Bioinformatics 1. Sepp Hochreiter. Biology, Sequences, Phylogenetics Part 4. Bioinformatics 1: Biology, Sequences, Phylogenetics
|
|
|
- Ethan Benson
- 5 years ago
- Views:
Transcription
1 Bioinformatics 1 Biology, Sequences, Phylogenetics Part 4 Sepp Hochreiter
2 Klausur Mo Zeit: 15:30 17:00 Raum: HS14 Anmeldung Kusss
3 Contents Methods and Bootstrapping of Maximum Methods Methods
4 Motivation central field in biology: relation between species in form of a tree Root: beginning of life Leaves: taxa (current species) Branches: relationship is ancestor of'' between nodes Node: split of a species into two phylogeny (phylo = tribe and genesis) Cladistic trees: conserved characters Phenetic trees: measure of distance between the leaves of the tree (distance as a whole and not based on single features) Phenetic problems: simultaneous development of features and different evolution rates Convergent evolution e.g. finding the best form in water
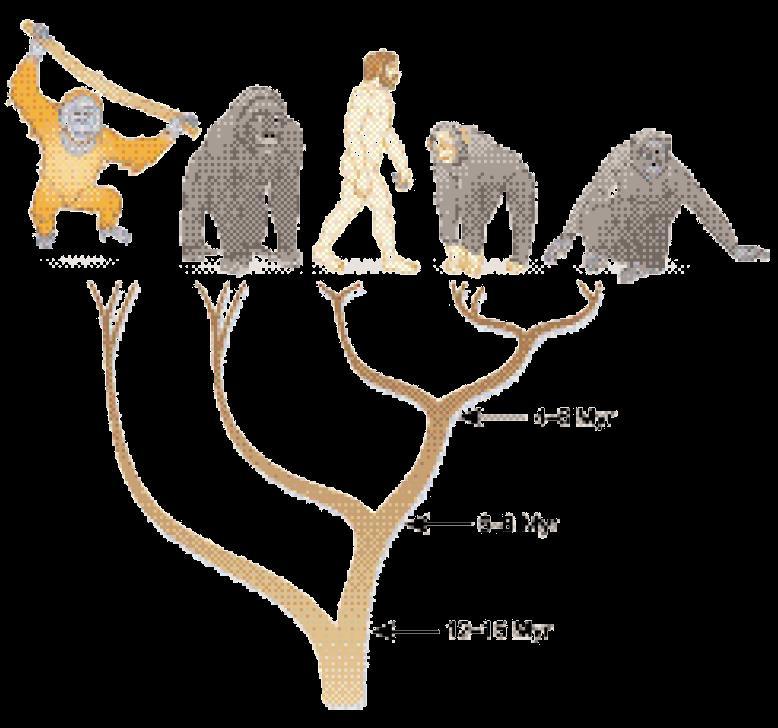
5 Motivation
6 Motivation
7 Motivation

8 Molecular Not characteristics like wings, feathers, (morphological) differences between organisms: RNA, DNA, and proteins α-hemoglobin Molecular phylogenetics is more precise can also distinguish bacteria or viruses connects all species by DNA uses mathematical and statistical methods is model-based as mutations can be modeled detects remote homologies

9 Molecular Difficulties in constructing a phylogenetic tree: different mutation rates horizontal transfer of genetic material (Horizontal Gene Transfer, Glycosyl Hydrolase from E.coli to B.subtilis) Species tree Gene tree
10 Molecular Branches: time in number of mutations molecular clock: same evolution / mutation rate number of substitution: Poisson distribution mutation rate: equally distributed over the sequence
11 Methods choose the sequences e.g. rrna (RNA of ribosomes) and mitochondrial genes (in most organism, enough mutations) pairwise and multiple sequence alignments method for constructing a phylogenetic tree distance-based maximum parsimony maximum likelihood Maximum parsimony: strong sequence similarities (few trees), few sequences Distance based (CLUSTALW): less similarity, many sequences Maximum likelihood: very variable sequences, high computational costs
12 Methods Software Name Author URL PHYLIP Felsenstein 89,96 PAUP washington.edu/phylip.html Sinauer Asssociates
13 Maximum minimize number of mutations mutations are branches in the tree tree explains the evolution of the sequences surviving mutations are rare tree with minimal mutations is most likely explanation maximum parsimony PHYLIP programs: DNAPARS, DNAPENNY, DNACOMP, DNAMOVE, and PROTPARS
14 Tree Length maximum parsimony tree: tree with smallest tree length tree length: number of substitutions in the tree Example protein triosephosphate isomerase for the taxa Human, Pig, Rye, Rice, and Chicken : Human ISPGMI Pig IGPGMI Rye ISAEQL Rice VSAEML Chicken ISPAMI If we focus on column 4: Human G Pig G Rye E Rice E Chicken A

 for a hypothetical")

15 Tree Length Fitch (71) alg. for computing the tree length with taxa at leaves: 1. Root node added to an arbitrary branch 2. bottom-up pass: sets of symbols (amino acids) for a hypothetical sequence at this node. Minimize the number mutations by maximal agreement of the subtrees avoid a mutation at the actual node In the first case is leave, the second case enforces a $m_ mutation, and the {12 third case avoids a mutation }$

16 Tree Length 3. top town pass: hypothetical sequences at the interior nodes; counts the number of mutations means that the formula holds for and for
17 Tree Length Non-informative columns: not used Columns with one symbol occur multiple an others only single (number of mutations independent of topology) Columns with only one symbol minimal number of substitutions: maximal subs., star tree (center: most frequent symbol): number of substitutions for the topology: consistency index : High values support the according tree as being plausible retention index rescaled consistency index
18 Tree Search few sequences: all trees and their length can be constructed for a larger number of sequences heuristics are used Branch and Bound (Hendy & Penny, 82) for 20 and more taxa: 1. This step determines the addition order of the taxa as follows. First, compute the core tree of three taxa with maximal length of all three taxa trees. Next the taxa is added to one of the three branches which leads to maximal tree length. For the tree with four branches we determine the next taxa which leads to maximal tree length. 2. This step determines an upper bound for the tree length by either distance based methods (neighbor joining) or heuristic search (stepwise addition algorithm).
19 Tree Search Branch and Bound (continued): 3. Start with the core tree of three taxa. 4. Construct new tree topologies by stepwise adding new taxa to the trees which do not possess a STOP mark. The next taxa is chosen according to the list in step 1 and added to each tree at all of its branches. Tree lengths are computed. 5. Assign STOP marks if upper bound is exceeded. Terminate if all trees possess a STOP mark. List of step 1 leads to many early STOP signals.

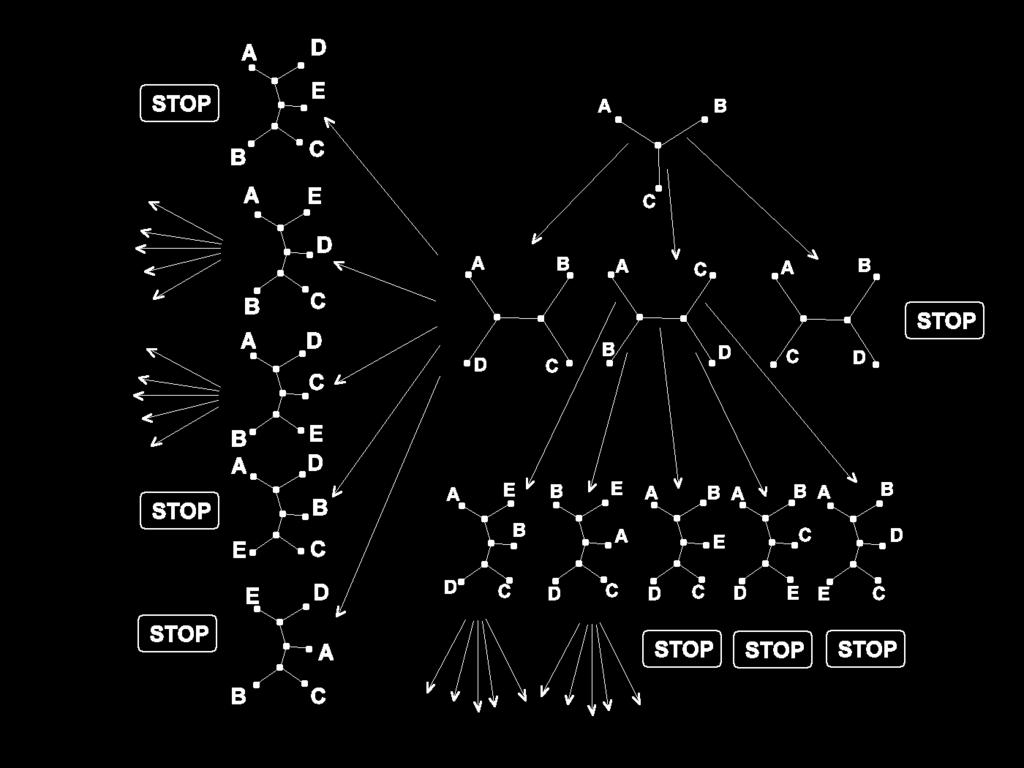
20 Tree Search
21 Tree Search Heuristics for Tree Search (Step 2 in previous algorithm) Stepwise Addition Algorithm: extend only tree with shortest length. If all taxa are inserted then perform branch swapping Branch Swapping: (1) neighbor interchange: two taxa connected to the same node, (2) subtree pruning and regrafting: remove small tree and connect it with the root to a branch, (3) bisection-reconnection: remove branch to obtain two trees and reconnect them by inserting a new branch where each branch of the subtrees can be connected in contrast to (2) where the root is connected
22 Tree Search Heuristics for Tree Search Branch and Bound Like: Use step 1. of the branch-and-bound algorithm to obtain the minimal tree (not maximal!). Upper local bounds U n for n taxa are constructed in this way. These upper bounds server for stopping signals.
23 Weighted / Bootstrapping Type of substitution is weighted according to PAM and BLOSUM matrices to address survival of substituition Bootstrapping - accesses the variability of the tree with respect to the data ( variance ) and identifies stable substructures - is possible because the temporal order of the alignment columns does not matter - cannot access the quality of a method but only its robustness
24 Inconsistency of Maximum True Tree Maximum
25 Inconsistency of Maximum a and b are similar to each other and match to 99% whereas c and d are not similar to any other sequence and only match 5% by chance (1 out of 20). Informative columns: only two symbols and each appears twice. Probabilities of informative columns and their rate: 90% of the cases of informative columns we observe c i = d i. Maximum parsimony will judge c and d as similar as a and b.
26 Distance-based Methods matrix of pairwise distances between the sequences A distance D is produced by a metric d (function) on objects x indexed by i,j,k: metric d must fulfill not all scoring schemes are a metric, e.g. the e-value
27 UPGMA Unweighted Pair Group Method using arithmetic Averages (constructive clustering method based on joining pairs of clusters) It works as follows: 1. each sequence i is a cluster c i with one element n i =1 and height l i = 0. Put all i into a list. 2. Select cluster pair (i,j) from the list with minimal D ij and create a new cluster c k by joining c i and c j with height l k = D ij / 2 and number of elements n k = n i + n j. 3. Compute the distances for c k : 4. Remove i and j from the list and add k to the list. If the list contains only one element then terminate else go to step 2.
28 UPGMA assumption of constant rate of evolution in different lineages bootstrap can evaluate the reliability to data variation Positive interior branches contribute to the quality of the tree
29 Least Squares minimize (D ij E ij ), where E ij is the sum of distances in the tree on the path from taxa i to taxa j (the path metric) The objective is Fitch and Margoliash, 1967, introduced weighted least squares: optimized under the constraint of nonnegative branch length
30 Least Squares If matrix A is the binary topology matrix with N(N-1)/2 rows, one for each D ij, and v columns for the v branches of the topology. In each row (i,j) all branches contained in the path from i to j are marked by 1 and all other branches are 0 l is the v -dimensional vector of branch weights, then least squares assumption: D ij deviates from E ij according to a Gaussian distribution ε ij with mean 0 and variance D 2 ij : maximum likelihood estimator (least squares) is Gaussianity assumption: justified by sufficient large sequences l i are Gaussian and, therefore, also D ij
31 Minimum The objective is the sum of branch length' l: Given an unbiased branch length estimator l The expected value of L is smallest for the true topology independent of the number of sequences (Rzhetsky and Nei, 1993) Minimum evolution is computational expensive
32 Neighbor The neighbor joining (Saitou and Nei, 1987) simplifies the minimum evolution method (for fewer than six taxa both methods give the same result) Neighbors: taxa that are connected by a single node Additive metric d: any four elements fulfill i j path metric (counting the branch weights) is an additive metric m k
33 Neighbor An additive metric can be represented by an unique additive tree construction of an additive tree where a node v is inserted: Path metric holds:
34 Neighbor objective of the neighbor joining algorithm is S the sum of all branch length' l ij starts with a star tree We assume N taxa with initial (star tree) objective S 0 : where the comes from the fact that, therefore is part of distances
35 Neighbor In the next step taxa 1 and 2 are joined and a new internal node Y is introduced and computed as set all paths from i to j containing equal to and solve for. These are all path' from nodes 1 and 2 to. Therefore paths start from nodes 1 and 2, each, giving paths. is in all node 1 paths and in all node 2 paths. The tail is in one node 1 and one node 2 path. Above equation is obtained by averaging over these equations for.
36 Neighbor similar as for the initial star tree inserted into the last equation:
37 Neighbor generalized from joining (1,2) to (k,l): net divergences r k are accumulated distances of k to all other taxa: giving is constant for all objectives, therefore an equivalent objective is If k and l are evolutionary neighbors but is large due to fast evolution of k and/or l, then r k and/or r l are large and Q kl small
38 Neighbor The algorithm: 1. Given start with a star tree where the taxa are the leaves. Put all taxa in a set of objects. 2. For each leave i compute 3. For each pair (i,j) compute 4. Determine the minimal Q ij. Join these (i,j) to new leave u. Compute new branch length and new distances of u: Delete i and j from the set of objects and add. Stop if the set of objects contains only u otherwise go to Step 1

39 Neighbor Neighbor joining: algorithm; for larger data sets The formula for Q ij accounts for differences in evolution rates The objective S is only minimized approximatively CLUSTALW uses neighbor-joining for multiple alignments
40 Distance we focus on nucleotides! substitution rates:
41 Distance Jukes Cantor Mutation probability: Identical positions of 2 seq. remain identical: different nucleotides will be identical: One changes and the other not: Two of these events:
42 Distance Jukes Cantor difference equation: continuous model: The solution for :
43 Distance Jukes Cantor The substitutions per position d for two sequences is 2r t Estimating q and inserting in above equation: estimate d. The variance of the estimate for d:
44 Distance Kimura group nucleotide pairs:
45 Distance Kimura transitional substitutions: transversional substitutions: equilibrium frequency of each nucleotide: 0.25 However occurrence of GC Drosophila mitochondrial DNA is 0.1
46 Distance Felsenstein / Tajima-Nei x ij : relative frequency of nucleotide pair (i,j)
47 Distance Tamura extends Kimura's model for GC content different from 0.5
48 Distance Hasegawa (HKY) hybrid of Kimuras and Felsenstein / Tajima-Nei: GC content and transition / transversion
49 Distance Tamura-Nei includes Hasegawa's model P 1 : proportion of transitional differences between A and G P 2 : proportion of transitional differences between T and C Q: proportion of transversional differences
50 Maximum Methods Tree probability: product of the mutation rates in each branch Mutation rate: product between substitution rate and branch length D: data, multiple alignment of N sequences (taxa) D k : N-dimensional vector at position k of the multiple alignment A: tree topology (see least squares) l: vector of branch length H: number of hidden nodes of the topology A : model for nucleotide substitution : set of letters (e.g. the amino acids) Hidden nodes are indexed from 1 to H taxa are indexed from H+1 to H+N Root node has index 1
51 Maximum Methods The likelihood of the tree at the k-th position: : prior probability of the root node assigned with : indicates an existing branch : probability of branch length between and hidden states are summed out Prior is estimated or given
52 Maximum Methods If is the Felsenstein / Tajima-Nei equal-input model, the branch length probabilities are For and we obtain Jukes-Cantor: reversible models: choice of the root does not matter because branch lengths count independent of their substitution direction
53 Maximum Methods Felsenstein's (81) pruning algorithm to compute (independent positions) recursive formula: : probability of a letter a at node i : computed by dynamic programming from leaves to root
54 Maximum Methods best tree: both the branch length' and the topology optimized branch length : gradient based / EM (expectation-maximization) tree topology: Felsenstein (81) growing (constructive) algorithm start with 3 taxa, at k-th taxa test all (2k-5) branches for insertion further optimized by local changing the topology tree topology: small N all topologies can be tested, then local changes similar to parsimony tree ML estimator is computationally expensive but unbiased (sequence length) and asymptotically efficient (minimal variance) fast heuristics: Strimmer and v. Haeseler (96): all topologies of 4 taxa then build the final tree (software:
55 Examples From triosephosphat isomerase of different species trees are constructed with PHYLIP (Phylogeny Inference Package) 3.5c
56 Examples Fitch-Margoliash
57 Examples Fitch-Margoliash with assumption of a molecular clock ( kitsch )
58 Examples neighbor joining
59 Examples UPGMA
60 Examples Fitch-Margoliash neighbor joining Kitsch UPGMA
61 Examples Phylogenetic tree based on triosephosphat isomerase for: Human Monkey Mouse Rat Cow Pig Goose Chicken Zebrafish Fruit FLY Rye Rice Corn Soybean Bacterium
62 Examples Fitch-Margoliash
63 Examples Fitch-Margoliash with assumption of a molecular clock ( kitsch )
64 Examples neighbor joining
65 Examples UPGMA
66 Examples Fitch-Margoliash neighbor joining Kitsch UPGMA

67 Examples relationship between humans and apes
68 Examples relationship between humans and apes
69 Examples
70 Three Anwers From where came the first human? Africa! Is Anna Anderson the tsar s daughter Anastasia? No! Are the neanderthals the ancestors of the humans? No! Separate Species Asiats Europeans Australians Africans Ancestor Modern Humans Neanderthals T T T 337 C C C C C C A A A T T T G G G A A A A A A T T T A A A T T T T T T G G G 106 T C C C T T 324 A A A G G G T T T C C C A A A A A A A A A T T T C C C C C C C C C A A A T C C 91 C C Prince Philip (Grand nephew zar) T C Carl Maucher (Grand nephew F. Schanzkowska) T C Anna Anderson
71 END
Constructing Evolutionary/Phylogenetic Trees
 Constructing Evolutionary/Phylogenetic Trees 2 broad categories: istance-based methods Ultrametric Additive: UPGMA Transformed istance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Constructing Evolutionary/Phylogenetic Trees 2 broad categories: istance-based methods Ultrametric Additive: UPGMA Transformed istance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Phylogenetic Tree Reconstruction
 I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
I519 Introduction to Bioinformatics, 2011 Phylogenetic Tree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & Computing, IUB Evolution theory Speciation Evolution of new organisms is driven
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
 Tree of Life iological Sequence nalysis Chapter http://tolweb.org/tree/ Phylogenetic Prediction ll organisms on Earth have a common ancestor. ll species are related. The relationship is called a phylogeny
Tree of Life iological Sequence nalysis Chapter http://tolweb.org/tree/ Phylogenetic Prediction ll organisms on Earth have a common ancestor. ll species are related. The relationship is called a phylogeny
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut
 Faculty of Veterinary Medicine Assiut") Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic analysis Phylogenetic Basics: Biological
"Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky
 MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
MOLECULAR PHYLOGENY "Nothing in biology makes sense except in the light of evolution Theodosius Dobzhansky EVOLUTION - theory that groups of organisms change over time so that descendeants differ structurally
Dr. Amira A. AL-Hosary
 Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Phylogenetic analysis Amira A. AL-Hosary PhD of infectious diseases Department of Animal Medicine (Infectious Diseases) Faculty of Veterinary Medicine Assiut University-Egypt Phylogenetic Basics: Biological
Constructing Evolutionary/Phylogenetic Trees
 Constructing Evolutionary/Phylogenetic Trees 2 broad categories: Distance-based methods Ultrametric Additive: UPGMA Transformed Distance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
Constructing Evolutionary/Phylogenetic Trees 2 broad categories: Distance-based methods Ultrametric Additive: UPGMA Transformed Distance Neighbor-Joining Character-based Maximum Parsimony Maximum Likelihood
POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics
 POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics - in deriving a phylogeny our goal is simply to reconstruct the historical relationships between a group of taxa. - before we review the
POPULATION GENETICS Winter 2005 Lecture 17 Molecular phylogenetics - in deriving a phylogeny our goal is simply to reconstruct the historical relationships between a group of taxa. - before we review the
Phylogenetics: Building Phylogenetic Trees
 1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
1 Phylogenetics: Building Phylogenetic Trees COMP 571 Luay Nakhleh, Rice University 2 Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary model should
Phylogenetics: Distance Methods. COMP Spring 2015 Luay Nakhleh, Rice University
 Phylogenetics: Distance Methods COMP 571 - Spring 2015 Luay Nakhleh, Rice University Outline Evolutionary models and distance corrections Distance-based methods Evolutionary Models and Distance Correction
Phylogenetics: Distance Methods COMP 571 - Spring 2015 Luay Nakhleh, Rice University Outline Evolutionary models and distance corrections Distance-based methods Evolutionary Models and Distance Correction
Phylogenetic Analysis. Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center
 Phylogenetic Analysis Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center Outline Basic Concepts Tree Construction Methods Distance-based methods
Phylogenetic Analysis Han Liang, Ph.D. Assistant Professor of Bioinformatics and Computational Biology UT MD Anderson Cancer Center Outline Basic Concepts Tree Construction Methods Distance-based methods
Phylogenetics: Building Phylogenetic Trees. COMP Fall 2010 Luay Nakhleh, Rice University
 Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
Phylogenetics: Building Phylogenetic Trees COMP 571 - Fall 2010 Luay Nakhleh, Rice University Four Questions Need to be Answered What data should we use? Which method should we use? Which evolutionary
Phylogenetic inference
 Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Letter to the Editor. Department of Biology, Arizona State University
 Letter to the Editor Traditional Phylogenetic Reconstruction Methods Reconstruct Shallow and Deep Evolutionary Relationships Equally Well Michael S. Rosenberg and Sudhir Kumar Department of Biology, Arizona
Letter to the Editor Traditional Phylogenetic Reconstruction Methods Reconstruct Shallow and Deep Evolutionary Relationships Equally Well Michael S. Rosenberg and Sudhir Kumar Department of Biology, Arizona
Phylogenetics. BIOL 7711 Computational Bioscience
 Consortium for Comparative Genomics! University of Colorado School of Medicine Phylogenetics BIOL 7711 Computational Bioscience Biochemistry and Molecular Genetics Computational Bioscience Program Consortium
Consortium for Comparative Genomics! University of Colorado School of Medicine Phylogenetics BIOL 7711 Computational Bioscience Biochemistry and Molecular Genetics Computational Bioscience Program Consortium
9/30/11. Evolution theory. Phylogenetic Tree Reconstruction. Phylogenetic trees (binary trees) Phylogeny (phylogenetic tree)
 Phylogeny (phylogenetic tree)") I9 Introduction to Bioinformatics, 0 Phylogenetic ree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & omputing, IUB Evolution theory Speciation Evolution of new organisms is driven by
I9 Introduction to Bioinformatics, 0 Phylogenetic ree Reconstruction Yuzhen Ye (yye@indiana.edu) School of Informatics & omputing, IUB Evolution theory Speciation Evolution of new organisms is driven by
DNA Phylogeny. Signals and Systems in Biology Kushal EE, IIT Delhi
 DNA Phylogeny Signals and Systems in Biology Kushal Shah @ EE, IIT Delhi Phylogenetics Grouping and Division of organisms Keeps changing with time Splitting, hybridization and termination Cladistics :
DNA Phylogeny Signals and Systems in Biology Kushal Shah @ EE, IIT Delhi Phylogenetics Grouping and Division of organisms Keeps changing with time Splitting, hybridization and termination Cladistics :
Molecular phylogeny - Using molecular sequences to infer evolutionary relationships. Tore Samuelsson Feb 2016
 Molecular phylogeny - Using molecular sequences to infer evolutionary relationships Tore Samuelsson Feb 2016 Molecular phylogeny is being used in the identification and characterization of new pathogens,
Molecular phylogeny - Using molecular sequences to infer evolutionary relationships Tore Samuelsson Feb 2016 Molecular phylogeny is being used in the identification and characterization of new pathogens,
Phylogenetic Trees. Phylogenetic Trees Five. Phylogeny: Inference Tool. Phylogeny Terminology. Picture of Last Quagga. Importance of Phylogeny 5.
 Five Sami Khuri Department of Computer Science San José State University San José, California, USA sami.khuri@sjsu.edu v Distance Methods v Character Methods v Molecular Clock v UPGMA v Maximum Parsimony
Five Sami Khuri Department of Computer Science San José State University San José, California, USA sami.khuri@sjsu.edu v Distance Methods v Character Methods v Molecular Clock v UPGMA v Maximum Parsimony
Estimating Phylogenies (Evolutionary Trees) II. Biol4230 Thurs, March 2, 2017 Bill Pearson Jordan 6-057
 II. Biol4230 Thurs, March 2, 2017 Bill Pearson Jordan 6-057") Estimating Phylogenies (Evolutionary Trees) II Biol4230 Thurs, March 2, 2017 Bill Pearson wrp@virginia.edu 4-2818 Jordan 6-057 Tree estimation strategies: Parsimony?no model, simply count minimum number
Estimating Phylogenies (Evolutionary Trees) II Biol4230 Thurs, March 2, 2017 Bill Pearson wrp@virginia.edu 4-2818 Jordan 6-057 Tree estimation strategies: Parsimony?no model, simply count minimum number
Phylogenetic analyses. Kirsi Kostamo
 Phylogenetic analyses Kirsi Kostamo The aim: To construct a visual representation (a tree) to describe the assumed evolution occurring between and among different groups (individuals, populations, species,
Phylogenetic analyses Kirsi Kostamo The aim: To construct a visual representation (a tree) to describe the assumed evolution occurring between and among different groups (individuals, populations, species,
Phylogeny and Evolution. Gina Cannarozzi ETH Zurich Institute of Computational Science
 Phylogeny and Evolution Gina Cannarozzi ETH Zurich Institute of Computational Science History Aristotle (384-322 BC) classified animals. He found that dolphins do not belong to the fish but to the mammals.
Phylogeny and Evolution Gina Cannarozzi ETH Zurich Institute of Computational Science History Aristotle (384-322 BC) classified animals. He found that dolphins do not belong to the fish but to the mammals.
Michael Yaffe Lecture #5 (((A,B)C)D) Database Searching & Molecular Phylogenetics A B C D B C D
C)D) Database Searching & Molecular Phylogenetics A B C D B C D") 7.91 Lecture #5 Database Searching & Molecular Phylogenetics Michael Yaffe B C D B C D (((,B)C)D) Outline Distance Matrix Methods Neighbor-Joining Method and Related Neighbor Methods Maximum Likelihood
7.91 Lecture #5 Database Searching & Molecular Phylogenetics Michael Yaffe B C D B C D (((,B)C)D) Outline Distance Matrix Methods Neighbor-Joining Method and Related Neighbor Methods Maximum Likelihood
Phylogenetic Trees. What They Are Why We Do It & How To Do It. Presented by Amy Harris Dr Brad Morantz
 Phylogenetic Trees What They Are Why We Do It & How To Do It Presented by Amy Harris Dr Brad Morantz Overview What is a phylogenetic tree Why do we do it How do we do it Methods and programs Parallels
Phylogenetic Trees What They Are Why We Do It & How To Do It Presented by Amy Harris Dr Brad Morantz Overview What is a phylogenetic tree Why do we do it How do we do it Methods and programs Parallels
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Distance Methods Character Methods
Algorithms in Bioinformatics Sami Khuri Department of Computer Science San José State University San José, California, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Distance Methods Character Methods
Theory of Evolution Charles Darwin
 Theory of Evolution Charles arwin 858-59: Origin of Species 5 year voyage of H.M.S. eagle (83-36) Populations have variations. Natural Selection & Survival of the fittest: nature selects best adapted varieties
Theory of Evolution Charles arwin 858-59: Origin of Species 5 year voyage of H.M.S. eagle (83-36) Populations have variations. Natural Selection & Survival of the fittest: nature selects best adapted varieties
Molecular phylogeny How to infer phylogenetic trees using molecular sequences
 Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 2009 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 2009 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
EVOLUTIONARY DISTANCES
 EVOLUTIONARY DISTANCES FROM STRINGS TO TREES Luca Bortolussi 1 1 Dipartimento di Matematica ed Informatica Università degli studi di Trieste luca@dmi.units.it Trieste, 14 th November 2007 OUTLINE 1 STRINGS:
EVOLUTIONARY DISTANCES FROM STRINGS TO TREES Luca Bortolussi 1 1 Dipartimento di Matematica ed Informatica Università degli studi di Trieste luca@dmi.units.it Trieste, 14 th November 2007 OUTLINE 1 STRINGS:
Molecular phylogeny How to infer phylogenetic trees using molecular sequences
 Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 200 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Molecular phylogeny How to infer phylogenetic trees using molecular sequences ore Samuelsson Nov 200 Applications of phylogenetic methods Reconstruction of evolutionary history / Resolving taxonomy issues
Inferring phylogeny. Today s topics. Milestones of molecular evolution studies Contributions to molecular evolution
 Today s topics Inferring phylogeny Introduction! Distance methods! Parsimony method!"#$%&'(!)* +,-.'/01!23454(6!7!2845*0&4'9#6!:&454(6 ;?@AB=C?DEF Overview of phylogenetic inferences Methodology Methods
Today s topics Inferring phylogeny Introduction! Distance methods! Parsimony method!"#$%&'(!)* +,-.'/01!23454(6!7!2845*0&4'9#6!:&454(6 ;?@AB=C?DEF Overview of phylogenetic inferences Methodology Methods
BINF6201/8201. Molecular phylogenetic methods
 BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
BINF60/80 Molecular phylogenetic methods 0-7-06 Phylogenetics Ø According to the evolutionary theory, all life forms on this planet are related to one another by descent. Ø Traditionally, phylogenetics
C3020 Molecular Evolution. Exercises #3: Phylogenetics
 C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
C3020 Molecular Evolution Exercises #3: Phylogenetics Consider the following sequences for five taxa 1-5 and the known outgroup O, which has the ancestral states (note that sequence 3 has changed from
Week 5: Distance methods, DNA and protein models
 Week 5: Distance methods, DNA and protein models Genome 570 February, 2016 Week 5: Distance methods, DNA and protein models p.1/69 A tree and the expected distances it predicts E A 0.08 0.05 0.06 0.03
Week 5: Distance methods, DNA and protein models Genome 570 February, 2016 Week 5: Distance methods, DNA and protein models p.1/69 A tree and the expected distances it predicts E A 0.08 0.05 0.06 0.03
Molecular Phylogenetics (part 1 of 2) Computational Biology Course João André Carriço
 Computational Biology Course João André Carriço") Molecular Phylogenetics (part 1 of 2) Computational Biology Course João André Carriço jcarrico@fm.ul.pt Charles Darwin (1809-1882) Charles Darwin s tree of life in Notebook B, 1837-1838 Ernst Haeckel (1934-1919)
Molecular Phylogenetics (part 1 of 2) Computational Biology Course João André Carriço jcarrico@fm.ul.pt Charles Darwin (1809-1882) Charles Darwin s tree of life in Notebook B, 1837-1838 Ernst Haeckel (1934-1919)
Multiple Sequence Alignment. Sequences
 Multiple Sequence Alignment Sequences > YOR020c mstllksaksivplmdrvlvqrikaqaktasglylpe knveklnqaevvavgpgftdangnkvvpqvkvgdqvl ipqfggstiklgnddevilfrdaeilakiakd > crassa mattvrsvksliplldrvlvqrvkaeaktasgiflpe
Multiple Sequence Alignment Sequences > YOR020c mstllksaksivplmdrvlvqrikaqaktasglylpe knveklnqaevvavgpgftdangnkvvpqvkvgdqvl ipqfggstiklgnddevilfrdaeilakiakd > crassa mattvrsvksliplldrvlvqrvkaeaktasgiflpe
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Evolutionary Tree Analysis. Overview
 CSI/BINF 5330 Evolutionary Tree Analysis Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Backgrounds Distance-Based Evolutionary Tree Reconstruction Character-Based
CSI/BINF 5330 Evolutionary Tree Analysis Young-Rae Cho Associate Professor Department of Computer Science Baylor University Overview Backgrounds Distance-Based Evolutionary Tree Reconstruction Character-Based
Maximum Likelihood Until recently the newest method. Popularized by Joseph Felsenstein, Seattle, Washington.
 Maximum Likelihood This presentation is based almost entirely on Peter G. Fosters - "The Idiot s Guide to the Zen of Likelihood in a Nutshell in Seven Days for Dummies, Unleashed. http://www.bioinf.org/molsys/data/idiots.pdf
Maximum Likelihood This presentation is based almost entirely on Peter G. Fosters - "The Idiot s Guide to the Zen of Likelihood in a Nutshell in Seven Days for Dummies, Unleashed. http://www.bioinf.org/molsys/data/idiots.pdf
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Algorithmic Methods Well-defined methodology Tree reconstruction those that are well-defined enough to be carried out by a computer. Felsenstein 2004,
 Tracing the Evolution of Numerical Phylogenetics: History, Philosophy, and Significance Adam W. Ferguson Phylogenetic Systematics 26 January 2009 Inferring Phylogenies Historical endeavor Darwin- 1837
Tracing the Evolution of Numerical Phylogenetics: History, Philosophy, and Significance Adam W. Ferguson Phylogenetic Systematics 26 January 2009 Inferring Phylogenies Historical endeavor Darwin- 1837
Lecture 27. Phylogeny methods, part 4 (Models of DNA and protein change) p.1/26
 p.1/26") Lecture 27. Phylogeny methods, part 4 (Models of DNA and protein change) Joe Felsenstein Department of Genome Sciences and Department of Biology Lecture 27. Phylogeny methods, part 4 (Models of DNA and
Lecture 27. Phylogeny methods, part 4 (Models of DNA and protein change) Joe Felsenstein Department of Genome Sciences and Department of Biology Lecture 27. Phylogeny methods, part 4 (Models of DNA and
Molecular Evolution and Phylogenetic Tree Reconstruction
 1 4 Molecular Evolution and Phylogenetic Tree Reconstruction 3 2 5 1 4 2 3 5 Orthology, Paralogy, Inparalogs, Outparalogs Phylogenetic Trees Nodes: species Edges: time of independent evolution Edge length
1 4 Molecular Evolution and Phylogenetic Tree Reconstruction 3 2 5 1 4 2 3 5 Orthology, Paralogy, Inparalogs, Outparalogs Phylogenetic Trees Nodes: species Edges: time of independent evolution Edge length
FUNDAMENTALS OF MOLECULAR EVOLUTION
 FUNDAMENTALS OF MOLECULAR EVOLUTION Second Edition Dan Graur TELAVIV UNIVERSITY Wen-Hsiung Li UNIVERSITY OF CHICAGO SINAUER ASSOCIATES, INC., Publishers Sunderland, Massachusetts Contents Preface xiii
FUNDAMENTALS OF MOLECULAR EVOLUTION Second Edition Dan Graur TELAVIV UNIVERSITY Wen-Hsiung Li UNIVERSITY OF CHICAGO SINAUER ASSOCIATES, INC., Publishers Sunderland, Massachusetts Contents Preface xiii
Phylogeny: traditional and Bayesian approaches
 Phylogeny: traditional and Bayesian approaches 5-Feb-2014 DEKM book Notes from Dr. B. John Holder and Lewis, Nature Reviews Genetics 4, 275-284, 2003 1 Phylogeny A graph depicting the ancestor-descendent
Phylogeny: traditional and Bayesian approaches 5-Feb-2014 DEKM book Notes from Dr. B. John Holder and Lewis, Nature Reviews Genetics 4, 275-284, 2003 1 Phylogeny A graph depicting the ancestor-descendent
Improving divergence time estimation in phylogenetics: more taxa vs. longer sequences
 Mathematical Statistics Stockholm University Improving divergence time estimation in phylogenetics: more taxa vs. longer sequences Bodil Svennblad Tom Britton Research Report 2007:2 ISSN 650-0377 Postal
Mathematical Statistics Stockholm University Improving divergence time estimation in phylogenetics: more taxa vs. longer sequences Bodil Svennblad Tom Britton Research Report 2007:2 ISSN 650-0377 Postal
What is Phylogenetics
 What is Phylogenetics Phylogenetics is the area of research concerned with finding the genetic connections and relationships between species. The basic idea is to compare specific characters (features)
What is Phylogenetics Phylogenetics is the area of research concerned with finding the genetic connections and relationships between species. The basic idea is to compare specific characters (features)
Additive distances. w(e), where P ij is the path in T from i to j. Then the matrix [D ij ] is said to be additive.
![Additive distances. w(e), where P ij is the path in T from i to j. Then the matrix [D ij ] is said to be additive.](/thumbs/92/110439366.jpg "Additive distances. w(e), where P ij is the path in T from i to j. Then the matrix [D ij ] is said to be additive.") Additive distances Let T be a tree on leaf set S and let w : E R + be an edge-weighting of T, and assume T has no nodes of degree two. Let D ij = e P ij w(e), where P ij is the path in T from i to j. Then
Additive distances Let T be a tree on leaf set S and let w : E R + be an edge-weighting of T, and assume T has no nodes of degree two. Let D ij = e P ij w(e), where P ij is the path in T from i to j. Then
Lecture 24. Phylogeny methods, part 4 (Models of DNA and protein change) p.1/22
 p.1/22") Lecture 24. Phylogeny methods, part 4 (Models of DNA and protein change) Joe Felsenstein Department of Genome Sciences and Department of Biology Lecture 24. Phylogeny methods, part 4 (Models of DNA and
Lecture 24. Phylogeny methods, part 4 (Models of DNA and protein change) Joe Felsenstein Department of Genome Sciences and Department of Biology Lecture 24. Phylogeny methods, part 4 (Models of DNA and
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Effects of Gap Open and Gap Extension Penalties
 Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Brigham Young University BYU ScholarsArchive All Faculty Publications 200-10-01 Effects of Gap Open and Gap Extension Penalties Hyrum Carroll hyrumcarroll@gmail.com Mark J. Clement clement@cs.byu.edu See
Phylogene)cs. IMBB 2016 BecA- ILRI Hub, Nairobi May 9 20, Joyce Nzioki
cs. IMBB 2016 BecA- ILRI Hub, Nairobi May 9 20, Joyce Nzioki") Phylogene)cs IMBB 2016 BecA- ILRI Hub, Nairobi May 9 20, 2016 Joyce Nzioki Phylogenetics The study of evolutionary relatedness of organisms. Derived from two Greek words:» Phle/Phylon: Tribe/Race» Genetikos:
Phylogene)cs IMBB 2016 BecA- ILRI Hub, Nairobi May 9 20, 2016 Joyce Nzioki Phylogenetics The study of evolutionary relatedness of organisms. Derived from two Greek words:» Phle/Phylon: Tribe/Race» Genetikos:
Phylogenies Scores for Exhaustive Maximum Likelihood and Parsimony Scores Searches
 Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
Int. J. Bioinformatics Research and Applications, Vol. x, No. x, xxxx Phylogenies Scores for Exhaustive Maximum Likelihood and s Searches Hyrum D. Carroll, Perry G. Ridge, Mark J. Clement, Quinn O. Snell
Phylogenetic trees 07/10/13
 Phylogenetic trees 07/10/13 A tree is the only figure to occur in On the Origin of Species by Charles Darwin. It is a graphical representation of the evolutionary relationships among entities that share
Phylogenetic trees 07/10/13 A tree is the only figure to occur in On the Origin of Species by Charles Darwin. It is a graphical representation of the evolutionary relationships among entities that share
Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM)
") Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Efficiencies of maximum likelihood methods of phylogenetic inferences when different substitution models are used
 Molecular Phylogenetics and Evolution 31 (2004) 865 873 MOLECULAR PHYLOGENETICS AND EVOLUTION www.elsevier.com/locate/ympev Efficiencies of maximum likelihood methods of phylogenetic inferences when different
Molecular Phylogenetics and Evolution 31 (2004) 865 873 MOLECULAR PHYLOGENETICS AND EVOLUTION www.elsevier.com/locate/ympev Efficiencies of maximum likelihood methods of phylogenetic inferences when different
A (short) introduction to phylogenetics
 introduction to phylogenetics") A (short) introduction to phylogenetics Thibaut Jombart, Marie-Pauline Beugin MRC Centre for Outbreak Analysis and Modelling Imperial College London Genetic data analysis with PR Statistics, Millport Field
A (short) introduction to phylogenetics Thibaut Jombart, Marie-Pauline Beugin MRC Centre for Outbreak Analysis and Modelling Imperial College London Genetic data analysis with PR Statistics, Millport Field
7. Tests for selection
 Sequence analysis and genomics 7. Tests for selection Dr. Katja Nowick Group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute for Brain Research www. nowicklab.info
Sequence analysis and genomics 7. Tests for selection Dr. Katja Nowick Group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute for Brain Research www. nowicklab.info
Theory of Evolution. Charles Darwin
 Theory of Evolution harles arwin 858-59: Origin of Species 5 year voyage of H.M.S. eagle (8-6) Populations have variations. Natural Selection & Survival of the fittest: nature selects best adapted varieties
Theory of Evolution harles arwin 858-59: Origin of Species 5 year voyage of H.M.S. eagle (8-6) Populations have variations. Natural Selection & Survival of the fittest: nature selects best adapted varieties
Sequence Analysis 17: lecture 5. Substitution matrices Multiple sequence alignment
 Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
The Phylogenetic Handbook
 The Phylogenetic Handbook A Practical Approach to DNA and Protein Phylogeny Edited by Marco Salemi University of California, Irvine and Katholieke Universiteit Leuven, Belgium and Anne-Mieke Vandamme Rega
The Phylogenetic Handbook A Practical Approach to DNA and Protein Phylogeny Edited by Marco Salemi University of California, Irvine and Katholieke Universiteit Leuven, Belgium and Anne-Mieke Vandamme Rega
How to read and make phylogenetic trees Zuzana Starostová
 How to read and make phylogenetic trees Zuzana Starostová How to make phylogenetic trees? Workflow: obtain DNA sequence quality check sequence alignment calculating genetic distances phylogeny estimation
How to read and make phylogenetic trees Zuzana Starostová How to make phylogenetic trees? Workflow: obtain DNA sequence quality check sequence alignment calculating genetic distances phylogeny estimation
Lecture 4. Models of DNA and protein change. Likelihood methods
 Lecture 4. Models of DNA and protein change. Likelihood methods Joe Felsenstein Department of Genome Sciences and Department of Biology Lecture 4. Models of DNA and protein change. Likelihood methods p.1/36
Lecture 4. Models of DNA and protein change. Likelihood methods Joe Felsenstein Department of Genome Sciences and Department of Biology Lecture 4. Models of DNA and protein change. Likelihood methods p.1/36
Introduction to MEGA
 Introduction to MEGA Download at: http://www.megasoftware.net/index.html Thomas Randall, PhD tarandal@email.unc.edu Manual at: www.megasoftware.net/mega4 Use of phylogenetic analysis software tools Bioinformatics
Introduction to MEGA Download at: http://www.megasoftware.net/index.html Thomas Randall, PhD tarandal@email.unc.edu Manual at: www.megasoftware.net/mega4 Use of phylogenetic analysis software tools Bioinformatics
Phylogeny: building the tree of life
 Phylogeny: building the tree of life Dr. Fayyaz ul Amir Afsar Minhas Department of Computer and Information Sciences Pakistan Institute of Engineering & Applied Sciences PO Nilore, Islamabad, Pakistan
Phylogeny: building the tree of life Dr. Fayyaz ul Amir Afsar Minhas Department of Computer and Information Sciences Pakistan Institute of Engineering & Applied Sciences PO Nilore, Islamabad, Pakistan
CS5263 Bioinformatics. Guest Lecture Part II Phylogenetics
 CS5263 Bioinformatics Guest Lecture Part II Phylogenetics Up to now we have focused on finding similarities, now we start focusing on differences (dissimilarities leading to distance measures). Identifying
CS5263 Bioinformatics Guest Lecture Part II Phylogenetics Up to now we have focused on finding similarities, now we start focusing on differences (dissimilarities leading to distance measures). Identifying
Michael Yaffe Lecture #4 (((A,B)C)D) Database Searching & Molecular Phylogenetics A B C D B C D
C)D) Database Searching & Molecular Phylogenetics A B C D B C D") 7.91 Lecture #4 Database Searching & Molecular Phylogenetics Michael Yaffe A B C D A B C D (((A,B)C)D) Outline FASTA, Blast searching, Smith-Waterman Psi-Blast Review of enomic DNA structure Substitution
7.91 Lecture #4 Database Searching & Molecular Phylogenetics Michael Yaffe A B C D A B C D (((A,B)C)D) Outline FASTA, Blast searching, Smith-Waterman Psi-Blast Review of enomic DNA structure Substitution
CSCI1950 Z Computa4onal Methods for Biology Lecture 5
 CSCI1950 Z Computa4onal Methods for Biology Lecture 5 Ben Raphael February 6, 2009 hip://cs.brown.edu/courses/csci1950 z/ Alignment vs. Distance Matrix Mouse: ACAGTGACGCCACACACGT Gorilla: CCTGCGACGTAACAAACGC
CSCI1950 Z Computa4onal Methods for Biology Lecture 5 Ben Raphael February 6, 2009 hip://cs.brown.edu/courses/csci1950 z/ Alignment vs. Distance Matrix Mouse: ACAGTGACGCCACACACGT Gorilla: CCTGCGACGTAACAAACGC
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Cladistics and Bioinformatics Questions 2013
 AP Biology Name Cladistics and Bioinformatics Questions 2013 1. The following table shows the percentage similarity in sequences of nucleotides from a homologous gene derived from five different species
AP Biology Name Cladistics and Bioinformatics Questions 2013 1. The following table shows the percentage similarity in sequences of nucleotides from a homologous gene derived from five different species
Inferring phylogeny. Constructing phylogenetic trees. Tõnu Margus. Bioinformatics MTAT
 Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
Inferring phylogeny Constructing phylogenetic trees Tõnu Margus Contents What is phylogeny? How/why it is possible to infer it? Representing evolutionary relationships on trees What type questions questions
(Stevens 1991) 1. morphological characters should be assumed to be quantitative unless demonstrated otherwise
 1. morphological characters should be assumed to be quantitative unless demonstrated otherwise") Bot 421/521 PHYLOGENETIC ANALYSIS I. Origins A. Hennig 1950 (German edition) Phylogenetic Systematics 1966 B. Zimmerman (Germany, 1930 s) C. Wagner (Michigan, 1920-2000) II. Characters and character states
Bot 421/521 PHYLOGENETIC ANALYSIS I. Origins A. Hennig 1950 (German edition) Phylogenetic Systematics 1966 B. Zimmerman (Germany, 1930 s) C. Wagner (Michigan, 1920-2000) II. Characters and character states
Understanding relationship between homologous sequences
 Molecular Evolution Molecular Evolution How and when were genes and proteins created? How old is a gene? How can we calculate the age of a gene? How did the gene evolve to the present form? What selective
Molecular Evolution Molecular Evolution How and when were genes and proteins created? How old is a gene? How can we calculate the age of a gene? How did the gene evolve to the present form? What selective
Kei Takahashi and Masatoshi Nei
 Efficiencies of Fast Algorithms of Phylogenetic Inference Under the Criteria of Maximum Parsimony, Minimum Evolution, and Maximum Likelihood When a Large Number of Sequences Are Used Kei Takahashi and
Efficiencies of Fast Algorithms of Phylogenetic Inference Under the Criteria of Maximum Parsimony, Minimum Evolution, and Maximum Likelihood When a Large Number of Sequences Are Used Kei Takahashi and
METHODS FOR DETERMINING PHYLOGENY. In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task.
 Chapter 12 (Strikberger) Molecular Phylogenies and Evolution METHODS FOR DETERMINING PHYLOGENY In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task. Modern
Chapter 12 (Strikberger) Molecular Phylogenies and Evolution METHODS FOR DETERMINING PHYLOGENY In Chapter 11, we discovered that classifying organisms into groups was, and still is, a difficult task. Modern
Phylogeny. November 7, 2017
 Phylogeny November 7, 2017 Phylogenetics Phylon = tribe/race, genetikos = relative to birth Phylogenetics: study of evolutionary relationships among organisms, sequences, or anything in between Related
Phylogeny November 7, 2017 Phylogenetics Phylon = tribe/race, genetikos = relative to birth Phylogenetics: study of evolutionary relationships among organisms, sequences, or anything in between Related
Bioinformatics. Scoring Matrices. David Gilbert Bioinformatics Research Centre
 Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Bioinformatics 1 -- lecture 9. Phylogenetic trees Distance-based tree building Parsimony
 ioinformatics -- lecture 9 Phylogenetic trees istance-based tree building Parsimony (,(,(,))) rees can be represented in "parenthesis notation". Each set of parentheses represents a branch-point (bifurcation),
ioinformatics -- lecture 9 Phylogenetic trees istance-based tree building Parsimony (,(,(,))) rees can be represented in "parenthesis notation". Each set of parentheses represents a branch-point (bifurcation),
Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM).
.") 1 Bioinformatics: In-depth PROBABILITY & STATISTICS Spring Semester 2011 University of Zürich and ETH Zürich Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM). Dr. Stefanie Muff
1 Bioinformatics: In-depth PROBABILITY & STATISTICS Spring Semester 2011 University of Zürich and ETH Zürich Lecture 4: Evolutionary models and substitution matrices (PAM and BLOSUM). Dr. Stefanie Muff
Bio 1B Lecture Outline (please print and bring along) Fall, 2007
 Fall, 2007") Bio 1B Lecture Outline (please print and bring along) Fall, 2007 B.D. Mishler, Dept. of Integrative Biology 2-6810, bmishler@berkeley.edu Evolution lecture #5 -- Molecular genetics and molecular evolution
Bio 1B Lecture Outline (please print and bring along) Fall, 2007 B.D. Mishler, Dept. of Integrative Biology 2-6810, bmishler@berkeley.edu Evolution lecture #5 -- Molecular genetics and molecular evolution
Phylogenetic Trees. How do the changes in gene sequences allow us to reconstruct the evolutionary relationships between related species?
 Why? Phylogenetic Trees How do the changes in gene sequences allow us to reconstruct the evolutionary relationships between related species? The saying Don t judge a book by its cover. could be applied
Why? Phylogenetic Trees How do the changes in gene sequences allow us to reconstruct the evolutionary relationships between related species? The saying Don t judge a book by its cover. could be applied
C.DARWIN ( )
") C.DARWIN (1809-1882) LAMARCK Each evolutionary lineage has evolved, transforming itself, from a ancestor appeared by spontaneous generation DARWIN All organisms are historically interconnected. Their relationships
C.DARWIN (1809-1882) LAMARCK Each evolutionary lineage has evolved, transforming itself, from a ancestor appeared by spontaneous generation DARWIN All organisms are historically interconnected. Their relationships
Intraspecific gene genealogies: trees grafting into networks
 Intraspecific gene genealogies: trees grafting into networks by David Posada & Keith A. Crandall Kessy Abarenkov Tartu, 2004 Article describes: Population genetics principles Intraspecific genetic variation
Intraspecific gene genealogies: trees grafting into networks by David Posada & Keith A. Crandall Kessy Abarenkov Tartu, 2004 Article describes: Population genetics principles Intraspecific genetic variation
Lecture Notes: Markov chains
 Computational Genomics and Molecular Biology, Fall 5 Lecture Notes: Markov chains Dannie Durand At the beginning of the semester, we introduced two simple scoring functions for pairwise alignments: a similarity
Computational Genomics and Molecular Biology, Fall 5 Lecture Notes: Markov chains Dannie Durand At the beginning of the semester, we introduced two simple scoring functions for pairwise alignments: a similarity
Molecular Evolution & Phylogenetics
 Molecular Evolution & Phylogenetics Heuristics based on tree alterations, maximum likelihood, Bayesian methods, statistical confidence measures Jean-Baka Domelevo Entfellner Learning Objectives know basic
Molecular Evolution & Phylogenetics Heuristics based on tree alterations, maximum likelihood, Bayesian methods, statistical confidence measures Jean-Baka Domelevo Entfellner Learning Objectives know basic
Phylogeny Estimation and Hypothesis Testing using Maximum Likelihood
 Phylogeny Estimation and Hypothesis Testing using Maximum Likelihood For: Prof. Partensky Group: Jimin zhu Rama Sharma Sravanthi Polsani Xin Gong Shlomit klopman April. 7. 2003 Table of Contents Introduction...3
Phylogeny Estimation and Hypothesis Testing using Maximum Likelihood For: Prof. Partensky Group: Jimin zhu Rama Sharma Sravanthi Polsani Xin Gong Shlomit klopman April. 7. 2003 Table of Contents Introduction...3
1 ATGGGTCTC 2 ATGAGTCTC
 We need an optimality criterion to choose a best estimate (tree) Other optimality criteria used to choose a best estimate (tree) Parsimony: begins with the assumption that the simplest hypothesis that
We need an optimality criterion to choose a best estimate (tree) Other optimality criteria used to choose a best estimate (tree) Parsimony: begins with the assumption that the simplest hypothesis that
CS5238 Combinatorial methods in bioinformatics 2003/2004 Semester 1. Lecture 8: Phylogenetic Tree Reconstruction: Distance Based - October 10, 2003
 CS5238 Combinatorial methods in bioinformatics 2003/2004 Semester 1 Lecture 8: Phylogenetic Tree Reconstruction: Distance Based - October 10, 2003 Lecturer: Wing-Kin Sung Scribe: Ning K., Shan T., Xiang
CS5238 Combinatorial methods in bioinformatics 2003/2004 Semester 1 Lecture 8: Phylogenetic Tree Reconstruction: Distance Based - October 10, 2003 Lecturer: Wing-Kin Sung Scribe: Ning K., Shan T., Xiang
Minimum evolution using ordinary least-squares is less robust than neighbor-joining
 Minimum evolution using ordinary least-squares is less robust than neighbor-joining Stephen J. Willson Department of Mathematics Iowa State University Ames, IA 50011 USA email: swillson@iastate.edu November
Minimum evolution using ordinary least-squares is less robust than neighbor-joining Stephen J. Willson Department of Mathematics Iowa State University Ames, IA 50011 USA email: swillson@iastate.edu November
Lecture 11 Friday, October 21, 2011
 Lecture 11 Friday, October 21, 2011 Phylogenetic tree (phylogeny) Darwin and classification: In the Origin, Darwin said that descent from a common ancestral species could explain why the Linnaean system
Lecture 11 Friday, October 21, 2011 Phylogenetic tree (phylogeny) Darwin and classification: In the Origin, Darwin said that descent from a common ancestral species could explain why the Linnaean system
Molecular Evolution, course # Final Exam, May 3, 2006
 Molecular Evolution, course #27615 Final Exam, May 3, 2006 This exam includes a total of 12 problems on 7 pages (including this cover page). The maximum number of points obtainable is 150, and at least
Molecular Evolution, course #27615 Final Exam, May 3, 2006 This exam includes a total of 12 problems on 7 pages (including this cover page). The maximum number of points obtainable is 150, and at least
Massachusetts Institute of Technology Computational Evolutionary Biology, Fall, 2005 Notes for November 7: Molecular evolution
 Massachusetts Institute of Technology 6.877 Computational Evolutionary Biology, Fall, 2005 Notes for November 7: Molecular evolution 1. Rates of amino acid replacement The initial motivation for the neutral
Massachusetts Institute of Technology 6.877 Computational Evolutionary Biology, Fall, 2005 Notes for November 7: Molecular evolution 1. Rates of amino acid replacement The initial motivation for the neutral
Phylogenetics: Parsimony and Likelihood. COMP Spring 2016 Luay Nakhleh, Rice University
 Phylogenetics: Parsimony and Likelihood COMP 571 - Spring 2016 Luay Nakhleh, Rice University The Problem Input: Multiple alignment of a set S of sequences Output: Tree T leaf-labeled with S Assumptions
Phylogenetics: Parsimony and Likelihood COMP 571 - Spring 2016 Luay Nakhleh, Rice University The Problem Input: Multiple alignment of a set S of sequences Output: Tree T leaf-labeled with S Assumptions
Maximum Likelihood Tree Estimation. Carrie Tribble IB Feb 2018
 Maximum Likelihood Tree Estimation Carrie Tribble IB 200 9 Feb 2018 Outline 1. Tree building process under maximum likelihood 2. Key differences between maximum likelihood and parsimony 3. Some fancy extras
Maximum Likelihood Tree Estimation Carrie Tribble IB 200 9 Feb 2018 Outline 1. Tree building process under maximum likelihood 2. Key differences between maximum likelihood and parsimony 3. Some fancy extras
Consistency Index (CI)
") Consistency Index (CI) minimum number of changes divided by the number required on the tree. CI=1 if there is no homoplasy negatively correlated with the number of species sampled Retention Index (RI)
Consistency Index (CI) minimum number of changes divided by the number required on the tree. CI=1 if there is no homoplasy negatively correlated with the number of species sampled Retention Index (RI)
Reconstruire le passé biologique modèles, méthodes, performances, limites
 Reconstruire le passé biologique modèles, méthodes, performances, limites Olivier Gascuel Centre de Bioinformatique, Biostatistique et Biologie Intégrative C3BI USR 3756 Institut Pasteur & CNRS Reconstruire
Reconstruire le passé biologique modèles, méthodes, performances, limites Olivier Gascuel Centre de Bioinformatique, Biostatistique et Biologie Intégrative C3BI USR 3756 Institut Pasteur & CNRS Reconstruire
08/21/2017 BLAST. Multiple Sequence Alignments: Clustal Omega
 BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
BLAST Multiple Sequence Alignments: Clustal Omega What does basic BLAST do (e.g. what is input sequence and how does BLAST look for matches?) Susan Parrish McDaniel College Multiple Sequence Alignments
The Generalized Neighbor Joining method
 The Generalized Neighbor Joining method Ruriko Yoshida Dept. of Mathematics Duke University Joint work with Dan Levy and Lior Pachter www.math.duke.edu/ ruriko data mining 1 Challenge We would like to
The Generalized Neighbor Joining method Ruriko Yoshida Dept. of Mathematics Duke University Joint work with Dan Levy and Lior Pachter www.math.duke.edu/ ruriko data mining 1 Challenge We would like to
arxiv:q-bio/ v1 [q-bio.pe] 27 May 2005
![arxiv:q-bio/ v1 [q-bio.pe] 27 May 2005](/thumbs/74/69965840.jpg "arxiv:q-bio/ v1 [q-bio.pe] 27 May 2005") Maximum Likelihood Jukes-Cantor Triplets: Analytic Solutions arxiv:q-bio/0505054v1 [q-bio.pe] 27 May 2005 Benny Chor Michael D. Hendy Sagi Snir December 21, 2017 Abstract Complex systems of polynomial
Maximum Likelihood Jukes-Cantor Triplets: Analytic Solutions arxiv:q-bio/0505054v1 [q-bio.pe] 27 May 2005 Benny Chor Michael D. Hendy Sagi Snir December 21, 2017 Abstract Complex systems of polynomial
Phylogeny and Molecular Evolution. Introduction
 Phylogeny and Molecular Evolution Introduction 1 2/62 3/62 Credit Serafim Batzoglou (UPGMA slides) http://www.stanford.edu/class/cs262/slides Notes by Nir Friedman, Dan Geiger, Shlomo Moran, Ron Shamir,
Phylogeny and Molecular Evolution Introduction 1 2/62 3/62 Credit Serafim Batzoglou (UPGMA slides) http://www.stanford.edu/class/cs262/slides Notes by Nir Friedman, Dan Geiger, Shlomo Moran, Ron Shamir,