Simulation Methods II
|
|
|
- Jonas Hudson
- 5 years ago
- Views:
Transcription
1 Simulation Methods II Maria Fyta Institute for Computational Physics Universität Stuttgart Summer Term 2018
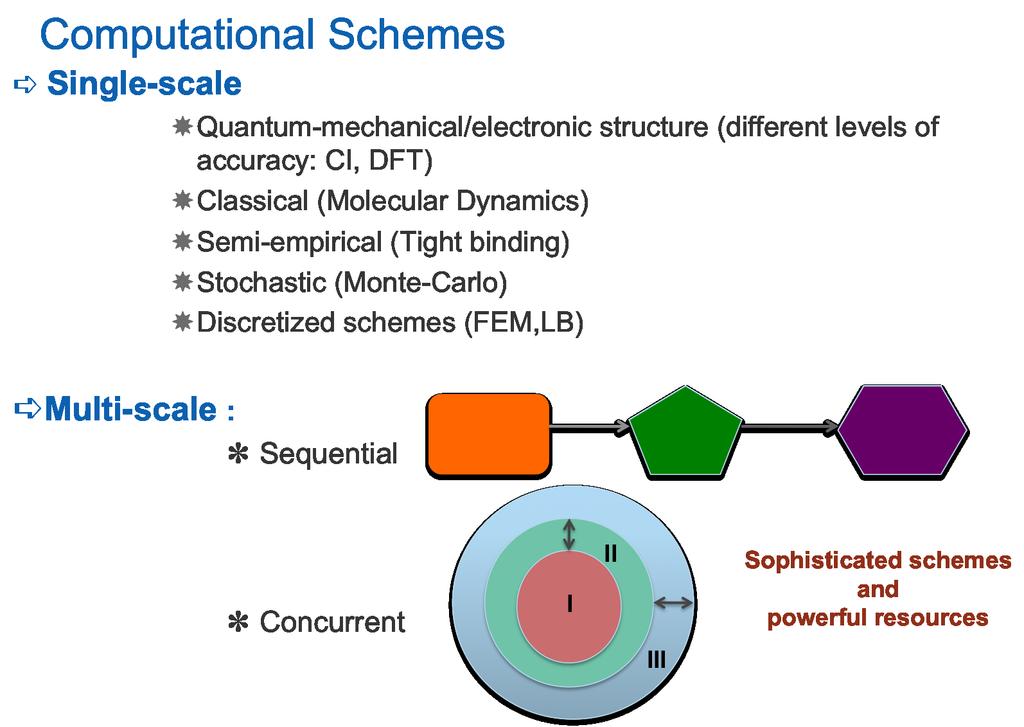
2 SM II - contents First principles methods Hartree-Fock and beyond Density-funtional-theory Ab initio Molecular Dynamics, forces and electronic structure QM/MM Classical force-fields, pair potentials Water models (implicit, explicit solvation) Hydrodynamic schemes lattice-boltzmann Brownian Dynamics Dissipative Particle Dynamics (DPD) Stochastic Rotation Dynamics (SRD) Free energy methods Overview of coarse-graining and multiscale methods M.Fyta 2/20
3 Course where & when Thursdays, 11:30-13:00, ICP Seminar room Tutorials Tutors: Miriam Kohagen David Sean New worksheets every other week handed out every 2 nd Thursday Discussion meetings and tutorials; Thursdays, 14:00-15:30 (?) ICP CIP-Pool Worksheets due every 2 nd Wednesday. First worksheet handed out on Wed First tutorial: week (19.04?) First worksheet due on at 13:00 Exam Prerequisite: 50% of the total points in the tutorials Oral examination at the end of the semester M.Fyta 3/20
4 Recommended Literature D. Frenkel and B. Smit, Understanding Molecular Simulation, Academic Press, San Diego, M.P. Allen and D.J. Tildesley, Computer Simulation of Liquids, Oxford Science Publications, Clarendon Press, Oxford, D. C. Rapaport, The Art of Molecular Dynamics Simulation, Cambridge University Press, D. P. Landau and K. Binder, A guide to Monte Carlo Simulations in Statistical Physics, Cambridge, M. E. J. Newman and G. T. Barkema, Monte Carlo Methods in Statistical Physics. Oxford University Press, J.M. Thijssen, Computational Physics, Cambridge (2007) S. Succi, The Lattice Boltzmann Equation for Fluid Dynamics and Beyond, Oxford Science Publ. (2001). M.E. Tuckermann, Statistical Mechanics: Theory and Moleculr Simulation, Oxford Graduate Texts (2010). M.O. Steinhauser, Computational Multiscale Modeling of Fluids and Solids, Springer, (2008). A. Leach, Molecular Modelling: Principles and Applications, Pearson Education Ltd. (2001). R.M. Martin, Electronic Stucture, Basic Theory and Practical Methods, Cambridge (2004). E. Kaxiras, Atomic and electronic structure of solids, Cambridge (2003). M.Fyta 4/20


5 Computational Physics Bridge theory and experiments Verify or guide experiments Involves different spatial and temporal scales Extraction of a wider range of properties, mechanical, thermodynamic, optical, electronic, etc... Accuracy vs. Efficiency! M.Fyta 5/20
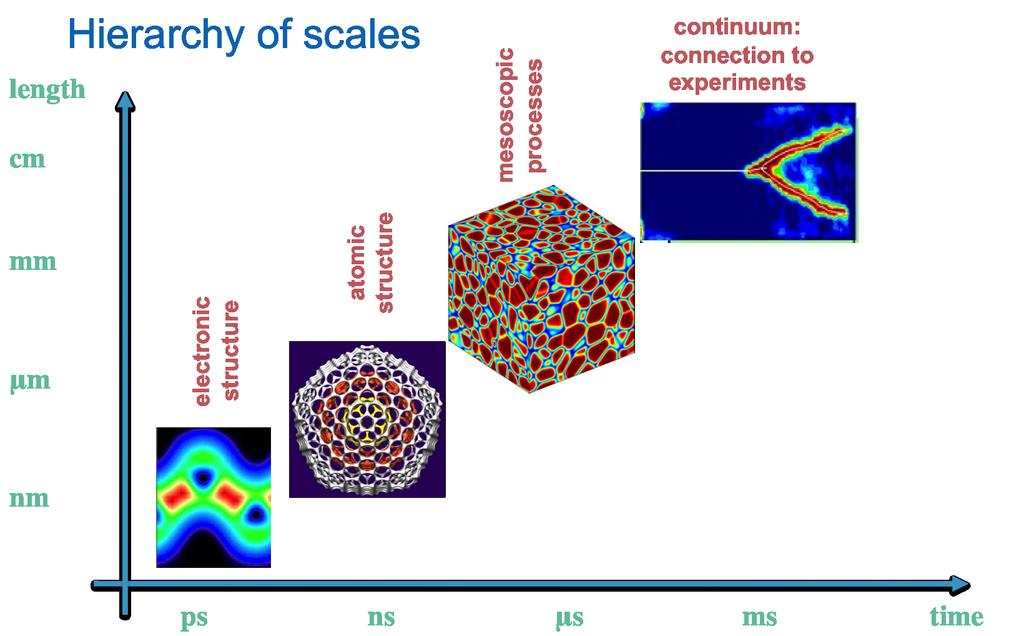
6 M.Fyta 6/20
7 M.Fyta 7/20
8 Introduction to electronic structure Source: commons.wikimedia.org Different properties according to atom type and number, i.e. number of electrons and their spatial and electronic configurations. M.Fyta 8/20

9 Electronic configuration Distribution of electrons in atoms/molecules in atomic/molecular orbitals Electrons described as moving independently in orbitals in an average field created by othe orbitals Electrons jump between configurations through emission/absorption of a photon Configurations are described by Slater determinants Source: chemistry.beloit.edu Energy associated with each electron is that of its orbital. Ground state: The configuration that corresponds to the lowest electronic energy. Excited state: Any other configuration. M.Fyta 9/20

10 Periodic table of the elements Source: chemistry.about.com Example for Carbon [C] 6 electrons: ground state = 1s 2 2s 2 2p 2 M.Fyta 10/20

11 Bulk vs. finite systems - electronic structure Source:
12 Bulk systems (crystalline/non-crystalline materials) Energy bands, band gap=(valence conduction) band energy Band structure (in k-space) Electronic density of states (edos) Example: Carbon, diamond [Dadsetani & Pourghazi, Diam. Rel. Mater., 15, 1695 (2006)]
 energy electronic density of states Example: Carbon, diamondoid [McIntosh et al, PRB, 70, 045401 (2001)] http://www.icp.uni-stuttgart.de M.")
13 Finite systems - (bio)molecules, clusters Distinct energy levels HOMO (highest occupied molecular orbital), LUMO (lowest unoccupied molecular orbial) band gap= (HOMO LUMO) energy electronic density of states Example: Carbon, diamondoid [McIntosh et al, PRB, 70, (2001)] M.Fyta 13/20
14 Electronic structure Probability distribution of electrons in chemical systems State of motion of electrons in an electrostatic field created by the nuclei Extraction of wavefunctions and associated energies through the Schrödinger equation: i t Ψ = ĤΨ time dependent EΨ = ĤΨ time independent Solves for: bonding and structure electronic, magnetic, and optical properties of materials chemistry and reactions. M.Fyta 14/20
15 Time-independent molecular Schrödinger equation ( ˆT e + V ee + V ek + ˆT k + V kk )Ψ(r, R) = EΨ(r, R) r, R: electron, nucleus coordinates ˆT e, ˆT k : electron, nucleus kinetic energy operator V ee : electron-electron repulsion V ek : electron-nuclear attraction V kk : nuclear-nuclear repulsion E: total molecular energy Ψ(r, R): total molecular wavefunction Born-Oppenheimer approximation Electrons much faster than nuclei separate nuclear from electronic motion Solve electronic and nuclear Schrödinger equation, Ψ e (r; R) and Ψ k (r) with Ψ = Ψ k Ψ e. M.Fyta 15/20
16 Approximations in electronic structure methods Common approximations: in the Hamiltonian, e.g. changing from a wavefunction-based to a density-based description of the electronic interaction simplification of the electronic interaction term in the description of the many-electron wavefunction Often the electronic wavefunction of a system is expanded in terms of Slater determinants, as a sum of anti-symmetric electron wavefunctions: Ψ el ( r 1, s 1, r 2, s 2,..., r N, s N ) = m 1,m 2,...,m N C m1,m 2,...,m N φ m1 ( r 1, s 1 )φ m2 ( r 2, s 2 )...φ mn ( r N, s N ) where r i, s 1 the cartesian coordinates and the spin components. The components φ mn ( r N, s N ) are one-electron orbitals. M.Fyta 16/20
17 Basis-sets wavefunctions represented as vectors compontents of vectors correspond to coefficients related to basis-set basis-sets: set of functions combined (typically in linear combinations) to create the wavefunctions of the system Choices Bulk systems: plane waves, atomic-like orbitals Molecules/finite systems: atomic-like orbitals Finite basis set ; computation: always an approximation The smaller the basis, the poorer the representation, i.e. accuracy of results The larger the basis, the larger the computational load. M.Fyta 17/20
18 Basis-sets Plane-waves Periodic functions Bloch s theorem for periodic solids: φ mn ( r N, s N ) = u n,k ( r)exp(i k r) Periodic u expanded in plane waves with expansion coefficients depending on the reciprocal lattice vectors: u n,k ( r) = c nk ( G)exp(i G r) G G max Atomic-like orbitals φ mn ( r N, s N ) = n D nmχ n ( r) Gaussian-type orbitals: χ ζ,n,l,m (r, θ, φ) = NY l,m (θ, φ)r 2n 2 l exp( ζr 2 ) χ ζ,lx,l y,l z (x, y, z) = Nx lx y ly z lz exp( ζr 2 ) the sum l x, l y, l z determines the orbital. M.Fyta 18/20
19 Optimization: wavefunctions and geometries numerical approximations of the wavefunction by successive iterations variational principle, convergence by minimizing the total energy: E Φ H Φ geometry optimization: nuclear forces computed at the end of wavefunction optimization process nuclei shifted along direction of computed forces new wavefunction(new positions) process until convergence: final geometry corresponds to global minimum of potential surface energy Self consistent field (SCF) Particles in the mean field created by the other particles Final field as computed from the wavefunction or charge density is self-consistent with the assumed initial field equations almost universally solved through an iterative method. M.Fyta 19/20
20 ab initio methods Solve Schrödinger s equation associated with the Hamiltonian of the system ab initio (first-principles): methods which use established laws of physics and do not include empirical or semi-empirical parameters; derived directly from theoretical principles, with no inclusion of experimental data Popular ab initio methods Hartree-Fock (Density functional theory) Møller-Plesset perturbation theory Multi-configurations self consistent field (MCSCF) Configuration interaction (CI), Multi-reference configuration interaction Coupled cluster (CC) Quantum Monte Carlo Reduced density matrix approaches M.Fyta 20/20
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry room B430, Chemicum 4th floor vesa.hanninen@helsinki.fi September 3, 2013 Introduction and theoretical backround September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry room B430, Chemicum 4th floor vesa.hanninen@helsinki.fi September 3, 2013 Introduction and theoretical backround September
Session 1. Introduction to Computational Chemistry. Computational (chemistry education) and/or (Computational chemistry) education
 and/or (Computational chemistry) education") Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
Session 1 Introduction to Computational Chemistry 1 Introduction to Computational Chemistry Computational (chemistry education) and/or (Computational chemistry) education First one: Use computational tools
AN INTRODUCTION TO QUANTUM CHEMISTRY. Mark S. Gordon Iowa State University
 AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
AN INTRODUCTION TO QUANTUM CHEMISTRY Mark S. Gordon Iowa State University 1 OUTLINE Theoretical Background in Quantum Chemistry Overview of GAMESS Program Applications 2 QUANTUM CHEMISTRY In principle,
Electron Correlation
 Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
Electron Correlation Levels of QM Theory HΨ=EΨ Born-Oppenheimer approximation Nuclear equation: H n Ψ n =E n Ψ n Electronic equation: H e Ψ e =E e Ψ e Single determinant SCF Semi-empirical methods Correlation
CHEM3023: Spins, Atoms and Molecules
 CHEM3023: Spins, Atoms and Molecules Lecture 5 The Hartree-Fock method C.-K. Skylaris Learning outcomes Be able to use the variational principle in quantum calculations Be able to construct Fock operators
CHEM3023: Spins, Atoms and Molecules Lecture 5 The Hartree-Fock method C.-K. Skylaris Learning outcomes Be able to use the variational principle in quantum calculations Be able to construct Fock operators
Chemistry 334 Part 2: Computational Quantum Chemistry
 Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Chemistry 334 Part 2: Computational Quantum Chemistry 1. Definition Louis Scudiero, Ben Shepler and Kirk Peterson Washington State University January 2006 Computational chemistry is an area of theoretical
Ab initio calculations for potential energy surfaces. D. Talbi GRAAL- Montpellier
 Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
Ab initio calculations for potential energy surfaces D. Talbi GRAAL- Montpellier A theoretical study of a reaction is a two step process I-Electronic calculations : techniques of quantum chemistry potential
MO Calculation for a Diatomic Molecule. /4 0 ) i=1 j>i (1/r ij )
 i=1 j>i (1/r ij )") MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
MO Calculation for a Diatomic Molecule Introduction The properties of any molecular system can in principle be found by looking at the solutions to the corresponding time independent Schrodinger equation
Exercise 1: Structure and dipole moment of a small molecule
 Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
Introduction to computational chemistry Exercise 1: Structure and dipole moment of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the dipole moment of a small
Condensed matter physics FKA091
 Condensed matter physics FKA091 Ermin Malic Department of Physics Chalmers University of Technology Henrik Johannesson Department of Physics University of Gothenburg Teaching assistants: Roland Jago &
Condensed matter physics FKA091 Ermin Malic Department of Physics Chalmers University of Technology Henrik Johannesson Department of Physics University of Gothenburg Teaching assistants: Roland Jago &
An Introduction to Quantum Chemistry and Potential Energy Surfaces. Benjamin G. Levine
 An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
An Introduction to Quantum Chemistry and Potential Energy Surfaces Benjamin G. Levine This Week s Lecture Potential energy surfaces What are they? What are they good for? How do we use them to solve chemical
Introduction to Electronic Structure Theory
 Introduction to Electronic Structure Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology June 2002 Last Revised: June 2003 1 Introduction The purpose of these
Introduction to Electronic Structure Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology June 2002 Last Revised: June 2003 1 Introduction The purpose of these
Computational Chemistry. An Introduction to Molecular Dynamic Simulations
 Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Computational Chemistry An Introduction to Molecular Dynamic Simulations Computational chemistry simulates chemical structures and reactions numerically, based in full or in part on the fundamental laws
Intro to ab initio methods
 Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Lecture 2 Part A Intro to ab initio methods Recommended reading: Leach, Chapters 2 & 3 for QM methods For more QM methods: Essentials of Computational Chemistry by C.J. Cramer, Wiley (2002) 1 ab initio
Quantum Mechanical Simulations
 Quantum Mechanical Simulations Prof. Yan Wang Woodruff School of Mechanical Engineering Georgia Institute of Technology Atlanta, GA 30332, U.S.A. yan.wang@me.gatech.edu Topics Quantum Monte Carlo Hartree-Fock
Quantum Mechanical Simulations Prof. Yan Wang Woodruff School of Mechanical Engineering Georgia Institute of Technology Atlanta, GA 30332, U.S.A. yan.wang@me.gatech.edu Topics Quantum Monte Carlo Hartree-Fock
Jack Simons, Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
2~:J~ -ryej- r- 2 Jr. A - f3. sr(djk nv~tor rn~ +~ rvjs (::-CJ) ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.
 ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.") .~, ~ I, sr(djk nv~tor rn~ +~ rvjs (::-CJ) ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.)1e'" 21t-ol Je C'...-------- lj-vi, J? Jr Jr \Ji 2~:J~ -ryej- r- 2 Jr A - f3 c _,~,= ~,.,w._..._.
.~, ~ I, sr(djk nv~tor rn~ +~ rvjs (::-CJ) ::;-1-.'--~ -. rhd. ('-.Ji.L.~ )- r'-d)c, -r/~ JJr - 2~d ~2-Jr fn'6.)1e'" 21t-ol Je C'...-------- lj-vi, J? Jr Jr \Ji 2~:J~ -ryej- r- 2 Jr A - f3 c _,~,= ~,.,w._..._.
Chemistry 4560/5560 Molecular Modeling Fall 2014
 Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Final Exam Name:. User s guide: 1. Read questions carefully and make sure you understand them before answering (if not, ask). 2. Answer only the question that is asked, not a different question. 3. Unless
Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule. Vesa Hänninen
 Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the electronic energy
Introduction to computational chemistry Exercise I: Structure and electronic energy of a small molecule Vesa Hänninen 1 Introduction In this exercise the equilibrium structure and the electronic energy
Quantum Chemical Simulations and Descriptors. Dr. Antonio Chana, Dr. Mosè Casalegno
 Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Quantum Chemical Simulations and Descriptors Dr. Antonio Chana, Dr. Mosè Casalegno Classical Mechanics: basics It models real-world objects as point particles, objects with negligible size. The motion
Part 2 Electronic Structure Theory
 HS 2013: Vorlesung Physikalische Chemie III Part 2 Electronic Structure Theory Lecture 6 Dr. Mar'n O. Steinhauser Fraunhofer Ins'tute for High- Speed Dynamics, Ernst- Mach- Ins'tut, EMI, Freiburg Email:
HS 2013: Vorlesung Physikalische Chemie III Part 2 Electronic Structure Theory Lecture 6 Dr. Mar'n O. Steinhauser Fraunhofer Ins'tute for High- Speed Dynamics, Ernst- Mach- Ins'tut, EMI, Freiburg Email:
Introduction to Hartree-Fock Molecular Orbital Theory
 Introduction to Hartree-Fock Molecular Orbital Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Origins of Mathematical Modeling in Chemistry Plato (ca. 428-347
Introduction to Hartree-Fock Molecular Orbital Theory C. David Sherrill School of Chemistry and Biochemistry Georgia Institute of Technology Origins of Mathematical Modeling in Chemistry Plato (ca. 428-347
Handbook of Computational Quantum Chemistry. DAVID B. COOK The Department of Chemistry, University of Sheffield
 Handbook of Computational Quantum Chemistry DAVID B. COOK The Department of Chemistry, University of Sheffield Oxford New York Tokyo OXFORD UNIVERSITY PRESS 1998 CONTENTS 1 Mechanics and molecules 1 1.1
Handbook of Computational Quantum Chemistry DAVID B. COOK The Department of Chemistry, University of Sheffield Oxford New York Tokyo OXFORD UNIVERSITY PRESS 1998 CONTENTS 1 Mechanics and molecules 1 1.1
Molecular Simulation I
 Molecular Simulation I Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
Molecular Simulation I Quantum Chemistry Classical Mechanics E = Ψ H Ψ ΨΨ U = E bond +E angle +E torsion +E non-bond Jeffry D. Madura Department of Chemistry & Biochemistry Center for Computational Sciences
1 Rayleigh-Schrödinger Perturbation Theory
 1 Rayleigh-Schrödinger Perturbation Theory All perturbative techniques depend upon a few simple assumptions. The first of these is that we have a mathematical expression for a physical quantity for which
1 Rayleigh-Schrödinger Perturbation Theory All perturbative techniques depend upon a few simple assumptions. The first of these is that we have a mathematical expression for a physical quantity for which
0 belonging to the unperturbed Hamiltonian H 0 are known
 Time Independent Perturbation Theory D Perturbation theory is used in two qualitatively different contexts in quantum chemistry. It allows one to estimate (because perturbation theory is usually employed
Time Independent Perturbation Theory D Perturbation theory is used in two qualitatively different contexts in quantum chemistry. It allows one to estimate (because perturbation theory is usually employed
Chemistry 2000 Lecture 1: Introduction to the molecular orbital theory
 Chemistry 2000 Lecture 1: Introduction to the molecular orbital theory Marc R. Roussel January 5, 2018 Marc R. Roussel Introduction to molecular orbitals January 5, 2018 1 / 24 Review: quantum mechanics
Chemistry 2000 Lecture 1: Introduction to the molecular orbital theory Marc R. Roussel January 5, 2018 Marc R. Roussel Introduction to molecular orbitals January 5, 2018 1 / 24 Review: quantum mechanics
VALENCE Hilary Term 2018
 VALENCE Hilary Term 2018 8 Lectures Prof M. Brouard Valence is the theory of the chemical bond Outline plan 1. The Born-Oppenheimer approximation 2. Bonding in H + 2 the LCAO approximation 3. Many electron
VALENCE Hilary Term 2018 8 Lectures Prof M. Brouard Valence is the theory of the chemical bond Outline plan 1. The Born-Oppenheimer approximation 2. Bonding in H + 2 the LCAO approximation 3. Many electron
This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry.
 1 Computational Chemistry (Quantum Chemistry) Primer This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry. TABLE OF CONTENTS Methods...1 Basis
1 Computational Chemistry (Quantum Chemistry) Primer This is a very succinct primer intended as supplementary material for an undergraduate course in physical chemistry. TABLE OF CONTENTS Methods...1 Basis
(1/2) M α 2 α, ˆTe = i. 1 r i r j, ˆV NN = α>β
 M α 2 α, ˆTe = i. 1 r i r j, ˆV NN = α>β") Chemistry 26 Spectroscopy Week # The Born-Oppenheimer Approximation, H + 2. Born-Oppenheimer approximation As for atoms, all information about a molecule is contained in the wave function Ψ, which is the
Chemistry 26 Spectroscopy Week # The Born-Oppenheimer Approximation, H + 2. Born-Oppenheimer approximation As for atoms, all information about a molecule is contained in the wave function Ψ, which is the
Density Functional Theory. Martin Lüders Daresbury Laboratory
 Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Density Functional Theory Martin Lüders Daresbury Laboratory Ab initio Calculations Hamiltonian: (without external fields, non-relativistic) impossible to solve exactly!! Electrons Nuclei Electron-Nuclei
Computational Methods. Chem 561
 Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Computational Methods Chem 561 Lecture Outline 1. Ab initio methods a) HF SCF b) Post-HF methods 2. Density Functional Theory 3. Semiempirical methods 4. Molecular Mechanics Computational Chemistry " Computational
Introduction to Computational Chemistry: Theory
 Introduction to Computational Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC andrew.gilbert@anu.edu.au 3023 Course Lectures Introduction Hartree Fock Theory Basis Sets Lecture 1 1 Introduction
Introduction to Computational Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC andrew.gilbert@anu.edu.au 3023 Course Lectures Introduction Hartree Fock Theory Basis Sets Lecture 1 1 Introduction
Introduction to Electronic Structure Theory
 CSC/PRACE Spring School in Computational Chemistry 2017 Introduction to Electronic Structure Theory Mikael Johansson http://www.iki.fi/~mpjohans Objective: To get familiarised with the, subjectively chosen,
CSC/PRACE Spring School in Computational Chemistry 2017 Introduction to Electronic Structure Theory Mikael Johansson http://www.iki.fi/~mpjohans Objective: To get familiarised with the, subjectively chosen,
CE 530 Molecular Simulation
 1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
1 CE 530 Molecular Simulation Lecture 14 Molecular Models David A. Kofke Department of Chemical Engineering SUNY Buffalo kofke@eng.buffalo.edu 2 Review Monte Carlo ensemble averaging, no dynamics easy
2 Electronic structure theory
 Electronic structure theory. Generalities.. Born-Oppenheimer approximation revisited In Sec..3 (lecture 3) the Born-Oppenheimer approximation was introduced (see also, for instance, [Tannor.]). We are
Electronic structure theory. Generalities.. Born-Oppenheimer approximation revisited In Sec..3 (lecture 3) the Born-Oppenheimer approximation was introduced (see also, for instance, [Tannor.]). We are
Introduction to Computational Quantum Chemistry: Theory
 Introduction to Computational Quantum Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC 3108 Course Lectures 2007 Introduction Hartree Fock Theory Configuration Interaction Lectures 1 Introduction
Introduction to Computational Quantum Chemistry: Theory Dr Andrew Gilbert Rm 118, Craig Building, RSC 3108 Course Lectures 2007 Introduction Hartree Fock Theory Configuration Interaction Lectures 1 Introduction
OVERVIEW OF QUANTUM CHEMISTRY METHODS
 OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
OVERVIEW OF QUANTUM CHEMISTRY METHODS Outline I Generalities Correlation, basis sets Spin II Wavefunction methods Hartree-Fock Configuration interaction Coupled cluster Perturbative methods III Density
Electronic structure calculations: fundamentals George C. Schatz Northwestern University
 Electronic structure calculations: fundamentals George C. Schatz Northwestern University Electronic Structure (often called Quantum Chemistry) calculations use quantum mechanics to determine the wavefunctions
Electronic structure calculations: fundamentals George C. Schatz Northwestern University Electronic Structure (often called Quantum Chemistry) calculations use quantum mechanics to determine the wavefunctions
Electron Correlation - Methods beyond Hartree-Fock
 Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Electron Correlation - Methods beyond Hartree-Fock how to approach chemical accuracy Alexander A. Auer Max-Planck-Institute for Chemical Energy Conversion, Mülheim September 4, 2014 MMER Summerschool 2014
Algorithms and Computational Aspects of DFT Calculations
 Algorithms and Computational Aspects of DFT Calculations Part I Juan Meza and Chao Yang High Performance Computing Research Lawrence Berkeley National Laboratory IMA Tutorial Mathematical and Computational
Algorithms and Computational Aspects of DFT Calculations Part I Juan Meza and Chao Yang High Performance Computing Research Lawrence Berkeley National Laboratory IMA Tutorial Mathematical and Computational
Introduction to Density Functional Theory
 1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
1 Introduction to Density Functional Theory 21 February 2011; V172 P.Ravindran, FME-course on Ab initio Modelling of solar cell Materials 21 February 2011 Introduction to DFT 2 3 4 Ab initio Computational
The Hartree-Fock approximation
 Contents The Born-Oppenheimer approximation Literature Quantum mechanics 2 - Lecture 7 November 21, 2012 Contents The Born-Oppenheimer approximation Literature 1 The Born-Oppenheimer approximation 2 3
Contents The Born-Oppenheimer approximation Literature Quantum mechanics 2 - Lecture 7 November 21, 2012 Contents The Born-Oppenheimer approximation Literature 1 The Born-Oppenheimer approximation 2 3
Study of Ozone in Tribhuvan University, Kathmandu, Nepal. Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal
 Study of Ozone in Tribhuvan University, Kathmandu, Nepal Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal 1 Country of the Mt Everest 2 View of the Mt Everest 3 4 5
Study of Ozone in Tribhuvan University, Kathmandu, Nepal Prof. S. Gurung Central Department of Physics, Tribhuvan University, Kathmandu, Nepal 1 Country of the Mt Everest 2 View of the Mt Everest 3 4 5
Introduction to Density Functional Theory
 Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Introduction to Density Functional Theory S. Sharma Institut für Physik Karl-Franzens-Universität Graz, Austria 19th October 2005 Synopsis Motivation 1 Motivation : where can one use DFT 2 : 1 Elementary
Summary of the new Modelling Vocabulary
 Summary of the new Modelling Vocabulary These two pages attempts to summarise in a concise manner the Modelling Vocabulary. What are Models? What are Simulations? Materials Models consist of Physics or
Summary of the new Modelling Vocabulary These two pages attempts to summarise in a concise manner the Modelling Vocabulary. What are Models? What are Simulations? Materials Models consist of Physics or
Wave function methods for the electronic Schrödinger equation
 Wave function methods for the electronic Schrödinger equation Zürich 2008 DFG Reseach Center Matheon: Mathematics in Key Technologies A7: Numerical Discretization Methods in Quantum Chemistry DFG Priority
Wave function methods for the electronic Schrödinger equation Zürich 2008 DFG Reseach Center Matheon: Mathematics in Key Technologies A7: Numerical Discretization Methods in Quantum Chemistry DFG Priority
André Schleife Department of Materials Science and Engineering
 André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
André Schleife Department of Materials Science and Engineering Yesterday you (should have) learned this: http://upload.wikimedia.org/wikipedia/commons/e/ea/ Simple_Harmonic_Motion_Orbit.gif 1. deterministic
The Overhauser Instability
 The Overhauser Instability Zoltán Radnai and Richard Needs TCM Group ESDG Talk 14th February 2007 Typeset by FoilTEX Introduction Hartree-Fock theory and Homogeneous Electron Gas Noncollinear spins and
The Overhauser Instability Zoltán Radnai and Richard Needs TCM Group ESDG Talk 14th February 2007 Typeset by FoilTEX Introduction Hartree-Fock theory and Homogeneous Electron Gas Noncollinear spins and
Electronic Structure Calculations and Density Functional Theory
 Electronic Structure Calculations and Density Functional Theory Rodolphe Vuilleumier Pôle de chimie théorique Département de chimie de l ENS CNRS Ecole normale supérieure UPMC Formation ModPhyChem Lyon,
Electronic Structure Calculations and Density Functional Theory Rodolphe Vuilleumier Pôle de chimie théorique Département de chimie de l ENS CNRS Ecole normale supérieure UPMC Formation ModPhyChem Lyon,
Electron Correlation Methods
 Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Electron Correlation Methods HF method: electron-electron interaction is replaced by an average interaction E HF c = E 0 E HF E 0 exact ground state energy E HF HF energy for a given basis set HF E c
Electric properties of molecules
 Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Electric properties of molecules For a molecule in a uniform electric fielde the Hamiltonian has the form: Ĥ(E) = Ĥ + E ˆµ x where we assume that the field is directed along the x axis and ˆµ x is the
Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012
 Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012 K. Kremer Max Planck Institute for Polymer Research, Mainz Overview Simulations, general considerations
Introduction to Computer Simulations of Soft Matter Methodologies and Applications Boulder July, 19-20, 2012 K. Kremer Max Planck Institute for Polymer Research, Mainz Overview Simulations, general considerations
Gustavus Adolphus College. Lab #5: Computational Chemistry
 CHE 372 Gustavus Adolphus College Lab #5: Computational Chemistry Introduction In this investigation we will apply the techniques of computational chemistry to several of the molecular systems that we
CHE 372 Gustavus Adolphus College Lab #5: Computational Chemistry Introduction In this investigation we will apply the techniques of computational chemistry to several of the molecular systems that we
Multi-reference Density Functional Theory. COLUMBUS Workshop Argonne National Laboratory 15 August 2005
 Multi-reference Density Functional Theory COLUMBUS Workshop Argonne National Laboratory 15 August 2005 Capt Eric V. Beck Air Force Institute of Technology Department of Engineering Physics 2950 Hobson
Multi-reference Density Functional Theory COLUMBUS Workshop Argonne National Laboratory 15 August 2005 Capt Eric V. Beck Air Force Institute of Technology Department of Engineering Physics 2950 Hobson
Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering
 Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering Outline PART 1: Fundamentals of Density functional theory (DFT)
Fundamentals and applications of Density Functional Theory Astrid Marthinsen PhD candidate, Department of Materials Science and Engineering Outline PART 1: Fundamentals of Density functional theory (DFT)
Chemistry, Physics and the Born- Oppenheimer Approximation
 Chemistry, Physics and the Born- Oppenheimer Approximation Hendrik J. Monkhorst Quantum Theory Project University of Florida Gainesville, FL 32611-8435 Outline 1. Structure of Matter 2. Shell Models 3.
Chemistry, Physics and the Born- Oppenheimer Approximation Hendrik J. Monkhorst Quantum Theory Project University of Florida Gainesville, FL 32611-8435 Outline 1. Structure of Matter 2. Shell Models 3.
FYS-6306 QUANTUM THEORY OF MOLECULES AND NANOSTRUCTURES
 i FYS-6306 QUANTUM THEORY OF MOLECULES AND NANOSTRUCTURES Credit units: 6 ECTS Lectures: 48 h Tapio Rantala, prof. Tue 10 12 SC203 SG219 8 10 SG312 FirstName.LastName@tut.fi http://www.tut.fi/~trantala/opetus/
i FYS-6306 QUANTUM THEORY OF MOLECULES AND NANOSTRUCTURES Credit units: 6 ECTS Lectures: 48 h Tapio Rantala, prof. Tue 10 12 SC203 SG219 8 10 SG312 FirstName.LastName@tut.fi http://www.tut.fi/~trantala/opetus/
Computational Chemistry. Ab initio methods seek to solve the Schrödinger equation.
 Theory Computational Chemistry Ab initio methods seek to solve the Schrödinger equation. Molecular orbital theory expresses the solution as a linear combination of atomic orbitals. Density functional theory
Theory Computational Chemistry Ab initio methods seek to solve the Schrödinger equation. Molecular orbital theory expresses the solution as a linear combination of atomic orbitals. Density functional theory
Set the initial conditions r i. Update neighborlist. Get new forces F i
 v Set the initial conditions r i ( t 0 ), v i ( t 0 ) Update neighborlist Quantum mechanical models Get new forces F i ( r i ) Solve the equations of motion numerically over time step Δt : r i ( t n )
v Set the initial conditions r i ( t 0 ), v i ( t 0 ) Update neighborlist Quantum mechanical models Get new forces F i ( r i ) Solve the equations of motion numerically over time step Δt : r i ( t n )
Lecture 10. Born-Oppenheimer approximation LCAO-MO application to H + The potential energy surface MOs for diatomic molecules. NC State University
 Chemistry 431 Lecture 10 Diatomic molecules Born-Oppenheimer approximation LCAO-MO application to H + 2 The potential energy surface MOs for diatomic molecules NC State University Born-Oppenheimer approximation
Chemistry 431 Lecture 10 Diatomic molecules Born-Oppenheimer approximation LCAO-MO application to H + 2 The potential energy surface MOs for diatomic molecules NC State University Born-Oppenheimer approximation
Yingwei Wang Computational Quantum Chemistry 1 Hartree energy 2. 2 Many-body system 2. 3 Born-Oppenheimer approximation 2
 Purdue University CHM 67300 Computational Quantum Chemistry REVIEW Yingwei Wang October 10, 2013 Review: Prof Slipchenko s class, Fall 2013 Contents 1 Hartree energy 2 2 Many-body system 2 3 Born-Oppenheimer
Purdue University CHM 67300 Computational Quantum Chemistry REVIEW Yingwei Wang October 10, 2013 Review: Prof Slipchenko s class, Fall 2013 Contents 1 Hartree energy 2 2 Many-body system 2 3 Born-Oppenheimer
Introduction to Computational Chemistry
 Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Introduction to Computational Chemistry Vesa Hänninen Laboratory of Physical Chemistry Chemicum 4th floor vesa.hanninen@helsinki.fi September 10, 2013 Lecture 3. Electron correlation methods September
Lecture 4: methods and terminology, part II
 So theory guys have got it made in rooms free of pollution. Instead of problems with the reflux, they have only solutions... In other words, experimentalists will likely die of cancer From working hard,
So theory guys have got it made in rooms free of pollution. Instead of problems with the reflux, they have only solutions... In other words, experimentalists will likely die of cancer From working hard,
Chemistry 3502/4502. Final Exam Part I. May 14, 2005
 Chemistry 3502/4502 Final Exam Part I May 14, 2005 1. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle (e) The
Chemistry 3502/4502 Final Exam Part I May 14, 2005 1. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle (e) The
Handbook of Computational Quantum Chemistry
 Handbook of Computational Quantum Chemistry David B. Cook Dept. of Chemistry University of Sheffield DOVER PUBLICATIONS, INC. Mineola, New York F Contents 1 Mechanics and molecules 1 1.1 1.2 1.3 1.4 1.5
Handbook of Computational Quantum Chemistry David B. Cook Dept. of Chemistry University of Sheffield DOVER PUBLICATIONS, INC. Mineola, New York F Contents 1 Mechanics and molecules 1 1.1 1.2 1.3 1.4 1.5
Density Functional Theory
 Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Density Functional Theory Iain Bethune EPCC ibethune@epcc.ed.ac.uk Overview Background Classical Atomistic Simulation Essential Quantum Mechanics DFT: Approximations and Theory DFT: Implementation using
Preface Introduction to the electron liquid
 Table of Preface page xvii 1 Introduction to the electron liquid 1 1.1 A tale of many electrons 1 1.2 Where the electrons roam: physical realizations of the electron liquid 5 1.2.1 Three dimensions 5 1.2.2
Table of Preface page xvii 1 Introduction to the electron liquid 1 1.1 A tale of many electrons 1 1.2 Where the electrons roam: physical realizations of the electron liquid 5 1.2.1 Three dimensions 5 1.2.2
Molecular Aggregation
 Molecular Aggregation Structure Analysis and Molecular Simulation of Crystals and Liquids ANGELO GAVEZZOTTI University of Milano OXFORD UNIVERSITY PRESS Contents PART I FUNDAMENTALS 1 The molecule: structure,
Molecular Aggregation Structure Analysis and Molecular Simulation of Crystals and Liquids ANGELO GAVEZZOTTI University of Milano OXFORD UNIVERSITY PRESS Contents PART I FUNDAMENTALS 1 The molecule: structure,
DFT calculations of NMR indirect spin spin coupling constants
 DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
DFT calculations of NMR indirect spin spin coupling constants Dalton program system Program capabilities Density functional theory Kohn Sham theory LDA, GGA and hybrid theories Indirect NMR spin spin coupling
QUANTUM CHEMISTRY FOR TRANSITION METALS
 QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
QUANTUM CHEMISTRY FOR TRANSITION METALS Outline I Introduction II Correlation Static correlation effects MC methods DFT III Relativity Generalities From 4 to 1 components Effective core potential Outline
Exam 4 Review. Exam Review: A exam review sheet for exam 4 will be posted on the course webpage. Additionally, a practice exam will also be posted.
 Chem 4502 Quantum Mechanics & Spectroscopy (Jason Goodpaster) Exam 4 Review Exam Review: A exam review sheet for exam 4 will be posted on the course webpage. Additionally, a practice exam will also be
Chem 4502 Quantum Mechanics & Spectroscopy (Jason Goodpaster) Exam 4 Review Exam Review: A exam review sheet for exam 4 will be posted on the course webpage. Additionally, a practice exam will also be
Three Most Important Topics (MIT) Today
 Today") Three Most Important Topics (MIT) Today Electrons in periodic potential Energy gap nearly free electron Bloch Theorem Energy gap tight binding Chapter 1 1 Electrons in Periodic Potential We now know the
Three Most Important Topics (MIT) Today Electrons in periodic potential Energy gap nearly free electron Bloch Theorem Energy gap tight binding Chapter 1 1 Electrons in Periodic Potential We now know the
Electronic Structure of Crystalline Solids
 Electronic Structure of Crystalline Solids Computing the electronic structure of electrons in solid materials (insulators, conductors, semiconductors, superconductors) is in general a very difficult problem
Electronic Structure of Crystalline Solids Computing the electronic structure of electrons in solid materials (insulators, conductors, semiconductors, superconductors) is in general a very difficult problem
Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs
 Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs Founded in 1993, Q-Chem strives to bring its customers state-ofthe-art methods and algorithms for performing quantum chemistry calculations. Cutting-edge
Q-Chem 5: Facilitating Worldwide Scientific Breakthroughs Founded in 1993, Q-Chem strives to bring its customers state-ofthe-art methods and algorithms for performing quantum chemistry calculations. Cutting-edge
Electron States of Diatomic Molecules
 IISER Pune March 2018 Hamiltonian for a Diatomic Molecule The hamiltonian for a diatomic molecule can be considered to be made up of three terms Ĥ = ˆT N + ˆT el + ˆV where ˆT N is the kinetic energy operator
IISER Pune March 2018 Hamiltonian for a Diatomic Molecule The hamiltonian for a diatomic molecule can be considered to be made up of three terms Ĥ = ˆT N + ˆT el + ˆV where ˆT N is the kinetic energy operator
Ch 125a Problem Set 1
 Ch 5a Problem Set Due Monday, Oct 5, 05, am Problem : Bra-ket notation (Dirac notation) Bra-ket notation is a standard and convenient way to describe quantum state vectors For example, φ is an abstract
Ch 5a Problem Set Due Monday, Oct 5, 05, am Problem : Bra-ket notation (Dirac notation) Bra-ket notation is a standard and convenient way to describe quantum state vectors For example, φ is an abstract
Computational Physics. J. M. Thijssen
 Computational Physics J. M. Thijssen Delft University of Technology CAMBRIDGE UNIVERSITY PRESS Contents Preface xi 1 Introduction 1 1.1 Physics and computational physics 1 1.2 Classical mechanics and statistical
Computational Physics J. M. Thijssen Delft University of Technology CAMBRIDGE UNIVERSITY PRESS Contents Preface xi 1 Introduction 1 1.1 Physics and computational physics 1 1.2 Classical mechanics and statistical
Progress toward a Monte Carlo Simulation of the Ice VI-VII Phase Transition
 Progress toward a Monte Carlo Simulation of the Ice VI-VII Phase Transition Christina Gower 2010 NSF/REU PROJECT Physics Department University of Notre Dame Advisor: Dr. Kathie E. Newman August 6, 2010
Progress toward a Monte Carlo Simulation of the Ice VI-VII Phase Transition Christina Gower 2010 NSF/REU PROJECT Physics Department University of Notre Dame Advisor: Dr. Kathie E. Newman August 6, 2010
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY
 DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
DENSITY FUNCTIONAL THEORY FOR NON-THEORISTS JOHN P. PERDEW DEPARTMENTS OF PHYSICS AND CHEMISTRY TEMPLE UNIVERSITY A TUTORIAL FOR PHYSICAL SCIENTISTS WHO MAY OR MAY NOT HATE EQUATIONS AND PROOFS REFERENCES
Jack Simons Henry Eyring Scientist and Professor Chemistry Department University of Utah
 1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
1. Born-Oppenheimer approx.- energy surfaces 2. Mean-field (Hartree-Fock) theory- orbitals 3. Pros and cons of HF- RHF, UHF 4. Beyond HF- why? 5. First, one usually does HF-how? 6. Basis sets and notations
Lec20 Fri 3mar17
 564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
564-17 Lec20 Fri 3mar17 [PDF]GAUSSIAN 09W TUTORIAL www.molcalx.com.cn/wp-content/uploads/2015/01/gaussian09w_tutorial.pdf by A Tomberg - Cited by 8 - Related articles GAUSSIAN 09W TUTORIAL. AN INTRODUCTION
v(r i r j ) = h(r i )+ 1 N
 = h(r i )+ 1 N") Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
Chapter 1 Hartree-Fock Theory 1.1 Formalism For N electrons in an external potential V ext (r), the many-electron Hamiltonian can be written as follows: N H = [ p i i=1 m +V ext(r i )]+ 1 N N v(r i r j
Molecular Dynamics for Microsystems. Introduction Evgenii B. Rudnyi and Jan G. Korvink IMTEK Albert Ludwig University Freiburg, Germany
 Molecular Dynamics for Microsystems. Introduction Evgenii B. Rudnyi and Jan G. Korvink IMTEK Albert Ludwig University Freiburg, Germany Preliminaries Learning Goals Why Molecular Simulation? Molecular
Molecular Dynamics for Microsystems. Introduction Evgenii B. Rudnyi and Jan G. Korvink IMTEK Albert Ludwig University Freiburg, Germany Preliminaries Learning Goals Why Molecular Simulation? Molecular
Dept of Mechanical Engineering MIT Nanoengineering group
 1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
1 Dept of Mechanical Engineering MIT Nanoengineering group » To calculate all the properties of a molecule or crystalline system knowing its atomic information: Atomic species Their coordinates The Symmetry
Translation Symmetry, Space Groups, Bloch functions, Fermi energy
 Translation Symmetry, Space Groups, Bloch functions, Fermi energy Roberto Orlando and Silvia Casassa Università degli Studi di Torino July 20, 2015 School Ab initio Modelling of Solids (UniTo) Symmetry
Translation Symmetry, Space Groups, Bloch functions, Fermi energy Roberto Orlando and Silvia Casassa Università degli Studi di Torino July 20, 2015 School Ab initio Modelling of Solids (UniTo) Symmetry
Lecture B6 Molecular Orbital Theory. Sometimes it's good to be alone.
 Lecture B6 Molecular Orbital Theory Sometimes it's good to be alone. Covalent Bond Theories 1. VSEPR (valence shell electron pair repulsion model). A set of empirical rules for predicting a molecular geometry
Lecture B6 Molecular Orbital Theory Sometimes it's good to be alone. Covalent Bond Theories 1. VSEPR (valence shell electron pair repulsion model). A set of empirical rules for predicting a molecular geometry
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Chemistry 3502/4502. Final Exam Part I. May 14, 2005
 Advocacy chit Chemistry 350/450 Final Exam Part I May 4, 005. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle
Advocacy chit Chemistry 350/450 Final Exam Part I May 4, 005. For which of the below systems is = where H is the Hamiltonian operator and T is the kinetic-energy operator? (a) The free particle
Introduction to Hartree-Fock calculations in Spartan
 EE5 in 2008 Hannes Jónsson Introduction to Hartree-Fock calculations in Spartan In this exercise, you will get to use state of the art software for carrying out calculations of wavefunctions for molecues,
EE5 in 2008 Hannes Jónsson Introduction to Hartree-Fock calculations in Spartan In this exercise, you will get to use state of the art software for carrying out calculations of wavefunctions for molecues,
Quantum Chemistry Methods
 1 Quantum Chemistry Methods T. Helgaker, Department of Chemistry, University of Oslo, Norway The electronic Schrödinger equation Hartree Fock theory self-consistent field theory basis functions and basis
1 Quantum Chemistry Methods T. Helgaker, Department of Chemistry, University of Oslo, Norway The electronic Schrödinger equation Hartree Fock theory self-consistent field theory basis functions and basis
Multi-Scale Modeling from First Principles
 m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
m mm Multi-Scale Modeling from First Principles μm nm m mm μm nm space space Predictive modeling and simulations must address all time and Continuum Equations, densityfunctional space scales Rate Equations
CHEM 6343 Advanced Computational Chemistry. Elfi Kraka, 231 FOSC, ext ,
 CHEM 6343 Advanced Computational Chemistry Class location: Lectures, time and location: Lab times and location: Instructor: Elfi Kraka, 231 FOSC, ext 8-2480, ekraka@smu.edu http://smu.edu/catco/ Office
CHEM 6343 Advanced Computational Chemistry Class location: Lectures, time and location: Lab times and location: Instructor: Elfi Kraka, 231 FOSC, ext 8-2480, ekraka@smu.edu http://smu.edu/catco/ Office
Chemistry 2. Lecture 1 Quantum Mechanics in Chemistry
 Chemistry 2 Lecture 1 Quantum Mechanics in Chemistry Your lecturers 8am Assoc. Prof Timothy Schmidt Room 315 timothy.schmidt@sydney.edu.au 93512781 12pm Assoc. Prof. Adam J Bridgeman Room 222 adam.bridgeman@sydney.edu.au
Chemistry 2 Lecture 1 Quantum Mechanics in Chemistry Your lecturers 8am Assoc. Prof Timothy Schmidt Room 315 timothy.schmidt@sydney.edu.au 93512781 12pm Assoc. Prof. Adam J Bridgeman Room 222 adam.bridgeman@sydney.edu.au
Introduction to first-principles modelling and CASTEP
 to first-principles modelling and Phil Hasnip to + Atomistic Simulations If we know what the bonding in a material is beforehand, then we can often find good expressions for the forces between atoms, e.g.
to first-principles modelling and Phil Hasnip to + Atomistic Simulations If we know what the bonding in a material is beforehand, then we can often find good expressions for the forces between atoms, e.g.
Module 6 1. Density functional theory
 Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Module 6 1. Density functional theory Updated May 12, 2016 B A DDFT C K A bird s-eye view of density-functional theory Authors: Klaus Capelle G http://arxiv.org/abs/cond-mat/0211443 R https://trac.cc.jyu.fi/projects/toolbox/wiki/dft
Calculations of band structures
 Chemistry and Physics at Albany Planning for the Future Calculations of band structures using wave-function based correlation methods Elke Pahl Centre of Theoretical Chemistry and Physics Institute of
Chemistry and Physics at Albany Planning for the Future Calculations of band structures using wave-function based correlation methods Elke Pahl Centre of Theoretical Chemistry and Physics Institute of
Computational Chemistry I
 Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
Computational Chemistry I Text book Cramer: Essentials of Quantum Chemistry, Wiley (2 ed.) Chapter 3. Post Hartree-Fock methods (Cramer: chapter 7) There are many ways to improve the HF method. Most of
APPENDIX 2. Semiempirical PM3 (Parametrization Method #3) When analytical solutions cannot be obtained, Schrödinger equation may be solved:
 When analytical solutions cannot be obtained, Schrödinger equation may be solved:") WRNING NOTICE: The experiments described in these materials are potentially hazardous and require a high level of safety training, special facilities and equipment, and supervision by appropriate individuals.
WRNING NOTICE: The experiments described in these materials are potentially hazardous and require a high level of safety training, special facilities and equipment, and supervision by appropriate individuals.
Lecture 5: More about one- Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory.
 Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem
Lecture 5: More about one- determinant wave functions Final words about the Hartree-Fock theory. First step above it by the Møller-Plesset perturbation theory. Items from Lecture 4 Could the Koopmans theorem