMPS Performance Benchmark on VSC-1 and VSC-2 Daniel Tunega and Roland Šolc Institute of Soil Research, University of Natural Resources and Life Sciences VSC meeting, Neusiedl am See, February 27-28,
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Heat Conduction in Double-walled Carbon Nanotubes with
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 015 Supporting Information Heat Conduction in Double-walled Carbon Nanotubes with Intertube
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 015 Supporting Information Heat Conduction in Double-walled Carbon Nanotubes with Intertube
Why Heteroepitaxy? Difficult to Model Multiple Species Dislocations and point defects important Species Flux important
 Why Heteroepitaxy? Difficult to Model Multiple Species Dislocations and point defects important Species Flux important The Silicon Age is Ending New materials and growth required Useful in design of Devices
Why Heteroepitaxy? Difficult to Model Multiple Species Dislocations and point defects important Species Flux important The Silicon Age is Ending New materials and growth required Useful in design of Devices
Part 2: Molecular Dynamics. Literature History Practical Issues
 Part 2: Molecular Dynamics Literature History Practical Issues 1 Literature A. K. Hartmann Big Practical Guide to Computer Simulations (World Scientific, 2015) M. P. Allen and D.J. Tildesley Computer Simulations
Part 2: Molecular Dynamics Literature History Practical Issues 1 Literature A. K. Hartmann Big Practical Guide to Computer Simulations (World Scientific, 2015) M. P. Allen and D.J. Tildesley Computer Simulations
Hyeyoung Shin a, Tod A. Pascal ab, William A. Goddard III abc*, and Hyungjun Kim a* Korea
 The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
Kinetic lattice Monte Carlo simulations of diffusion processes in Si and SiGe alloys
 Kinetic lattice Monte Carlo simulations of diffusion processes in Si and SiGe alloys, Scott Dunham Department of Electrical Engineering Multiscale Modeling Hierarchy Configuration energies and transition
Kinetic lattice Monte Carlo simulations of diffusion processes in Si and SiGe alloys, Scott Dunham Department of Electrical Engineering Multiscale Modeling Hierarchy Configuration energies and transition
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular Dynamics of Covalent Crystals
 Molecular Dynamics of Covalent Crystals J. Hahn and H.-R. Trebin Institut für Theoretische und Angewandte Physik, Universität Stuttgart, D-70550 Stuttgart, Germany Abstract. A molecular mechanics-like
Molecular Dynamics of Covalent Crystals J. Hahn and H.-R. Trebin Institut für Theoretische und Angewandte Physik, Universität Stuttgart, D-70550 Stuttgart, Germany Abstract. A molecular mechanics-like
Teoría del Funcional de la Densidad (Density Functional Theory)
") Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
Teoría del Funcional de la Densidad (Density Functional Theory) Motivation: limitations of the standard approach based on the wave function. The electronic density n(r) as the key variable: Functionals
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
MOLECULAR DYNAMIC SIMULATION OF WATER VAPOR INTERACTION WITH VARIOUS TYPES OF PORES USING HYBRID COMPUTING STRUCTURES
 MOLECULAR DYNAMIC SIMULATION OF WATER VAPOR INTERACTION WITH VARIOUS TYPES OF PORES USING HYBRID COMPUTING STRUCTURES V.V. Korenkov 1,3, a, E.G. Nikonov 1, b, M. Popovičová 2, с 1 Joint Institute for Nuclear
MOLECULAR DYNAMIC SIMULATION OF WATER VAPOR INTERACTION WITH VARIOUS TYPES OF PORES USING HYBRID COMPUTING STRUCTURES V.V. Korenkov 1,3, a, E.G. Nikonov 1, b, M. Popovičová 2, с 1 Joint Institute for Nuclear
Chem 1140; Molecular Modeling
 P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
P. Wipf 1 Chem 1140 $E = -! (q a + q b )" ab S ab + ab!k
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
PH 548 Atomistic Simulation Techniques
 PH 548 Atomistic Simulation Techniques Lectures: Lab: 2-0-2-6 Tuesday 12-1 PM Thursday 12-1 PM Monday 2-5 PM P. K. Padmanabhan PH548: Two Excellent Books How to do it? 1 2 F 12 F 23 3 F i = F ij j i F
PH 548 Atomistic Simulation Techniques Lectures: Lab: 2-0-2-6 Tuesday 12-1 PM Thursday 12-1 PM Monday 2-5 PM P. K. Padmanabhan PH548: Two Excellent Books How to do it? 1 2 F 12 F 23 3 F i = F ij j i F
ReaxFF MgH Reactive Force Field for Magnesium Hydride Systems
 J. Phys. Chem. A 2005, 109, 851-859 851 ReaxFF MgH Reactive Force Field for Magnesium Hydride Systems Sam Cheung, Wei-Qiao Deng, Adri C. T. van Duin, and William A. Goddard III* Materials and Process Simulation
J. Phys. Chem. A 2005, 109, 851-859 851 ReaxFF MgH Reactive Force Field for Magnesium Hydride Systems Sam Cheung, Wei-Qiao Deng, Adri C. T. van Duin, and William A. Goddard III* Materials and Process Simulation
The split-charge method:
 Split charge equilibration: A charge transfer method for electrolytes and other non-metallic materials The split-charge method: Martin H. Müser Materialwissenschaften und Werkstoffwissenschaften Motivation
Split charge equilibration: A charge transfer method for electrolytes and other non-metallic materials The split-charge method: Martin H. Müser Materialwissenschaften und Werkstoffwissenschaften Motivation
Au-C Au-Au. g(r) r/a. Supplementary Figures
 r/a. Supplementary Figures") g(r) Supplementary Figures 60 50 40 30 20 10 0 Au-C Au-Au 2 4 r/a 6 8 Supplementary Figure 1 Radial bond distributions for Au-C and Au-Au bond. The zero density regime between the first two peaks in g
g(r) Supplementary Figures 60 50 40 30 20 10 0 Au-C Au-Au 2 4 r/a 6 8 Supplementary Figure 1 Radial bond distributions for Au-C and Au-Au bond. The zero density regime between the first two peaks in g
The wavefunction that describes a bonding pair of electrons:
 4.2. Molecular Properties from VB Theory a) Bonding and Bond distances The wavefunction that describes a bonding pair of electrons: Ψ b = a(h 1 ) + b(h 2 ) where h 1 and h 2 are HAOs on adjacent atoms
4.2. Molecular Properties from VB Theory a) Bonding and Bond distances The wavefunction that describes a bonding pair of electrons: Ψ b = a(h 1 ) + b(h 2 ) where h 1 and h 2 are HAOs on adjacent atoms
sharing or transferring electrons between atoms covalent ionic polar covalent Quantitative description: Quantum mechanics
 Chapter. 3 Chemical Bonding: The Classical Description Two or more atoms approach -> their electrons interact and form new arrangements of electrons with lower total potential energy than isolated atoms
Chapter. 3 Chemical Bonding: The Classical Description Two or more atoms approach -> their electrons interact and form new arrangements of electrons with lower total potential energy than isolated atoms
Molecular Dynamics Simulation of a Nanoconfined Water Film
 Molecular Dynamics Simulation of a Nanoconfined Water Film Kyle Lindquist, Shu-Han Chao May 7, 2013 1 Introduction The behavior of water confined in nano-scale environment is of interest in many applications.
Molecular Dynamics Simulation of a Nanoconfined Water Film Kyle Lindquist, Shu-Han Chao May 7, 2013 1 Introduction The behavior of water confined in nano-scale environment is of interest in many applications.
Advanced Electronic Structure Theory Density functional theory. Dr Fred Manby
 Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Advanced Electronic Structure Theory Density functional theory Dr Fred Manby fred.manby@bris.ac.uk http://www.chm.bris.ac.uk/pt/manby/ 6 Strengths of DFT DFT is one of many theories used by (computational)
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory.
 Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
Walter Kohn was awarded with the Nobel Prize in Chemistry in 1998 for his development of the density functional theory. Walter Kohn receiving his Nobel Prize from His Majesty the King at the Stockholm
What is Classical Molecular Dynamics?
 What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
What is Classical Molecular Dynamics? Simulation of explicit particles (atoms, ions,... ) Particles interact via relatively simple analytical potential functions Newton s equations of motion are integrated
Molecular Dynamics and Accelerated Molecular Dynamics
 Molecular Dynamics and Accelerated Molecular Dynamics Arthur F. Voter Theoretical Division National Laboratory Lecture 3 Tutorial Lecture Series Institute for Pure and Applied Mathematics (IPAM) UCLA September
Molecular Dynamics and Accelerated Molecular Dynamics Arthur F. Voter Theoretical Division National Laboratory Lecture 3 Tutorial Lecture Series Institute for Pure and Applied Mathematics (IPAM) UCLA September
ReaxFF force fields. Development of a transferable empirical method for atomic-scale simulations of chemical reactions
 ReaxFF force fields Development of a transferable empirical method for atomic-scale simulations of chemical reactions Adri van Duin, Kimberley Chenoweth 2 and Bill Goddard 2 : Department of Mechanical
ReaxFF force fields Development of a transferable empirical method for atomic-scale simulations of chemical reactions Adri van Duin, Kimberley Chenoweth 2 and Bill Goddard 2 : Department of Mechanical
Atomistic Insights into the Conversion Reaction in Iron Fluoride: A Dynamically Adaptive Force Field Approach
 pubs.acs.org/jacs Atomistic Insights into the Conversion Reaction in Iron Fluoride: A Dynamically Adaptive Force Field Approach Ying Ma and Stephen H. Garofalini* Interfacial Molecular Science Laboratory,
pubs.acs.org/jacs Atomistic Insights into the Conversion Reaction in Iron Fluoride: A Dynamically Adaptive Force Field Approach Ying Ma and Stephen H. Garofalini* Interfacial Molecular Science Laboratory,
Universal Repulsive Contribution to the. Solvent-Induced Interaction Between Sizable, Curved Hydrophobes: Supporting Information
 Universal Repulsive Contribution to the Solvent-Induced Interaction Between Sizable, Curved Hydrophobes: Supporting Information B. Shadrack Jabes, Dusan Bratko, and Alenka Luzar Department of Chemistry,
Universal Repulsive Contribution to the Solvent-Induced Interaction Between Sizable, Curved Hydrophobes: Supporting Information B. Shadrack Jabes, Dusan Bratko, and Alenka Luzar Department of Chemistry,
k θ (θ θ 0 ) 2 angles r i j r i j
 2 angles r i j r i j") 1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
1 Force fields 1.1 Introduction The term force field is slightly misleading, since it refers to the parameters of the potential used to calculate the forces (via gradient) in molecular dynamics simulations.
Nucleation rate (m -3 s -1 ) Radius of water nano droplet (Å) 1e+00 1e-64 1e-128 1e-192 1e-256
 Radius of water nano droplet (Å) 1e+00 1e-64 1e-128 1e-192 1e-256") Supplementary Figures Nucleation rate (m -3 s -1 ) 1e+00 1e-64 1e-128 1e-192 1e-256 Calculated R in bulk water Calculated R in droplet Modified CNT 20 30 40 50 60 70 Radius of water nano droplet (Å) Supplementary
Supplementary Figures Nucleation rate (m -3 s -1 ) 1e+00 1e-64 1e-128 1e-192 1e-256 Calculated R in bulk water Calculated R in droplet Modified CNT 20 30 40 50 60 70 Radius of water nano droplet (Å) Supplementary
Multiple time step Monte Carlo simulations: Application to charged systems with Ewald summation
 JOURNAL OF CHEMICAL PHYSICS VOLUME 11, NUMBER 1 1 JULY 004 Multiple time step Monte Carlo simulations: Application to charged systems with Ewald summation Katarzyna Bernacki a) Department of Chemistry
JOURNAL OF CHEMICAL PHYSICS VOLUME 11, NUMBER 1 1 JULY 004 Multiple time step Monte Carlo simulations: Application to charged systems with Ewald summation Katarzyna Bernacki a) Department of Chemistry
Direct Molecular Simulation of Hypersonic Flows
 Direct Molecular Simulation of Hypersonic Flows Thomas E. Schwartzentruber Aerospace Engineering & Mechanics University of Minnesota UMN Students and Researchers: Savio Poovathingal 1 Paul Norman 3 Chonglin
Direct Molecular Simulation of Hypersonic Flows Thomas E. Schwartzentruber Aerospace Engineering & Mechanics University of Minnesota UMN Students and Researchers: Savio Poovathingal 1 Paul Norman 3 Chonglin
Melting line of the Lennard-Jones system, infinite size, and full potential
 THE JOURNAL OF CHEMICAL PHYSICS 127, 104504 2007 Melting line of the Lennard-Jones system, infinite size, and full potential Ethan A. Mastny a and Juan J. de Pablo b Chemical and Biological Engineering
THE JOURNAL OF CHEMICAL PHYSICS 127, 104504 2007 Melting line of the Lennard-Jones system, infinite size, and full potential Ethan A. Mastny a and Juan J. de Pablo b Chemical and Biological Engineering
MD simulation: output
 Properties MD simulation: output Trajectory of atoms positions: e. g. diffusion, mass transport velocities: e. g. v-v autocorrelation spectrum Energies temperature displacement fluctuations Mean square
Properties MD simulation: output Trajectory of atoms positions: e. g. diffusion, mass transport velocities: e. g. v-v autocorrelation spectrum Energies temperature displacement fluctuations Mean square
A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane
 A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane Supplementary Information: Simulation Procedure and Physical Property Analysis Simulation Procedure The molecular
A Molecular Dynamics Simulation of a Homogeneous Organic-Inorganic Hybrid Silica Membrane Supplementary Information: Simulation Procedure and Physical Property Analysis Simulation Procedure The molecular
Development of a ReaxFF Reactive Force Field for Glycine and Application to Solvent Effect and Tautomerization
 J. Phys. Chem. B 2011, 115, 249 261 249 Development of a ReaxFF Reactive Force Field for Glycine and Application to Solvent Effect and Tautomerization Obaidur Rahaman, Adri C. T. van Duin, William A. Goddard
J. Phys. Chem. B 2011, 115, 249 261 249 Development of a ReaxFF Reactive Force Field for Glycine and Application to Solvent Effect and Tautomerization Obaidur Rahaman, Adri C. T. van Duin, William A. Goddard
Molecular Dynamics. What to choose in an integrator The Verlet algorithm Boundary Conditions in Space and time Reading Assignment: F&S Chapter 4
 Molecular Dynamics What to choose in an integrator The Verlet algorithm Boundary Conditions in Space and time Reading Assignment: F&S Chapter 4 MSE485/PHY466/CSE485 1 The Molecular Dynamics (MD) method
Molecular Dynamics What to choose in an integrator The Verlet algorithm Boundary Conditions in Space and time Reading Assignment: F&S Chapter 4 MSE485/PHY466/CSE485 1 The Molecular Dynamics (MD) method
Chemical Bonding. Chemical Bonding 20/03/2015. The atomic radius increases from right to left. The atomic radius increases from top to bottom
 Chemical Bonding Atomic Radius: This distance from the nucleus to the outermost electron. Chemical Bonding Chemistry 11 Two factors must be taken into consideration in explaining this periodic trend: Increasing
Chemical Bonding Atomic Radius: This distance from the nucleus to the outermost electron. Chemical Bonding Chemistry 11 Two factors must be taken into consideration in explaining this periodic trend: Increasing
Force Fields in Molecular Mechanics
 Force Fields in Molecular Mechanics Rajarshi Guha (9915607) and Rajesh Sardar (9915610) March 21, 2001 1 Introduction With the advent of computers chemists have realized the utility of carrying out simulations
Force Fields in Molecular Mechanics Rajarshi Guha (9915607) and Rajesh Sardar (9915610) March 21, 2001 1 Introduction With the advent of computers chemists have realized the utility of carrying out simulations
Thermal decomposition of RDX from reactive molecular dynamics
 THE JOURNAL OF CHEMICAL PHYSICS 122, 054502 2005 Thermal decomposition of RDX from reactive molecular dynamics Alejandro Strachan a) and Edward M. Kober Theoretical Division, Los Alamos National Laboratory,
THE JOURNAL OF CHEMICAL PHYSICS 122, 054502 2005 Thermal decomposition of RDX from reactive molecular dynamics Alejandro Strachan a) and Edward M. Kober Theoretical Division, Los Alamos National Laboratory,
Electronic Structure Theory for Periodic Systems: The Concepts. Christian Ratsch
 Electronic Structure Theory for Periodic Systems: The Concepts Christian Ratsch Institute for Pure and Applied Mathematics and Department of Mathematics, UCLA Motivation There are 10 20 atoms in 1 mm 3
Electronic Structure Theory for Periodic Systems: The Concepts Christian Ratsch Institute for Pure and Applied Mathematics and Department of Mathematics, UCLA Motivation There are 10 20 atoms in 1 mm 3
arxiv:cond-mat/ v1 [cond-mat.stat-mech] 8 Oct 1996
![arxiv:cond-mat/ v1 [cond-mat.stat-mech] 8 Oct 1996](/thumbs/72/66812395.jpg "arxiv:cond-mat/ v1 [cond-mat.stat-mech] 8 Oct 1996") December 21, 2013 arxiv:cond-mat/9610066v1 [cond-mat.stat-mech] 8 Oct 1996 Some Finite Size Effects in Simulations of Glass Dynamics Jürgen Horbach, Walter Kob, Kurt Binder Institut für Physik, Johannes
December 21, 2013 arxiv:cond-mat/9610066v1 [cond-mat.stat-mech] 8 Oct 1996 Some Finite Size Effects in Simulations of Glass Dynamics Jürgen Horbach, Walter Kob, Kurt Binder Institut für Physik, Johannes
Accelerated Quantum Molecular Dynamics
 Accelerated Quantum Molecular Dynamics Enrique Martinez, Christian Negre, Marc J. Cawkwell, Danny Perez, Arthur F. Voter and Anders M. N. Niklasson Outline Quantum MD Current approaches Challenges Extended
Accelerated Quantum Molecular Dynamics Enrique Martinez, Christian Negre, Marc J. Cawkwell, Danny Perez, Arthur F. Voter and Anders M. N. Niklasson Outline Quantum MD Current approaches Challenges Extended
Electronic structure and transport in silicon nanostructures with non-ideal bonding environments
 Purdue University Purdue e-pubs Other Nanotechnology Publications Birck Nanotechnology Center 9-15-2008 Electronic structure and transport in silicon nanostructures with non-ideal bonding environments
Purdue University Purdue e-pubs Other Nanotechnology Publications Birck Nanotechnology Center 9-15-2008 Electronic structure and transport in silicon nanostructures with non-ideal bonding environments
Density Functional Theory - II part
 Density Functional Theory - II part antonino.polimeno@unipd.it Overview From theory to practice Implementation Functionals Local functionals Gradient Others From theory to practice From now on, if not
Density Functional Theory - II part antonino.polimeno@unipd.it Overview From theory to practice Implementation Functionals Local functionals Gradient Others From theory to practice From now on, if not
Journal of Theoretical Physics
 1 Journal of Theoretical Physics Founded and Edited by M. Apostol 53 (2000) ISSN 1453-4428 Ionization potential for metallic clusters L. C. Cune and M. Apostol Department of Theoretical Physics, Institute
1 Journal of Theoretical Physics Founded and Edited by M. Apostol 53 (2000) ISSN 1453-4428 Ionization potential for metallic clusters L. C. Cune and M. Apostol Department of Theoretical Physics, Institute
Heidelberg Molecular Modelling Summer School The Challenges of Transition Metal Systems
 Heidelberg Molecular Modelling Summer School The Challenges of Transition Metal Systems Dr Rob Deeth Inorganic Computational Chemistry Group University of Warwick UK verview Is molecular modelling of TM
Heidelberg Molecular Modelling Summer School The Challenges of Transition Metal Systems Dr Rob Deeth Inorganic Computational Chemistry Group University of Warwick UK verview Is molecular modelling of TM
Biomolecules are dynamic no single structure is a perfect model
 Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Molecular Dynamics Simulations of Biomolecules References: A. R. Leach Molecular Modeling Principles and Applications Prentice Hall, 2001. M. P. Allen and D. J. Tildesley "Computer Simulation of Liquids",
Non-bonded interactions
 speeding up the number-crunching continued Marcus Elstner and Tomáš Kubař December 3, 2013 why care? number of individual pair-wise interactions bonded interactions proportional to N: O(N) non-bonded interactions
speeding up the number-crunching continued Marcus Elstner and Tomáš Kubař December 3, 2013 why care? number of individual pair-wise interactions bonded interactions proportional to N: O(N) non-bonded interactions
c 2012 Society for Industrial and Applied Mathematics
 SIAM J. SCI. COMPUT. Vol. 34, No. 1, pp. C1 C23 c 2012 Society for Industrial and Applied Mathematics REACTIVE MOLECULAR DYNAMICS: NUMERICAL METHODS AND ALGORITHMIC TECHNIQUES HASAN METIN AKTULGA, SAGAR
SIAM J. SCI. COMPUT. Vol. 34, No. 1, pp. C1 C23 c 2012 Society for Industrial and Applied Mathematics REACTIVE MOLECULAR DYNAMICS: NUMERICAL METHODS AND ALGORITHMIC TECHNIQUES HASAN METIN AKTULGA, SAGAR
Electron Configurations and the Periodic Table
 Electron Configurations and the Periodic Table The periodic table can be used as a guide for electron configurations. The period number is the value of n. Groups 1A and 2A have the s-orbital filled. Groups
Electron Configurations and the Periodic Table The periodic table can be used as a guide for electron configurations. The period number is the value of n. Groups 1A and 2A have the s-orbital filled. Groups
MatSci 331 Homework 4 Molecular Dynamics and Monte Carlo: Stress, heat capacity, quantum nuclear effects, and simulated annealing
 MatSci 331 Homework 4 Molecular Dynamics and Monte Carlo: Stress, heat capacity, quantum nuclear effects, and simulated annealing Due Thursday Feb. 21 at 5pm in Durand 110. Evan Reed In this homework,
MatSci 331 Homework 4 Molecular Dynamics and Monte Carlo: Stress, heat capacity, quantum nuclear effects, and simulated annealing Due Thursday Feb. 21 at 5pm in Durand 110. Evan Reed In this homework,
CHEMISTRY 4021/8021 MIDTERM EXAM 1 SPRING 2014
 CHEMISTRY 4021/8021 Q1) Propose a simple, united-atom molecular mechanics force-field needed to generate a potential energy surface for an isolated molecule of acetone (Me 2 CO). I.e., provide an energy
CHEMISTRY 4021/8021 Q1) Propose a simple, united-atom molecular mechanics force-field needed to generate a potential energy surface for an isolated molecule of acetone (Me 2 CO). I.e., provide an energy
Lecture February 8-10, NiCHx
 Lecture 16-17 February 8-10, 2011 Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course number: Ch120a
Lecture 16-17 February 8-10, 2011 Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy Course number: Ch120a
Dislocations in graphene
 Dislocations in graphene M. Ortiz California Institute of Technology In collaboration with: M.P. Ariza, Universidad de Sevilla Symposium on Multiscale Dislocation Dynamics UCSD, La Jolla, January 16-17,
Dislocations in graphene M. Ortiz California Institute of Technology In collaboration with: M.P. Ariza, Universidad de Sevilla Symposium on Multiscale Dislocation Dynamics UCSD, La Jolla, January 16-17,
Molecular Dynamics Simulation Study of the Ionic Mobility of OH Using the OSS2 Model
 1154 Bull. Korean Chem. Soc. 2006, Vol. 27, No. 8 Song Hi Lee Molecular Dynamics Simulation Study of the Ionic Mobility of OH Using the OSS2 Model Song Hi Lee Department of Chemistry, Kyungsung University,
1154 Bull. Korean Chem. Soc. 2006, Vol. 27, No. 8 Song Hi Lee Molecular Dynamics Simulation Study of the Ionic Mobility of OH Using the OSS2 Model Song Hi Lee Department of Chemistry, Kyungsung University,
Ab initio molecular dynamics simulation on temperature-dependent properties of Al Si liquid alloy
 INSTITUTE OF PHYSICSPUBLISHING JOURNAL OFPHYSICS: CONDENSED MATTER J. Phys.: Condens. Matter 16 (4) 57 514 PII: S953-8984(4)7691-8 Ab initio molecular dynamics simulation on temperature-dependent properties
INSTITUTE OF PHYSICSPUBLISHING JOURNAL OFPHYSICS: CONDENSED MATTER J. Phys.: Condens. Matter 16 (4) 57 514 PII: S953-8984(4)7691-8 Ab initio molecular dynamics simulation on temperature-dependent properties
Chapter 7. Generally, the electronic structure of atoms correlates w. the prop. of the elements
 Chapter 7 Periodic Properties of the Elements I) Development of the P.T. Generally, the electronic structure of atoms correlates w. the prop. of the elements - reflected by the arrangement of the elements
Chapter 7 Periodic Properties of the Elements I) Development of the P.T. Generally, the electronic structure of atoms correlates w. the prop. of the elements - reflected by the arrangement of the elements
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Lecture outline: Chapter 7 Periodic properties
 Lecture outline: Chapter 7 Periodic properties 1. Electrostatic effects 2. Atomic size 3. Ionization energy 4. Electron affinity it 5. Summarize some periodic properties 1 Some important terms Electron
Lecture outline: Chapter 7 Periodic properties 1. Electrostatic effects 2. Atomic size 3. Ionization energy 4. Electron affinity it 5. Summarize some periodic properties 1 Some important terms Electron