Comparative Genomics Background and Strategies. Nitya Sharma, Emily Rogers, Kanika Arora, Zhiming Zhao, Yun Gyeong Lee
|
|
|
- Roxanne Marshall
- 5 years ago
- Views:
Transcription
1 Comparative Genomics Background and Strategies Nitya Sharma, Emily Rogers, Kanika Arora, Zhiming Zhao, Yun Gyeong Lee
2 Introduction



3 Why comparative genomes? h"p:// h"p:// h"p://genome.ucsc.edu/cgi bin/hggateway?org=human&db=hg18&hgsid=
4 Why comparative genomes? Genome information Pan genome Core genome Pathogenome Genome evolution Carriage strain vs virulent strain
5 Genome Structure Small scale: nucleotide Large scale: Gene Synteny: physical co-localization of genetic loci on the same chromosome within an individual or species. Chromosomes (unichromosome; multichromosome)
6 Genome Evolution Local events: point mutations, small insertions and deletions Large scale events: Gene content: indel Gene order: translocation, transposition Gene orientation: inversion Gene number: duplication Chromosome fusion and fission
7 Large scale genome evolution h"p://
8 Signed permutation model (genome evolution) Savva, 2003
9 Main Pipeline Protein/DNA Sequences from Gene Prediciton COG HGT Synteny Phylogenies Virulence Functional Annotation evolutionary history candidate genes/regions for further investigation of pathogenicity

10 Clusters of Orthologous Groups of Proteins (COGs)
11 Main Pipeline Protein / DNA Sequences from Gene Prediciton COG HGT Synteny Phylogenies Virulence Functional Annotation evolutionary history candidate genes/regions for further investigation of pathogenicity

12 Orthologs vs Paralogs Homolog: A gene related to a second gene by descent from a common ancestral DNA sequence. Orthologs are genes in different species that evolved from a common ancestral gene by speciation. Orthologs typically occupy the same functional niche in different species Paralogs are genes evolved by duplication within a genome. Paralogs tend to evolve towards functional diversification
13 Clusters of Orthologous Group of Proteins Represents an attempt on a phylogenetic classification of the proteins encoded in complete genomes Each COG includes proteins that are connected through vertical evolutionary descent Serves as a platform for: Functional annotation of newly sequenced genome Studies of genome evolution
14 Clusters of Orthologous Groups of Proteins Database COGs were delineated by comparing protein sequences encoded in complete genomes, representing major phylogenetic lineages. Each COG consists of individual proteins or groups of paralogs from at least 3 lineages and thus corresponds to an ancient conserved domain. COG database
 Merge triangles with a common side to form COGs Identify multidomain proteins, separate domains and assign to different COGs Examination of large COGs using phylogenetic trees and")
15 Construction of COGs All-against-all sequence comparisons of proteins encoded in complete genome Detection and collapsing obvious paralogs Detect triangles of mutually consistent genome-specific best hits (BeTs) Merge triangles with a common side to form COGs Identify multidomain proteins, separate domains and assign to different COGs Examination of large COGs using phylogenetic trees and splitting them into two or more smaller groups
16 Goal: To look for differential distribution of COGs in different strains of Neisseria meningitidis and use this data to determine the phylogeny Approach: Create a comprehensive list of COGs for Neisseria gonorrhoea (FA 1090), and different strains of Neisseria meningitidis, and create a presence/absence matrix of COGs for each of the strain N. meningitidis strains to be used: Z2491*, MC58*, FAM18, α14, α153, α275 and our strain * List of COGs for these strains present in COG database
17 Protein sequences from a strain BLAST COG Database List of COGs Comprehensive List of COGs Presence / Absence Matrix Phylogenetic Tree
18 Searching for Horizontal Gene Transfer Events Emily Rogers
19 Main Pipeline Protein / DNA Sequences from Gene Prediciton COG HGT Synteny Phylogenies Virulence Functional Annotation evolutionary history candidate genes/regions for further investigation of pathogenicity
20 What are horizontal gene transfers? Horizontal gene transfers are events where an organism acquires genetic material from another organism that is not its ancestor HGT events are believed to be a major phenomena between prokaryotes, and is common among unicellular eukaryotes Thus, we should expect Neisseria meningitidis to exhibit signs of horizontal gene transfer
21 Why do we care about HGT's? HGT's are important because they can mess up your phylogenies, since the history of a gene acquired laterally is not the history of the organism Also, in our investigation of virulence, we would like to investigate the origin of pathogenicity, if any virulent gene came from other similarly pathogenetic organism Horizontal (or lateral) gene transfer is a known method for the acquisition of a block of virulent genes known as pathogenicity islands (PAIs); HGT is what allows quantum leaps in the evolution of a bacteria that can drastically alter its phenotype

22 A tree illustrating HGT's
23 Illustration of HGT vs inheritance fibr/step.jsp
24 How can we detect HGT events? As mentioned earlier, methods can be either intrinsic (using information embodied in the gene of interest alone) or extrinsic (relying on outside knowledge); these are known as signature methods and phylogenetic methods We will be using both to uncover HGT information We will be using a combination of programs that predict potential HGT's and also comparisons to databases of HGT's predicted in other Neisseria meningitidis strains.
25 Programs We found three available on the command line that uses differing methods to predict HGT's These different methods complement each other, and gives us a breadth of predicted HGT's and also a level of confidence on any agreements Available methods for identifying horizontal transfer generally rely on finding anomalies in either nucleotide composition or phylogenetic relationships with orthologous proteins The three we found and will be using are UCSD's Darkhorse, EMBL's alien_hunter, and CodonW

26 DarkHorse
27 DarkHorse Darkhorse works by selecting potential ortholog matches from a reference amino acid database It then uses these matches to calculate something it calls a lineage probability index (LPI) score LPI scores are inversely proportional to the phylogenetic distance between database match sequences and the query genome. Candidates having low LPI scores are likely to have been horizontally transferred, since they are not highly conserved among closely related organisms.
28 alien_hunter
29 alien_hunter alien_hunter is another program that searches for HGT's It uses Interpolated Variable Order Motifs (IVOM's), a novel computational method introduced by the authors "An IVOM approach exploits compositional biases using variable order motif distributions and captures more reliably the local composition of a sequence compared to fixed-order methods."
30 CodonW
31 Codon usage bias and CodonW Although the genetic code is redundant, often with more than one three letter code specifying a protein, most proteins do not use all possibly synonymous codons equally Literature has shown that more highly expressed proteins tend to have optimized their translational efficiency such that they prefer certain codons for a given amino acid CodonW analyses sequences in order to give their statistics of codon usage bias This is handy to get a feel for the general codon bias, and to detect any unusual deviations from it that may indicate HGT's CodonW also calculates G+C content, which may be another indicator of abnormal gene lineage and is linked with a particular genome s codon usage bias
32 Databases Once we have a set of three programs' predictions, we can then compare them with databases of predicted HGT's of other Neisseria meningitidis strains DarkHorse's DB contains pre-computed predictions for N. meningitidis , FAM18, MC58 using its LPI index IBM's Bioinformatics and Pattern Discovery Group's HGT- DB contains predictions for strains MC58 and Z2491 A codon usage program called CAICal has a database containing strains FAM18, MC58 and Z2491 using unusual codon usage These putative HGT genes can be reciprocally blasted against our set of predictions to see if our genes have any match with other strains, and if other strains have any predictions we missed
33 Proposed HGT pipeline DarkHorse Candidate HGT among diff. phyl. Compare HGT across granularities G E N E S Alien_hunter HGT candidates Compare Compare w/ HGT db s CodonW Codon usage stats Genes with atypical codon/gc usage List of HGTs and support Virulence (Nitya) Phylogenies (Yun)
34 Genome Alignment and Visualization
35 Main Pipeline Protein / DNA Sequences from Gene Prediciton COG HGT Synteny Phylogenies Virulence Functional Annotation evolutionary history candidate genes/regions for further investigation of pathogenicity
36 Large scale genome evolution h"p://
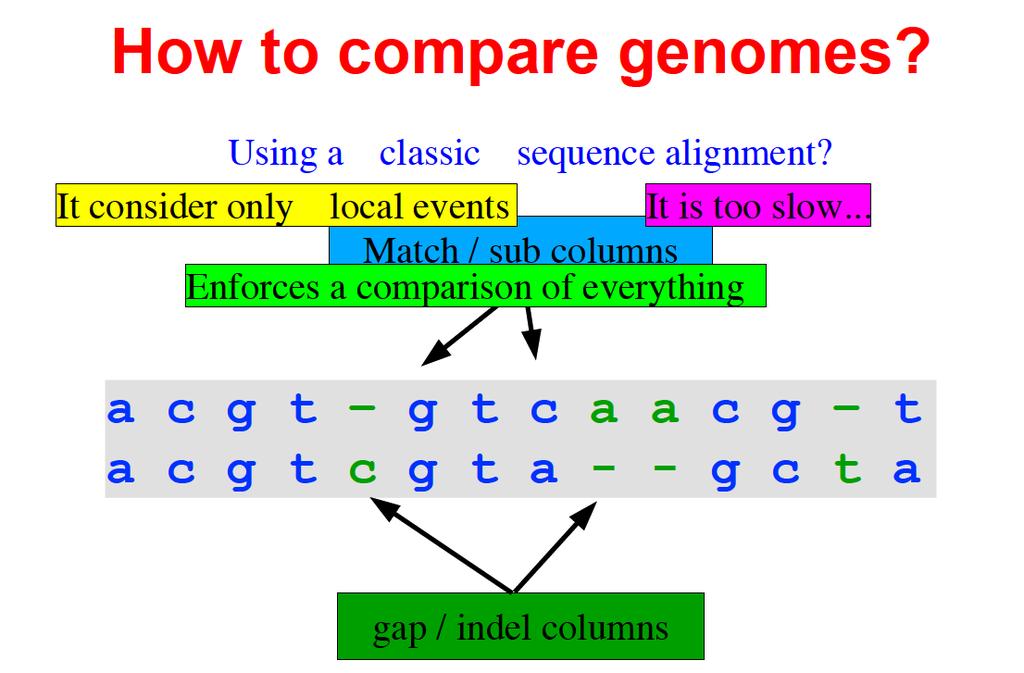
37 How to align genomes? h"p://
38 Genome Alignment Computation: time and space Genome large scale evolution: rearrangement, inversion

39 Tools for Genome Alignment and Visualization Jayaraj, 2005
40 Genome Alignment Pairwise: MUMmer (Maximum Unique Match).1999, Steven Salzberg's group, also Glimmer. Multiple: MAUVE (Multiple Alignment of Conserved Genomic Sequence with Rearrangements)
41 MUMmer Maximal Unique Matcher (MUM) match exact match of a minimum length maximal cannot be extended in either direction without a mismatch unique occurs only once in both sequences (MUM)
42 MUMmer: MUM, MAM, MEM MUM : maximal unique match MAM : maximal almost-unique match MEM : maximal exact match Reference Query h"p://

43 h"p://


44 B Translocation Inversion Insertion A B Output: 2D plot h"p://mummer.sourceforge.net/manual/alignmenttypes.pdf h"p:// A

45 MUMmer - VISTA Reference genome: Neisseria mengingitidis Z Neisseria meningitidis MC58 2- Neisseria gonorrhoeae FA1090
46 MAUVE Multiple Alignment of Conserved Genomic Sequence with Rearrangements LCB: locally collinear blocks (many anchors) Genomic distance: based on the gene order (or LCB) GRIMM, can infer genomic phylogeny.
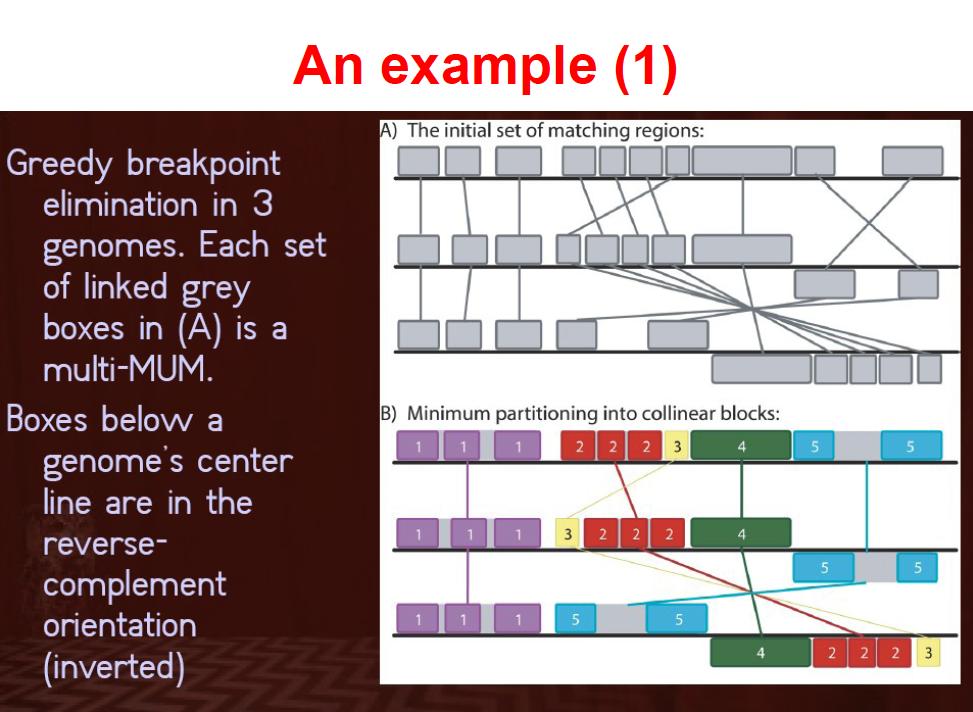
47 h"p://
48 MAUVE - GRIMM Signed permutation Genomic distance Genomic phylogeny Reversal distance
49 Reversal distance (rearrangement distance) Software: MGR, GPAPPA, GRIMM web sever. Bourque and Pevzner, 2002

50 Pipeline MUMmer Sequences VISTA Synteny Virulence MAUVE
51 Main Pipeline Protein / DNA Sequences from Gene Prediciton COG HGT Synteny Phylogenies Virulence Functional Annotation evolutionary history candidate genes/regions for further investigation of pathogenicity

52 Phylogeny tree Purpose: To summarize the key aspects of a reconstructed evolutionary history by providing simple representation.
53 Maximum parsimony based on 23 proteins; Brown et al. 2001
54 Main Goals 1. Find out evolution of Neisseria Meningitidis 2. Discover relatedness between Neisseria Meningitidis strains
55 Main questions before we 1.Which data to use? starting analysis 2.Which method to use? 3.Which tests to perform to assess the robustness of the prediction of particular tree features? 4.What is the state-of-the-art in phylogenetic analysis tool for this type of data?
56 1. Which data to use? 1) 16S rrna What is 16S rrna? -16S rrna is 1542 nt long component of the small prokaryotic ribosomal subunit Why 16S rrna? - Derived from common ancestor - It s highly conserved region in all prokaryotes
57 1. Which data to use? 2) CoGs binary result - HGT result Result From CoGs Result From HGT Result From CoGs-HGT Why? CoGs = Clusters of Orthologous Groups of proteins HGT= Horizontal Gene Transfer
58 What data to use? MLST(Multi Locus Sequence Typing) A nucleotide sequence based approach for the unambiguous characterisation of isolates of bacteria and other organisms via the internet. To provide a portable, accurate, and highly discriminating typing system Helpful for the typing of bacterial pathogens
59 Methods of phylogenetic reconstruction Distance based Maximum parsimony Maximum likelihood Pairwise evolutionary distances computed for all taxa Tree constructed using algorithm based on relationships between distances Algorithmic: UPGMA Neighbor-joining Optimality criteria Least Squares Minimum Evolution Nucleotides or amino acids are considered as character states Best phylogeny is chosen as the one that minimizes the number of changes between character states Statistical method of phylogeny reconstruction Explicit model for how data set generated -nucleotide or amino acid substitution Find topology that maximizes the probability of the data given the model and the parameter values (estimated from data) one tree a set of trees a set of tree
60 UPGMA (unweighted pair group method with arithmetic mean) Simplest method -uses sequential clustering algorithm Results in ultrameric trees equal distances from root to all tips Based on assumption of strict rate constancy among lineages Rely on the overly strict assumption of rate constancy but it is conceptually important Neighbor-joining Star decomposition identification of neighbors that sequentially minimize the total length of the tree Extremely fast and efficient method Tends to perform fairly well in simulation studies Greedy Algorithm so can get stuck in local optima Produces only one tree and does not give any idea of how many other trees are equally well or almost as supported by the data To find a starting tree that other methods (e.g. minimum evolution) will evaluate to find the best tree
61 Maximum parsimony method -The best tree is chosen as the one that requires the smallest number of changes between characters -Based on a logically coherent and biologically plausible model of evolution -Useful for certain types of molecular data e.g. insertions and deletions -Provides several ways to evaluate the support for the topologies produced -Gives incorrect topologies when backward substitutions are present (common with nucleotides) and when the number of sites is fairly small /when rate of substitution varies substantially across lineages -Long branch attraction long branches (and short branches) tend to group together on reconstructed tree -Difficult to treat the results in a statistical framework Maximum likelihood -Statistically very well defined -Extremely slow method (computationally expensive method) -Method estimates branch lengths not topology so may give wrong topology -Based on explicit models of evolution -Uses all sequence information (characters) -Requires expert user input for model and parameter selection
62 3. Which tests to perform to assess the robustness of the prediction of How confident are we of this tree? Do Bootstrap particular tree features? What is boostsrap sampling? Bootstrap is sampling with replacement from a sample. Bootstrap is sampling within a sample. The name may come from phrase pull up by your own bootstraps which mean rely on your own resources'. What are the assumption of Bootstrap? Your sample is a valid representative of the population Bootstrap method will take sampling with replacement from the sample. Each sub sampling is independent and identical distribution (i.i.d.). In other word, it assumes that the sub samples come from the same distribution of the population, but each sample is drawn independently from the other samples.



63 Bootstrap Ex. Pseudosample Data Re-sampling Sample Data n replicates Inferred Tree Bootstrap Value Bootstrap Trees 63 (D.Graur and W.Li, 2000)

64 4.Which tool is the state-of-the-art SplitTree 4 in phylogenetic analysis?
65 Software SplitsTree4 Details Compute evolutionary networks from molecular sequence data (alignment of sequences, a distance matrix or a set of trees) Integrates a wide range of phylogenetic network and phylogenetic tree methods Compute a phylogenetic tree or network using many methods such as split decomposition, neighbor-net, consensus network, super networks methods or methods for computing hybridization or simple recombination networks. Why SplitsTree? Phylogenetic networks are more useful for reticulate events than phylogenetic trees.
66 Software SplitsTree4
67 Software MEGA 4.0 Feature Input Data :DNA, Protein, Pairwise distance matrix Sequence Alignment Construction Tree-making Methods Distance Matrix Viewer Tree Explorers
68 Pipeline 7 loci seq. (MLST Database) MEGA4 SplitsTree4
69 Virulence
70 Main Pipeline Protein / DNA Sequences from Gene Prediciton COG HGT Synteny Phylogenies Virulence Functional Annotation evolutionary history candidate genes/regions for further investigation of pathogenicity
71 N. meningitidis Gram-negative Pangenome is open Colonizes the nasopharynx and can enter the bloodstream (bypassing the epithelial barrier) Septicaemia Meningitis via BBB crossing Accidental pathogen Non disease causing isolates (carriage) in about 10% of healthy population
72 Pathogenicity vs. Virulence Bacterial pathogen: any bacterium that has the capacity to cause disease ability to cause disease is called pathogenicity Virulence: provides a quan`ta`ve measure of pathogenicity or the likelihood of causing disease Virulence factors: proper`es (i.e. gene products) that enable a microorganism to establish itself on or within a host and enhance its poten`al to cause disease Pathogenicity Islands: comprise of large genomic regions that encode for various virulence factors

73 Polysaccharide Capsule Defining characteristic for serogroup classification A C, W-135, Y Most characterized virulence factor Involved in evading immune defense against complement-mediated lysis and opsonophagocytosis Necessary but NOT SUFFICIENT!
74 Virulence Factors Adherence Genes able to mediate adhesion to host nasopharynx epithelium Immune evasion mediate resistance of both phagocytosis and complement-mediated killing by expression of capsule Invasion Enzymes that mediate movement across epithelium Iron uptake systems mediate iron uptake from host and contribute virulence Protease Genes that encode to proteins that cleave antibodies to evade immune system response Toxin Modify or disrupt essential functions of eukaryotic cells Major toxin LOS

75 Virulence Factor DB - Virulence Factors divided by category, with lists of corresponding genes - Comparative pathogenomics of disease causing strains
76 Pathogenicity Islands Criteria Subclass of genomic islands (GI) that are defined by the following criteria: 1) Encodes for virulence factors 2) Present in pathogenic strains, absent in non-pathogenic strains of one species or a related species 3) Different G+C content and codon usage (remember HGT) 4) Large genomic regions 5) Fanked by insertion sequece (IS) and/or direct repeats elements and/or trna genes at boundaries sites of recombination 6) Unstable
77 Pathogenicity Islands Neisseria meningitidis MC58 IHT-A: Genes of serogroup B capsulation cluster and an adenine rrna methylase IHT-C: Three toxin/toxin-related homologs; a protein known to be immunogenic, one intact and three fragmented proteins previously associated with bacteriophage Neisseria meningitidis Z2491 No known PAIs cpai: Candidate PAI (PAI-like region overlapping genomic islands) homologous to IHT-A
78 PAIs Cannot determine virulence by the presence or absence of specific genes Loses its utility in investigating virulence in our context Found PAIs in N.meningitidis, but did not investigate carriage vs. virulent strains More later background research
79 What have we learned about virulence and pathogenicity from past research?

80 Shoen et al. 2008

81 Comparative Genomics 2008
82 Majority of candidate virulence genes are found in the core genome (shared by all), and are not virulent strain-specific Not just due to presence or absence of certain genes So, what is causing differences in virulence?
83 What have we learned about virulence from past research? What can we do differently?
84 Candidate causes of virulence variability Chromosomal rearrangements Affect expression breadth Insertion Sequences Small genetic differences in genes from core genome or between genomes of carriage and disease strains May influence pathogenic potential SNPs
85 Goal and Approach Goal: Use more fine tuned methods to compare carriage versus disease strains in N. meningitidis Approach 1: Determine whether the IS profile distribution discriminates carriage strains from virulent strains Approach 2: Whole genome association mapping (WGAM) in disease vs. carriage strains
86 Insertion Sequences Short DNA (about 2.5 kbps) sequence whose function is exclusively involved in mobility Can cause mutations as a result of their translocation Many IS elements can enhance expression of neighboring genes if inserted (Mahillan and Chandler 1998) Associated with bacterial pathogenesis and virulence Most have short terminal repeat sequences Composite transposon: two copies of certain ISs flanking a DNA segment causing mobility of whole region Upon insertion, most generate short directly repeated sequences (drs) of the target DNA
87 Insertion Sequences the phenotype of the recipient bacterium can be changed if the IS is inserted into a structural gene or if the insertion in front of a gene affects the expression of a downstream gene(s) mediate deletions, duplications, and inversions and cointegrate formation contributing to changes in the bacterial genome
88 IS element structure
 IS1655 (IS30 family) specific to N.")
89 IS Family Classification Similarities in genetic organization Relatedness of transposases Similar features of ends (terminal IRs) Fate of nucleotide sequence of their target sites Families of interest: IS110, IS3, IS30, IS5, ISNCY (Shoen et al. 2008) IS1655 (IS30 family) specific to N. meningitidis

90 IS info from gene prediction R IS info from all available strains Distribution by IS family Genome BLAST to VFDB Significantly different IS family in carriage vs. disease strains Positional info Genes around IS sequences within family HGT Flanking genes, interrupted genes, neighboring genes associated with IS interference, and Virulence genes Synteny
91 SNPs as markers for WGAM Haplotype: set of SNPs that are statistically associated haplotype block Use whole genome sequences of disease vs. carriage strains and look for increased variability in local haplotype structure If there is increased variations in virulent strains as compared to carriage strains, then such variations can be considered to be associated with the virulence Identify regions of high variability in virulent vs. carriage strains These regions can be used as pointers to direct further study of genes within and/or around the haplotype block
92 Conclusion
93 Main Pipeline Protein / DNA Sequences from Gene Prediciton COG HGT Synteny Phylogenies Virulence Functional Annotation evolutionary history candidate genes/regions for further investigation of pathogenicity
Comparative genomics: Overview & Tools + MUMmer algorithm
 Comparative genomics: Overview & Tools + MUMmer algorithm Urmila Kulkarni-Kale Bioinformatics Centre University of Pune, Pune 411 007. urmila@bioinfo.ernet.in Genome sequence: Fact file 1995: The first
Comparative genomics: Overview & Tools + MUMmer algorithm Urmila Kulkarni-Kale Bioinformatics Centre University of Pune, Pune 411 007. urmila@bioinfo.ernet.in Genome sequence: Fact file 1995: The first
Dr. Amira A. AL-Hosary
 Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut
 Faculty of Veterinary Medicine Assiut") Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics
 POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics - in deriving a phylogeny our goal is simply to reconstruct the historical relationships between a group of taxa. - before we review the
POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics - in deriving a phylogeny our goal is simply to reconstruct the historical relationships between a group of taxa. - before we review the
I519 Introduction to Bioinformatics, Genome Comparison. Yuzhen Ye School of Informatics & Computing, IUB
 I519 Introduction to Bioinformatics, 2015 Genome Comparison Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Whole genome comparison/alignment Build better phylogenies Identify polymorphism
I519 Introduction to Bioinformatics, 2015 Genome Comparison Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Whole genome comparison/alignment Build better phylogenies Identify polymorphism
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Bio 1B Lecture Outline (please print and bring along) Fall, 2007
 Fall, 2007") Bio 1B Lecture Outline (please print and bring along) Fall, 2007 B.D. Mishler, Dept. of Integrative Biology 2-6810, bmishler@berkeley.edu Evolution lecture #5 -- Molecular genetics and molecular evolution
Bio 1B Lecture Outline (please print and bring along) Fall, 2007 B.D. Mishler, Dept. of Integrative Biology 2-6810, bmishler@berkeley.edu Evolution lecture #5 -- Molecular genetics and molecular evolution
C3020 Molecular Evolution. Exercises #3: Phylogenetics
 C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
Inferring phylogeny. Constructing phylogenetic trees. Tõnu Margus. Bioinformatics MTAT
 Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Genômica comparativa. João Carlos Setubal IQ-USP outubro /5/2012 J. C. Setubal
 Genômica comparativa João Carlos Setubal IQ-USP outubro 2012 11/5/2012 J. C. Setubal 1 Comparative genomics There are currently (out/2012) 2,230 completed sequenced microbial genomes publicly available
Genômica comparativa João Carlos Setubal IQ-USP outubro 2012 11/5/2012 J. C. Setubal 1 Comparative genomics There are currently (out/2012) 2,230 completed sequenced microbial genomes publicly available
17 Non-collinear alignment Motivation A B C A B C A B C A B C D A C. This exposition is based on:
 17 Non-collinear alignment This exposition is based on: 1. Darling, A.E., Mau, B., Perna, N.T. (2010) progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5(6):e11147.
17 Non-collinear alignment This exposition is based on: 1. Darling, A.E., Mau, B., Perna, N.T. (2010) progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5(6):e11147.
RGP finder: prediction of Genomic Islands
 Training courses on MicroScope platform RGP finder: prediction of Genomic Islands Dynamics of bacterial genomes Gene gain Horizontal gene transfer Gene loss Deletion of one or several genes Duplication
Training courses on MicroScope platform RGP finder: prediction of Genomic Islands Dynamics of bacterial genomes Gene gain Horizontal gene transfer Gene loss Deletion of one or several genes Duplication
CHAPTERS 24-25: Evidence for Evolution and Phylogeny
 CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
CHAPTERS 24-25: Evidence for Evolution and Phylogeny 1. For each of the following, indicate how it is used as evidence of evolution by natural selection or shown as an evolutionary trend: a. Paleontology
Whole Genome Alignment. Adam Phillippy University of Maryland, Fall 2012
 Whole Genome Alignment Adam Phillippy University of Maryland, Fall 2012 Motivation cancergenome.nih.gov Breast cancer karyotypes www.path.cam.ac.uk Goal of whole-genome alignment } For two genomes, A and
Whole Genome Alignment Adam Phillippy University of Maryland, Fall 2012 Motivation cancergenome.nih.gov Breast cancer karyotypes www.path.cam.ac.uk Goal of whole-genome alignment } For two genomes, A and
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
Phylogenetic Tree Reconstruction
 I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
A Phylogenetic Network Construction due to Constrained Recombination
 A Phylogenetic Network Construction due to Constrained Recombination Mohd. Abdul Hai Zahid Research Scholar Research Supervisors: Dr. R.C. Joshi Dr. Ankush Mittal Department of Electronics and Computer
A Phylogenetic Network Construction due to Constrained Recombination Mohd. Abdul Hai Zahid Research Scholar Research Supervisors: Dr. R.C. Joshi Dr. Ankush Mittal Department of Electronics and Computer
Genetic Variation: The genetic substrate for natural selection. Horizontal Gene Transfer. General Principles 10/2/17.
 Genetic Variation: The genetic substrate for natural selection What about organisms that do not have sexual reproduction? Horizontal Gene Transfer Dr. Carol E. Lee, University of Wisconsin In prokaryotes:
Genetic Variation: The genetic substrate for natural selection What about organisms that do not have sexual reproduction? Horizontal Gene Transfer Dr. Carol E. Lee, University of Wisconsin In prokaryotes:
Intraspecific gene genealogies: trees grafting into networks
 Intraspecific gene genealogies: trees grafting into networks by David Posada & Keith A. Crandall Kessy Abarenkov Tartu, 2004 Article describes: Population genetics principles Intraspecific genetic variation
Intraspecific gene genealogies: trees grafting into networks by David Posada & Keith A. Crandall Kessy Abarenkov Tartu, 2004 Article describes: Population genetics principles Intraspecific genetic variation
METHODS FOR DETERMINING PHYLOGENY. In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task.
 Chapter 12 (Strikberger) Molecular Phylogenies and Evolution METHODS FOR DETERMINING PHYLOGENY In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task. Modern
Chapter 12 (Strikberger) Molecular Phylogenies and Evolution METHODS FOR DETERMINING PHYLOGENY In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task. Modern
Phylogenetic inference
 Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic relationship among S. castellii, S. cerevisiae and C. glabrata.
 Supplementary Note S2 Phylogenetic relationship among S. castellii, S. cerevisiae and C. glabrata. Phylogenetic trees reconstructed by a variety of methods from either single-copy orthologous loci (Class
Supplementary Note S2 Phylogenetic relationship among S. castellii, S. cerevisiae and C. glabrata. Phylogenetic trees reconstructed by a variety of methods from either single-copy orthologous loci (Class
Computational approaches for functional genomics
 Computational approaches for functional genomics Kalin Vetsigian October 31, 2001 The rapidly increasing number of completely sequenced genomes have stimulated the development of new methods for finding
Computational approaches for functional genomics Kalin Vetsigian October 31, 2001 The rapidly increasing number of completely sequenced genomes have stimulated the development of new methods for finding
Constructing Evolutionary/Phylogenetic Trees
 Constructing Evolutionary/Phylogenetic Trees 2 broad categories: istance-based methods Ultrametric Additive: UPGMA Transformed istance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Constructing Evolutionary/Phylogenetic Trees 2 broad categories: istance-based methods Ultrametric Additive: UPGMA Transformed istance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
SUPPLEMENTARY INFORMATION
 Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Supplementary information S3 (box) Methods Methods Genome weighting The currently available collection of archaeal and bacterial genomes has a highly biased distribution of isolates across taxa. For example,
Phylogenetics: Building Phylogenetic Trees
 1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
MiGA: The Microbial Genome Atlas
 December 12 th 2017 MiGA: The Microbial Genome Atlas Jim Cole Center for Microbial Ecology Dept. of Plant, Soil & Microbial Sciences Michigan State University East Lansing, Michigan U.S.A. Where I m From
December 12 th 2017 MiGA: The Microbial Genome Atlas Jim Cole Center for Microbial Ecology Dept. of Plant, Soil & Microbial Sciences Michigan State University East Lansing, Michigan U.S.A. Where I m From
2 Genome evolution: gene fusion versus gene fission
 2 Genome evolution: gene fusion versus gene fission Berend Snel, Peer Bork and Martijn A. Huynen Trends in Genetics 16 (2000) 9-11 13 Chapter 2 Introduction With the advent of complete genome sequencing,
2 Genome evolution: gene fusion versus gene fission Berend Snel, Peer Bork and Martijn A. Huynen Trends in Genetics 16 (2000) 9-11 13 Chapter 2 Introduction With the advent of complete genome sequencing,
Outline. I. Methods. II. Preliminary Results. A. Phylogeny Methods B. Whole Genome Methods C. Horizontal Gene Transfer
 Comparative Genomics Preliminary Results April 4, 2016 Juan Castro, Aroon Chande, Cheng Chen, Evan Clayton, Hector Espitia, Alli Gombolay, Walker Gussler, Ken Lee, Tyrone Lee, Hari Prasanna, Carlos Ruiz,
Comparative Genomics Preliminary Results April 4, 2016 Juan Castro, Aroon Chande, Cheng Chen, Evan Clayton, Hector Espitia, Alli Gombolay, Walker Gussler, Ken Lee, Tyrone Lee, Hari Prasanna, Carlos Ruiz,
Phylogenetics: Building Phylogenetic Trees. COMP Fall 2010 Luay Nakhleh, Rice University
 Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
BINF6201/8201. Molecular phylogenetic methods
 BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
The minimal prokaryotic genome. The minimal prokaryotic genome. The minimal prokaryotic genome. The minimal prokaryotic genome
 Dr. Dirk Gevers 1,2 1 Laboratorium voor Microbiologie 2 Bioinformatics & Evolutionary Genomics The bacterial species in the genomic era CTACCATGAAAGACTTGTGAATCCAGGAAGAGAGACTGACTGGGCAACATGTTATTCAG GTACAAAAAGATTTGGACTGTAACTTAAAAATGATCAAATTATGTTTCCCATGCATCAGG
Dr. Dirk Gevers 1,2 1 Laboratorium voor Microbiologie 2 Bioinformatics & Evolutionary Genomics The bacterial species in the genomic era CTACCATGAAAGACTTGTGAATCCAGGAAGAGAGACTGACTGGGCAACATGTTATTCAG GTACAAAAAGATTTGGACTGTAACTTAAAAATGATCAAATTATGTTTCCCATGCATCAGG
Comparative Bioinformatics Midterm II Fall 2004
 Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
Comparative Bioinformatics Midterm II Fall 2004 Objective Answer, part I: For each of the following, select the single best answer or completion of the phrase. (3 points each) 1. Deinococcus radiodurans
USING BLAST TO IDENTIFY PROTEINS THAT ARE EVOLUTIONARILY RELATED ACROSS SPECIES
 USING BLAST TO IDENTIFY PROTEINS THAT ARE EVOLUTIONARILY RELATED ACROSS SPECIES HOW CAN BIOINFORMATICS BE USED AS A TOOL TO DETERMINE EVOLUTIONARY RELATIONSHPS AND TO BETTER UNDERSTAND PROTEIN HERITAGE?
USING BLAST TO IDENTIFY PROTEINS THAT ARE EVOLUTIONARILY RELATED ACROSS SPECIES HOW CAN BIOINFORMATICS BE USED AS A TOOL TO DETERMINE EVOLUTIONARY RELATIONSHPS AND TO BETTER UNDERSTAND PROTEIN HERITAGE?
Phylogeny and systematics. Why are these disciplines important in evolutionary biology and how are they related to each other?
 Phylogeny and systematics Why are these disciplines important in evolutionary biology and how are they related to each other? Phylogeny and systematics Phylogeny: the evolutionary history of a species
Phylogeny and systematics Why are these disciplines important in evolutionary biology and how are they related to each other? Phylogeny and systematics Phylogeny: the evolutionary history of a species
Bioinformatics tools for phylogeny and visualization. Yanbin Yin
 Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
Bioinformatics tools for phylogeny and visualization Yanbin Yin 1 Homework assignment 5 1. Take the MAFFT alignment http://cys.bios.niu.edu/yyin/teach/pbb/purdue.cellwall.list.lignin.f a.aln as input and
"Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky
 MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
Microbes usually have few distinguishing properties that relate them, so a hierarchical taxonomy mainly has not been possible.
 Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Microbial Taxonomy. Slowly evolving molecules (e.g., rrna) used for large-scale structure; "fast- clock" molecules for fine-structure.
 used for large-scale structure; fast- clock molecules for fine-structure.") Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
SUPPLEMENTARY INFORMATION
 Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Supplementary information S1 (box). Supplementary Methods description. Prokaryotic Genome Database Archaeal and bacterial genome sequences were downloaded from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/)
Processes of Evolution
 15 Processes of Evolution Forces of Evolution Concept 15.4 Selection Can Be Stabilizing, Directional, or Disruptive Natural selection can act on quantitative traits in three ways: Stabilizing selection
15 Processes of Evolution Forces of Evolution Concept 15.4 Selection Can Be Stabilizing, Directional, or Disruptive Natural selection can act on quantitative traits in three ways: Stabilizing selection
C.DARWIN ( )
") C.DARWIN (1809-1882) LAMARCK Each evolutionary lineage has evolved, transforming itself, from a ancestor appeared by spontaneous generation DARWIN All organisms are historically interconnected. Their relationships
C.DARWIN (1809-1882) LAMARCK Each evolutionary lineage has evolved, transforming itself, from a ancestor appeared by spontaneous generation DARWIN All organisms are historically interconnected. Their relationships
Horizontal transfer and pathogenicity
 Horizontal transfer and pathogenicity Victoria Moiseeva Genomics, Master on Advanced Genetics UAB, Barcelona, 2014 INDEX Horizontal Transfer Horizontal gene transfer mechanisms Detection methods of HGT
Horizontal transfer and pathogenicity Victoria Moiseeva Genomics, Master on Advanced Genetics UAB, Barcelona, 2014 INDEX Horizontal Transfer Horizontal gene transfer mechanisms Detection methods of HGT
Chapter 27: Evolutionary Genetics
 Chapter 27: Evolutionary Genetics Student Learning Objectives Upon completion of this chapter you should be able to: 1. Understand what the term species means to biology. 2. Recognize the various patterns
Chapter 27: Evolutionary Genetics Student Learning Objectives Upon completion of this chapter you should be able to: 1. Understand what the term species means to biology. 2. Recognize the various patterns
Comparing whole genomes
 BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
Microbial Taxonomy. Microbes usually have few distinguishing properties that relate them, so a hierarchical taxonomy mainly has not been possible.
 Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Microbial Taxonomy Traditional taxonomy or the classification through identification and nomenclature of microbes, both "prokaryote" and eukaryote, has been in a mess we were stuck with it for traditional
Nature Genetics: doi: /ng Supplementary Figure 1. Icm/Dot secretion system region I in 41 Legionella species.
 Supplementary Figure 1 Icm/Dot secretion system region I in 41 Legionella species. Homologs of the effector-coding gene lega15 (orange) were found within Icm/Dot region I in 13 Legionella species. In four
Supplementary Figure 1 Icm/Dot secretion system region I in 41 Legionella species. Homologs of the effector-coding gene lega15 (orange) were found within Icm/Dot region I in 13 Legionella species. In four
I519 Introduction to Bioinformatics, Genome Comparison. Yuzhen Ye School of Informatics & Computing, IUB
 I519 Introduction to Bioinformatics, 2011 Genome Comparison Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Whole genome comparison/alignment Build better phylogenies Identify polymorphism
I519 Introduction to Bioinformatics, 2011 Genome Comparison Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Whole genome comparison/alignment Build better phylogenies Identify polymorphism
Molecular Phylogenetics (part 1 of 2) Computational Biology Course João André Carriço
 Computational Biology Course João André Carriço") Molecular Phylogenetics (part 1 of 2) Computational Biology Course João André Carriço jcarrico@fm.ul.pt Charles Darwin (1809-1882) Charles Darwin s tree of life in Notebook B, 1837-1838 Ernst Haeckel (1934-1919)
Molecular Phylogenetics (part 1 of 2) Computational Biology Course João André Carriço jcarrico@fm.ul.pt Charles Darwin (1809-1882) Charles Darwin s tree of life in Notebook B, 1837-1838 Ernst Haeckel (1934-1919)
Multiple Sequence Alignment. Sequences
 Multiple Sequence Alignment Sequences > YOR020c mstllksaksivplmdrvlvqrikaqaktasglylpe knveklnqaevvavgpgftdangnkvvpqvkvgdqvl ipqfggstiklgnddevilfrdaeilakiakd > crassa mattvrsvksliplldrvlvqrvkaeaktasgiflpe
Multiple Sequence Alignment Sequences > YOR020c mstllksaksivplmdrvlvqrikaqaktasglylpe knveklnqaevvavgpgftdangnkvvpqvkvgdqvl ipqfggstiklgnddevilfrdaeilakiakd > crassa mattvrsvksliplldrvlvqrvkaeaktasgiflpe
Bioinformatics Chapter 1. Introduction
 Bioinformatics Chapter 1. Introduction Outline! Biological Data in Digital Symbol Sequences! Genomes Diversity, Size, and Structure! Proteins and Proteomes! On the Information Content of Biological Sequences!
Bioinformatics Chapter 1. Introduction Outline! Biological Data in Digital Symbol Sequences! Genomes Diversity, Size, and Structure! Proteins and Proteomes! On the Information Content of Biological Sequences!
8/23/2014. Phylogeny and the Tree of Life
 Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Phylogeny and the Tree of Life Chapter 26 Objectives Explain the following characteristics of the Linnaean system of classification: a. binomial nomenclature b. hierarchical classification List the major
Bioinformatics 1. Sepp Hochreiter. Biology, Sequences, Phylogenetics Part 4. Bioinformatics 1: Biology, Sequences, Phylogenetics
 Bioinformatics 1 Biology, Sequences, Phylogenetics Part 4 Sepp Hochreiter Klausur Mo. 30.01.2011 Zeit: 15:30 17:00 Raum: HS14 Anmeldung Kusss Contents Methods and Bootstrapping of Maximum Methods Methods
Bioinformatics 1 Biology, Sequences, Phylogenetics Part 4 Sepp Hochreiter Klausur Mo. 30.01.2011 Zeit: 15:30 17:00 Raum: HS14 Anmeldung Kusss Contents Methods and Bootstrapping of Maximum Methods Methods
Microbial Taxonomy and the Evolution of Diversity
 19 Microbial Taxonomy and the Evolution of Diversity Copyright McGraw-Hill Global Education Holdings, LLC. Permission required for reproduction or display. 1 Taxonomy Introduction to Microbial Taxonomy
19 Microbial Taxonomy and the Evolution of Diversity Copyright McGraw-Hill Global Education Holdings, LLC. Permission required for reproduction or display. 1 Taxonomy Introduction to Microbial Taxonomy
Molecular phylogeny - Using molecular sequences to infer evolutionary relationships. Tore Samuelsson Feb 2016
 Molecular phylogeny - Using molecular sequences to infer evolutionary relationships Tore Samuelsson Feb 2016 Molecular phylogeny is being used in the identification and characterization of new pathogens,
Molecular phylogeny - Using molecular sequences to infer evolutionary relationships Tore Samuelsson Feb 2016 Molecular phylogeny is being used in the identification and characterization of new pathogens,
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
Consensus Methods. * You are only responsible for the first two
 Consensus Trees * consensus trees reconcile clades from different trees * consensus is a conservative estimate of phylogeny that emphasizes points of agreement * philosophy: agreement among data sets is
Consensus Trees * consensus trees reconcile clades from different trees * consensus is a conservative estimate of phylogeny that emphasizes points of agreement * philosophy: agreement among data sets is
Analysis of Gene Order Evolution beyond Single-Copy Genes
 Analysis of Gene Order Evolution beyond Single-Copy Genes Nadia El-Mabrouk Département d Informatique et de Recherche Opérationnelle Université de Montréal mabrouk@iro.umontreal.ca David Sankoff Department
Analysis of Gene Order Evolution beyond Single-Copy Genes Nadia El-Mabrouk Département d Informatique et de Recherche Opérationnelle Université de Montréal mabrouk@iro.umontreal.ca David Sankoff Department
Chapter 26: Phylogeny and the Tree of Life Phylogenies Show Evolutionary Relationships
 Chapter 26: Phylogeny and the Tree of Life You Must Know The taxonomic categories and how they indicate relatedness. How systematics is used to develop phylogenetic trees. How to construct a phylogenetic
Chapter 26: Phylogeny and the Tree of Life You Must Know The taxonomic categories and how they indicate relatedness. How systematics is used to develop phylogenetic trees. How to construct a phylogenetic
A. Incorrect! In the binomial naming convention the Kingdom is not part of the name.
 Microbiology Problem Drill 08: Classification of Microorganisms No. 1 of 10 1. In the binomial system of naming which term is always written in lowercase? (A) Kingdom (B) Domain (C) Genus (D) Specific
Microbiology Problem Drill 08: Classification of Microorganisms No. 1 of 10 1. In the binomial system of naming which term is always written in lowercase? (A) Kingdom (B) Domain (C) Genus (D) Specific
Constructing Evolutionary/Phylogenetic Trees
 Constructing Evolutionary/Phylogenetic Trees 2 broad categories: Distance-based methods Ultrametric Additive: UPGMA Transformed Distance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Constructing Evolutionary/Phylogenetic Trees 2 broad categories: Distance-based methods Ultrametric Additive: UPGMA Transformed Distance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
What is Phylogenetics
 What is Phylogenetics Phylogenetics is the area of research concerned with finding the genetic connections and relationships between species. The basic idea is to compare specific characters (features)
What is Phylogenetics Phylogenetics is the area of research concerned with finding the genetic connections and relationships between species. The basic idea is to compare specific characters (features)
Comparative Genomics Background & Strategy. Faction 2
 Comparative Genomics Background & Strategy Faction 2 Overview Introduction to comparative genomics Salmonella enterica subsp. enterica serovar Heidelberg Comparative Genomics Faction 2 Objectives Genomic
Comparative Genomics Background & Strategy Faction 2 Overview Introduction to comparative genomics Salmonella enterica subsp. enterica serovar Heidelberg Comparative Genomics Faction 2 Objectives Genomic
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment
 Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Genome Rearrangements In Man and Mouse. Abhinav Tiwari Department of Bioengineering
 Genome Rearrangements In Man and Mouse Abhinav Tiwari Department of Bioengineering Genome Rearrangement Scrambling of the order of the genome during evolution Operations on chromosomes Reversal Translocation
Genome Rearrangements In Man and Mouse Abhinav Tiwari Department of Bioengineering Genome Rearrangement Scrambling of the order of the genome during evolution Operations on chromosomes Reversal Translocation
Phylogeny: building the tree of life
 Phylogeny: building the tree of life Dr. Fayyaz ul Amir Afsar Minhas Department of Computer and Information Sciences Pakistan Institute of Engineering & Applied Sciences PO Nilore, Islamabad, Pakistan
Phylogeny: building the tree of life Dr. Fayyaz ul Amir Afsar Minhas Department of Computer and Information Sciences Pakistan Institute of Engineering & Applied Sciences PO Nilore, Islamabad, Pakistan
Bio 119 Bacterial Genomics 6/26/10
 BACTERIAL GENOMICS Reading in BOM-12: Sec. 11.1 Genetic Map of the E. coli Chromosome p. 279 Sec. 13.2 Prokaryotic Genomes: Sizes and ORF Contents p. 344 Sec. 13.3 Prokaryotic Genomes: Bioinformatic Analysis
BACTERIAL GENOMICS Reading in BOM-12: Sec. 11.1 Genetic Map of the E. coli Chromosome p. 279 Sec. 13.2 Prokaryotic Genomes: Sizes and ORF Contents p. 344 Sec. 13.3 Prokaryotic Genomes: Bioinformatic Analysis
EVOLUTIONARY DISTANCES
 EVOLUTIONARY DISTANCES FROM STRINGS TO TREES Luca Bortolussi 1 1 Dipartimento di Matematica ed Informatica Università degli studi di Trieste luca@dmi.units.it Trieste, 14 th November 2007 OUTLINE 1 STRINGS:
EVOLUTIONARY DISTANCES FROM STRINGS TO TREES Luca Bortolussi 1 1 Dipartimento di Matematica ed Informatica Università degli studi di Trieste luca@dmi.units.it Trieste, 14 th November 2007 OUTLINE 1 STRINGS:
Phylogenetics. Applications of phylogenetics. Unrooted networks vs. rooted trees. Outline
 Phylogenetics Todd Vision iology 522 March 26, 2007 pplications of phylogenetics Studying organismal or biogeographic history Systematics ating events in the fossil record onservation biology Studying
Phylogenetics Todd Vision iology 522 March 26, 2007 pplications of phylogenetics Studying organismal or biogeographic history Systematics ating events in the fossil record onservation biology Studying
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction. Lesser Tenrec (Echinops telfairi)
") Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
Phylogenetics - Orthology, phylogenetic experimental design and phylogeny reconstruction Lesser Tenrec (Echinops telfairi) Goals: 1. Use phylogenetic experimental design theory to select optimal taxa to
HORIZONTAL TRANSFER IN EUKARYOTES KIMBERLEY MC GRAIL FERNÁNDEZ GENOMICS
 HORIZONTAL TRANSFER IN EUKARYOTES KIMBERLEY MC GRAIL FERNÁNDEZ GENOMICS OVERVIEW INTRODUCTION MECHANISMS OF HGT IDENTIFICATION TECHNIQUES EXAMPLES - Wolbachia pipientis - Fungus - Plants - Drosophila ananassae
HORIZONTAL TRANSFER IN EUKARYOTES KIMBERLEY MC GRAIL FERNÁNDEZ GENOMICS OVERVIEW INTRODUCTION MECHANISMS OF HGT IDENTIFICATION TECHNIQUES EXAMPLES - Wolbachia pipientis - Fungus - Plants - Drosophila ananassae
Taxonomy. Content. How to determine & classify a species. Phylogeny and evolution
 Taxonomy Content Why Taxonomy? How to determine & classify a species Domains versus Kingdoms Phylogeny and evolution Why Taxonomy? Classification Arrangement in groups or taxa (taxon = group) Nomenclature
Taxonomy Content Why Taxonomy? How to determine & classify a species Domains versus Kingdoms Phylogeny and evolution Why Taxonomy? Classification Arrangement in groups or taxa (taxon = group) Nomenclature
Computational methods for predicting protein-protein interactions
 Computational methods for predicting protein-protein interactions Tomi Peltola T-61.6070 Special course in bioinformatics I 3.4.2008 Outline Biological background Protein-protein interactions Computational
Computational methods for predicting protein-protein interactions Tomi Peltola T-61.6070 Special course in bioinformatics I 3.4.2008 Outline Biological background Protein-protein interactions Computational
Phylogenetic Trees. What They Are Why We Do It & How To Do It. Presented by Amy Harris Dr Brad Morantz
 Phylogenetic Trees What They Are Why We Do It & How To Do It Presented by Amy Harris Dr Brad Morantz Overview What is a phylogenetic tree Why do we do it How do we do it Methods and programs Parallels
Phylogenetic Trees What They Are Why We Do It & How To Do It Presented by Amy Harris Dr Brad Morantz Overview What is a phylogenetic tree Why do we do it How do we do it Methods and programs Parallels
Elements of Bioinformatics 14F01 TP5 -Phylogenetic analysis
 Elements of Bioinformatics 14F01 TP5 -Phylogenetic analysis 10 December 2012 - Corrections - Exercise 1 Non-vertebrate chordates generally possess 2 homologs, vertebrates 3 or more gene copies; a Drosophila
Elements of Bioinformatics 14F01 TP5 -Phylogenetic analysis 10 December 2012 - Corrections - Exercise 1 Non-vertebrate chordates generally possess 2 homologs, vertebrates 3 or more gene copies; a Drosophila
BLAST Database Searching. BME 110: CompBio Tools Todd Lowe April 8, 2010
 BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
Stepping stones towards a new electronic prokaryotic taxonomy. The ultimate goal in taxonomy. Pragmatic towards diagnostics
 Stepping stones towards a new electronic prokaryotic taxonomy - MLSA - Dirk Gevers Different needs for taxonomy Describe bio-diversity Understand evolution of life Epidemiology Diagnostics Biosafety...
Stepping stones towards a new electronic prokaryotic taxonomy - MLSA - Dirk Gevers Different needs for taxonomy Describe bio-diversity Understand evolution of life Epidemiology Diagnostics Biosafety...
Comparative Genomics II
 Comparative Genomics II Advances in Bioinformatics and Genomics GEN 240B Jason Stajich May 19 Comparative Genomics II Slide 1/31 Outline Introduction Gene Families Pairwise Methods Phylogenetic Methods
Comparative Genomics II Advances in Bioinformatics and Genomics GEN 240B Jason Stajich May 19 Comparative Genomics II Slide 1/31 Outline Introduction Gene Families Pairwise Methods Phylogenetic Methods
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Vital Statistics Derived from Complete Genome Sequencing (for E. coli MG1655)
") We still consider the E. coli genome as a fairly typical bacterial genome, and given the extensive information available about this organism and it's lifestyle, the E. coli genome is a useful point of
We still consider the E. coli genome as a fairly typical bacterial genome, and given the extensive information available about this organism and it's lifestyle, the E. coli genome is a useful point of
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
CONTENTS. P A R T I Genomes 1. P A R T II Gene Transcription and Regulation 109
 CONTENTS ix Preface xv Acknowledgments xxi Editors and contributors xxiv A computational micro primer xxvi P A R T I Genomes 1 1 Identifying the genetic basis of disease 3 Vineet Bafna 2 Pattern identification
CONTENTS ix Preface xv Acknowledgments xxi Editors and contributors xxiv A computational micro primer xxvi P A R T I Genomes 1 1 Identifying the genetic basis of disease 3 Vineet Bafna 2 Pattern identification
Phylogenetic inference: from sequences to trees
 W ESTFÄLISCHE W ESTFÄLISCHE W ILHELMS -U NIVERSITÄT NIVERSITÄT WILHELMS-U ÜNSTER MM ÜNSTER VOLUTIONARY FUNCTIONAL UNCTIONAL GENOMICS ENOMICS EVOLUTIONARY Bioinformatics 1 Phylogenetic inference: from sequences
W ESTFÄLISCHE W ESTFÄLISCHE W ILHELMS -U NIVERSITÄT NIVERSITÄT WILHELMS-U ÜNSTER MM ÜNSTER VOLUTIONARY FUNCTIONAL UNCTIONAL GENOMICS ENOMICS EVOLUTIONARY Bioinformatics 1 Phylogenetic inference: from sequences
Chromosomal rearrangements in mammalian genomes : characterising the breakpoints. Claire Lemaitre
 PhD defense Chromosomal rearrangements in mammalian genomes : characterising the breakpoints Claire Lemaitre Laboratoire de Biométrie et Biologie Évolutive Université Claude Bernard Lyon 1 6 novembre 2008
PhD defense Chromosomal rearrangements in mammalian genomes : characterising the breakpoints Claire Lemaitre Laboratoire de Biométrie et Biologie Évolutive Université Claude Bernard Lyon 1 6 novembre 2008
Computational Biology: Basics & Interesting Problems
 Computational Biology: Basics & Interesting Problems Summary Sources of information Biological concepts: structure & terminology Sequencing Gene finding Protein structure prediction Sources of information
Computational Biology: Basics & Interesting Problems Summary Sources of information Biological concepts: structure & terminology Sequencing Gene finding Protein structure prediction Sources of information
Evolutionary Tree Analysis. Overview
 CSI/BINF 5330 Evolutionary Tree Analysis Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Backgrounds Distance-Based Evolutionary Tree Reconstruction Character-Based
CSI/BINF 5330 Evolutionary Tree Analysis Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Backgrounds Distance-Based Evolutionary Tree Reconstruction Character-Based
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
The Minimal-Gene-Set -Kapil PHY498BIO, HW 3
 The Minimal-Gene-Set -Kapil Rajaraman(rajaramn@uiuc.edu) PHY498BIO, HW 3 The number of genes in organisms varies from around 480 (for parasitic bacterium Mycoplasma genitalium) to the order of 100,000
The Minimal-Gene-Set -Kapil Rajaraman(rajaramn@uiuc.edu) PHY498BIO, HW 3 The number of genes in organisms varies from around 480 (for parasitic bacterium Mycoplasma genitalium) to the order of 100,000
1 ATGGGTCTC 2 ATGAGTCTC
 We need an optimality criterion to choose a best estimate (tree) Other optimality criteria used to choose a best estimate (tree) Parsimony: begins with the assumption that the simplest hypothesis that
We need an optimality criterion to choose a best estimate (tree) Other optimality criteria used to choose a best estimate (tree) Parsimony: begins with the assumption that the simplest hypothesis that
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Distance Methods Character Methods
Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Distance Methods Character Methods
Unsupervised Learning in Spectral Genome Analysis
 Unsupervised Learning in Spectral Genome Analysis Lutz Hamel 1, Neha Nahar 1, Maria S. Poptsova 2, Olga Zhaxybayeva 3, J. Peter Gogarten 2 1 Department of Computer Sciences and Statistics, University of
Unsupervised Learning in Spectral Genome Analysis Lutz Hamel 1, Neha Nahar 1, Maria S. Poptsova 2, Olga Zhaxybayeva 3, J. Peter Gogarten 2 1 Department of Computer Sciences and Statistics, University of
BMI/CS 776 Lecture #20 Alignment of whole genomes. Colin Dewey (with slides adapted from those by Mark Craven)
") BMI/CS 776 Lecture #20 Alignment of whole genomes Colin Dewey (with slides adapted from those by Mark Craven) 2007.03.29 1 Multiple whole genome alignment Input set of whole genome sequences genomes diverged
BMI/CS 776 Lecture #20 Alignment of whole genomes Colin Dewey (with slides adapted from those by Mark Craven) 2007.03.29 1 Multiple whole genome alignment Input set of whole genome sequences genomes diverged
On the identification and investigation of homologous gene families, with particular emphasis on the accuracy of multidomain families
 On the identification and investigation of homologous gene families, with particular emphasis on the accuracy of multidomain families Jacob M. Joseph August 2012 CMU-CB-12-103 Publisher: Lane Center for
On the identification and investigation of homologous gene families, with particular emphasis on the accuracy of multidomain families Jacob M. Joseph August 2012 CMU-CB-12-103 Publisher: Lane Center for
Molecular phylogeny How to infer phylogenetic trees using molecular sequences
 Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 200 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 200 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences
 Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 2009 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 2009 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Cladistics and Bioinformatics Questions 2013
 AP Biology Name Cladistics and Bioinformatics Questions 2013 1. The following table shows the percentage similarity in sequences of nucleotides from a homologous gene derived from five different species
AP Biology Name Cladistics and Bioinformatics Questions 2013 1. The following table shows the percentage similarity in sequences of nucleotides from a homologous gene derived from five different species
9/30/11. Evolution theory. Phylogenetic Tree Reconstruction. Phylogenetic trees (binary trees) Phylogeny (phylogenetic tree)
 Phylogeny (phylogenetic tree)") I9 Introduction to Bioinformatics, 0 Phylogenetic ree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & omputing, IUB Evolution theory Speciation Evolution of new organisms is driven by
I9 Introduction to Bioinformatics, 0 Phylogenetic ree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & omputing, IUB Evolution theory Speciation Evolution of new organisms is driven by