3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
|
|
|
- Drusilla Lloyd
- 5 years ago
- Views:
Transcription




1 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
2 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode genes. Protein structure prediction Sequence assembly Database searching 2

3 3

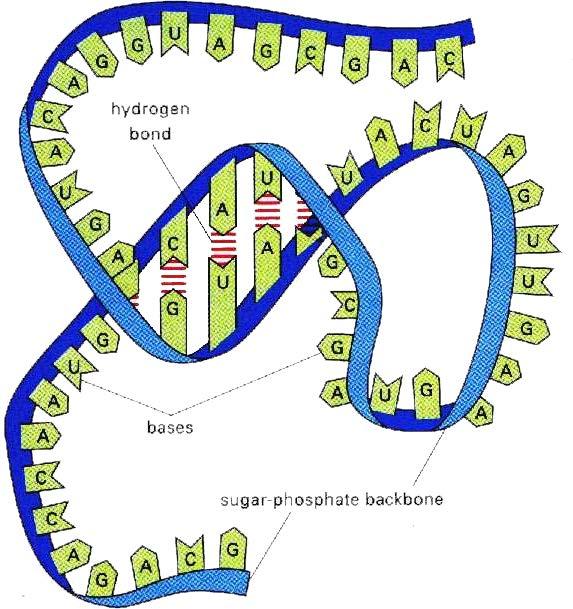

4 BIOLOGICAL SEQUENCES DNA RNA PROTEIN 4
5 EVOLUTION OF SEQUENCES All living organisms are related to each other through evolution. This means: any pair of organism, no matter how different, have a common ancestor sometime in the past, from which they evolved. Mutations and selection over long periods of time can result in considerable difference between present-day sequences derived from the same ancestral sequences. The base pair composition of the sequences can change due to point mutation (substitutions), and the sequence lengths can vary due to indels (insertions/deletions). 5
6 MUTATIONS / SUBSTITUTIONS 6

7 POINT MUTATIONS 7
8 DNA SEQUENCE EVOLUTION GGCTA Substitution GCGTA Deletion GGTA 8
9 DNA SEQUENCE EVOLUTION GGCTA Substitution GCGTA Deletion GGTA GTA GCTA CGTA GGGTA???? 9
10 DNA SEQUENCE EVOLUTION GGCTA Substitution Deletion Deletion GCGTA GGTA Insertion GTA GCTA CGTA GGGTA 10
11 SEARCHING THE DATABASE exact matches 11
12 SEARCHING THE DATABASE exact matches 12

13 SEARCHING THE DATABASE exact matches 13
14 SEARCHING THE DATABASE Searching databases is an important task in molecular biology. Searching databases for sequences tries to find similar sequences to the query sequence in the database. Such search amounts to aligning the query sequence to sequences in the database and returning results with good alignment score. Exact match approaches can be computationally very expensive which yields to usage of heuristic approaches. 14
15 SEQUENCE SIMILARITY Homology: deriving from a common ancestor-gene. Orthologous: homologous genes in different organisms. Paralogous: homologus genes in one organism that derive from gene duplication. Gene duplication: one gene is duplicated in multiple copies that can each evolve separately and assume new functions. 15

 gaps (correspond to indels:")
16 PRINCIPLES OF SEQUENCE ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity Mismatches (correspond to mutations) gaps (correspond to indels: insertions/deletions) 16
17 PRINCIPLES OF SEQUENCE ALIGNMENT Alignment can reveal homology between sequences Similarity is a descriptive term that tells about the degree of match between the two sequences. Sequence similarity does not always imply a common function. Conserved function does not always imply similarity at the sequence level. 17
18 PRINCIPLES OF SEQUENCE ALIGNMENT It is easier to detect homology when comparing protein sequences then when comparing nucleic acid sequences The probability of a match by chance is much higher in DNA sequences then in protein sequences. The genetic code is redundant: identical amino acids can be coded by different codons. The complex 3D structure of a protein, and hence its function, is determined by the amino acid sequence. Hence, conserving function leads to fewer changes in the amino acids than in the nucleotide sequence. 18

19 TYPES OF ALIGNMENT PAIRWISE ALIGNMENT used to find best-matching piecewise local or global alignment of two query sequences. MULTIPLE ALIGNMENT an extension to pairwise alignment to incorporate more then two sequences at a time it tries to align all of the sequences in a given query set. 19
20 GLOBAL VS LOCAL ALIGNMENT Global alignment tries to align the entire sequence, using as many characters as possible, up to both ends of each sequence. In local alignment, streches of sequences with the highest density of matches are aligned, generating one or more subalignments in the aligned sequences. LGPSSKQTGKGS-SRIWDN Global alignment LN-ITKSAGKGAIMRLGDA TGKG Local alignment AGKG
21 PAIRWISE SEQUENCE ALIGNMENT Dot matrix analysis The dynamic programming (or DP) algorithms Needleman-Wunch (1970) global alignment Smith-Waterman (1981) local alignment Word or k-tuple methods FASTA (Wilbur and Lipman, 1983) BLAST 21
22 DOT PLOT T H I S I S N O T R N A S E Q U E N C E W T H T H I S I S A D N A S E Q U E N C E Red rectangles are true matching of identical residue-pairs and green rectangles represent noise 22
23 DOT PLOT First described by Gibbs and McIntyre (1970) Sequence A is listed from left to right Sequence B is listed from up to down Starting from the first character of B, one moves accross all the characters in A and places a dot whenever the character in A is the same as the character in B The process is continued until all characters from both sequences are compared against each other Similar regions are revealed by diagonal rows of dots Isolated dots that are not on the diagonal represent random noise 23
24 DOT PLOT library(seqinr) seq1 = unlist( strsplit( "THISISADNASEQUENCE", split = "" ) ) seq2 = unlist( strsplit( "THISISNOTRNASEQUENCEWTH", split = "" ) ) dotplot( seq1, seq2, wsize=1, wstep=1, col=c("white","black")) There is a lot of background noise, so we need to add a filter 24
25 DOT PLOT library(seqinr) seq1 = unlist( strsplit( "THISISADNASEQUENCE", split = "" ) ) seq2 = unlist( strsplit( "THISISNOTRNASEQUENCEWTH", split = "" ) ) dotplot( seq1, seq2, wsize=4, wstep=1, nmatch=3, col=c("white","black")) We are using a sliding window filter and specifying the number of required matches per window (identity) 25
26 SCORING ALIGNMENTS Alignment of related sequences should give good scores compared with non-related alignments Genuine matches do not have to be identical The more similar the physiochemical properties of two residues, the greater the chance that the substitution is harmeless to protein function Such substitution should be penalized less then the one where physiochemical properties are more different 26
27 SCORING ALIGNMENTS Identity is the extend to which two sequences are invariant to each other. Percent Identity is obtained by taking the percentage of identical matches from the total length of sequence alignment. Taking into consideration the similarity between the amino acids, one can replace percent identity with percent similarity. 27
28 SUBSTITUTION MATRICES Describes the rate at which one character in a sequence changes to other character states over time. Provide scores for matches based on their occurences in aligned protein families. A dot is placed in the matrix only if a minimum similarity score is found. Can be used with the sliding window option that averages the score within the window and prints a dot only above a certain average score. 28
29 SUBSTITUTION MATRICES PAM Point Accepted Mutation (Margaret Dayhoff) Depicts the likelyhood of change from one amino acid to another based on observed mutations in 71 families of closely related proteins (85% identity) depicting homologus protein sequences during evolution. PAM250 corresponds to 250 amino acid replacements per 100 residues 29
 depicting homologus protein sequences during")

30 SUBSTITUTION MATRICES PAM Point Accepted Mutation (Margaret Dayhoff) Depicts the likelyhood of change from one amino acid to another based on observed mutations in 71 families of closely related proteins (85% identity) depicting homologus protein sequences during evolution. PAM1 gives the probability that a given amino acid will be replaced by any other particular amino acid after a given evolutionary interval, in this case 1 accepted point mutation per 100 amino acids PAM250 corresponds to 250 amino acid replacments per 100 residues 30
31 SUBSTITUTION MATRICES BLOSUM Blocks Amino Acid Substitution Matrix (Henikoff and Henikoff) Based on the observed amino acid substitutions in a large set ( 2000) of conserved amino acid patterns, called blocks. More then 500 protein families used to create the matrix. BLOSUM62 means that sequences clustered in the block were at least 62% identical. Allows for detection of more distantly related sequences. 31
32 SUBSTITUTION MATRICES BLOSUM Blocks Amino Acid Substitution Matrix (Henikoff and Henikoff) 32
33 WHICH MATRIX TO USE? For global alignment use PAM matrices Lower PAM matrices tend to find short alignments of highly similar regions. Higher PAM matrices will find weaker, longer alignments. For local alignments use BLOSUM matrices Higher number BLOSUM matrices are better for similar sequences. Low number BLOSUM matrices are better for distant sequences. 33
34 DYNAMIC PROGRAMMING Breaks down the alignment of sequences into small parts, considering all possible changes when moving from one pair of characters to the next. Finds the best or optimal alignments given an additive alignment score. May produce more then one optimal alignment. Global alignment uses Needleman-Wunch algorithm. Local alignment uses Smith-Waterman algorithm. 34
 is -1 http://www.nature.")
35 DYNAMIC PROGRAMMING Score measurement is determined by match award, mismatch penalty and gap penalty. The higher the score the better the alignment. Optimal alignment has the higest possible score given a substitution matrix and a set of gap penalties. match is 1 mismatch is -1 insertion deletion (gap) is
36 GAP PENALTY Used to score insertions and deletions. Optimal alignment maximizes the number of matches and minimizes the number of gaps. Adding gaps reduces mismatches. Affine gap penalties give a big penalty for each new gap, but a much smaller penalty for gap elongation. gap penalty: 10 gap elongation: 2 ACTCTTACGGGCATATTGCTAGCATTGGCTAGCCTCA ACTCTT-----CATATT-CTAGCA---GCTAGCCTCA penalties
37 DYNAMIC PROGRAMMING BASIC PRINCIPLES Creation of an alignment path matrix Stepwise calculation of score values Backtracking (evaluation of the optimal path) 37
38 NEEDLEMAN-WUNSCH 1. INITIALIZATION assign values for the first row and column the score of each cell is set to gap score multiplied by the distance from the origin match is 1 mismatch is -1 insertion deletion (gap) is -1

39 NEEDLEMAN-WUNSCH 2. FILL the entire matrix is filled with scores and pointers compute match score, vertical gap score and horizontal gap score assign the maximal value to the cell 1 2 D(i-1,j-1) D(i-1,j) 2 3 gap gap 1 score 3 D(i,j-1) D(i,j)

40 NEEDLEMAN-WUNSCH 2. FILL the entire matrix is filled with scores and pointers compute match score, vertical gap score and horizontal gap score assign the maximal value to the cell

41 NEEDLEMAN-WUNSCH 3. TRACEBACK traceback recovers the alignment from the matrix start at the bottom right and move in the direction of arrows until you arrive at the top left corner BIOINFORMATICS BIO-----MATICS
42 SMITH-WATERMAN Similar to Needleman-Wunsch where negative scoring matrix cells are set to 0. Backtracking starts with the highest scoring cell and continues until a cell with score zero or D(0,0) is encountered, yielding the highest scoring local alignment. 42
43 SMITH-WATERMAN Begin with maximal scoring element Follow pointers that gave max score for each element Continue until you reach an element with zero score Construct alignment from traceback path START HERE BIOINFORMATICS Local alignment show in red BIOMATICS 43

44 EXTENDED SMITH-WATERMAN Delete regions around best path Repeat backtracking START HERE BIOINFORMATICS Local alignment show BIOMATICS in red 44
45 K-TUPLE METHODS FASTA & BLAST Instead of comparing individual characters, k-tuple methods compare sequence patterns of words, called k-tuples. These patterns comprise of k consecutive matches in both sequences. Such methods are much faster than dynamic programming methods, but are also less sensitive. 45
46 K-TUPLE METHODS FASTA & BLAST For query BIOINFORMATICS, for k = 8, the set of k- tuples for query is: BIOINFOR IOINFORM OINFORMA INFORMAT NFORMATI FORMATIC ORMATICS How many k-tuples are there in a string of length n? 46
47 K-TUPLE METHODS FASTA & BLAST For query BIOINFORMATICS, for k = 8, the set of k- tuples for query is: BIOINFOR IOINFORM OINFORMA INFORMAT NFORMATI FORMATIC ORMATICS How many k-tuples are there in a string of length n? The answer is: n k
48 K-TUPLE METHODS FASTA & BLAST For query BIOINFORMATICS, for k = 8, the set of k- tuples for query is: BIOINFOR IOINFORM OINFORMA INFORMAT NFORMATI FORMATIC ORMATICS If not one query k-tuple is found from the target sequence, we can deduce that these two sequences are different from each other How many k-tuples are there in a string of length n? The answer is: n k
49 FASTA Program for rapid alignment of pairs of protein and DNA sequences. Uses local sequence alignment to find matches of similar database sequences. Main idea: Choose regions of the two sequences that look promising (have some degree of similarity). Compute local alignment using dynamic programming in these regions. 49
50 FASTA 1. Identify common k-words between two sequences. 2. Score diagonals with k-word matches, identify 10 best diagonals. 3. Rescore initial regions with a substitution matrix. 4. Join intial regions using gaps, penalize gaps. 5. Perform dynamic programming to find inital alignments. 50
51 FASTA Identify all exact matches of length k or greater between the two sequences. I: GCATCGGC J: CCATCGCCATCG Look up table k-word I Location k-word J Location AT 3 AT 3,9 CA 2 CA 2,8 CC 1,7 CG 5 CG 5,11 GC 1,7 GC 6 GG 6 TC 4 TC 4,10 51
52 FASTA k-tup matches can be depicted in a matrix; diagonals indicate matches that have the highest density of common words. Top ten matches are selected (initial regions). 52
53 FASTA Rescore top 10 diagonals using a substitution matrices. 53
54 FASTA Check if the initial regions can be joined to form an approximate alignment with gaps. Calculate the similarity score, penalize with gaps. 54
55 FASTA Uses Smith-Waterman algorithm to find an optimal score for alignment. 55
56 BLAST BASIC LOCAL ALIGNMENT SEARCH TOOL Retrieves homologus sequences from the database. Used to find best local alignment to a query sequences against the sequences in the database (both DNA and protein). Heuristic approach based on Smith-Waterman algorithm. Calculates the statistical significance of matches. 56
57 BLAST 57
58 BLAST 1. Compile a list of high-scoring words 2. Scan the database for instances of these words, called hits 3. Extend hits to differentiate random hits from meaningful hits 58
59 BLAST Create neighborhood words for each query word Compile a list of high scoring words of length w. 59
60 BLAST Create neighborhood words for each query word Compile a list of high scoring words of length w. 60
61 BLAST Compare the word list to a database and identify exact matches 61
62 BLAST Extend hits by summing residue pairs from both sides of the word boundary. Extension stops when score drops below a threshold of the best score yet observed. All extended hits above the minimum score are reported. 62
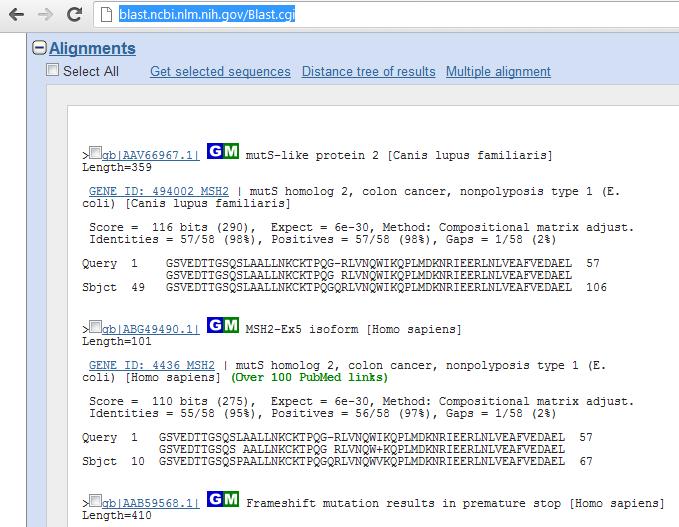
63 BLAST 63
64 WHY OR WHEN TO COMPARE TWO SEQUENCES Are they homologous / share common ancestor Do they share similar domains Identify exact locations to see common feature-active sites Compare a gene and its product to other genes and products 64
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Biochemistry 324 Bioinformatics. Pairwise sequence alignment
 Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Biochemistry 324 Bioinformatics Pairwise sequence alignment How do we compare genes/proteins? When we have sequenced a genome, we try and identify the function of unknown genes by finding a similar gene
Algorithms in Bioinformatics
 Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Algorithms in Bioinformatics Sami Khuri Department of omputer Science San José State University San José, alifornia, USA khuri@cs.sjsu.edu www.cs.sjsu.edu/faculty/khuri Pairwise Sequence Alignment Homology
Sara C. Madeira. Universidade da Beira Interior. (Thanks to Ana Teresa Freitas, IST for useful resources on this subject)
") Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Bioinformática Sequence Alignment Pairwise Sequence Alignment Universidade da Beira Interior (Thanks to Ana Teresa Freitas, IST for useful resources on this subject) 1 16/3/29 & 23/3/29 27/4/29 Outline
Sequence analysis and Genomics
 Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
Sequence analysis and Genomics October 12 th November 23 rd 2 PM 5 PM Prof. Peter Stadler Dr. Katja Nowick Katja: group leader TFome and Transcriptome Evolution Bioinformatics group Paul-Flechsig-Institute
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment
 Pairwise sequence alignment") Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Bioinformatics (GLOBEX, Summer 2015) Pairwise sequence alignment Substitution score matrices, PAM, BLOSUM Needleman-Wunsch algorithm (Global) Smith-Waterman algorithm (Local) BLAST (local, heuristic) E-value
Single alignment: Substitution Matrix. 16 march 2017
 Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
Single alignment: Substitution Matrix 16 march 2017 BLOSUM Matrix BLOSUM Matrix [2] (Blocks Amino Acid Substitution Matrices ) It is based on the amino acids substitutions observed in ~2000 conserved block
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I)
 Sequence pairwise alignment (I)") CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
CISC 889 Bioinformatics (Spring 2004) Sequence pairwise alignment (I) Contents Alignment algorithms Needleman-Wunsch (global alignment) Smith-Waterman (local alignment) Heuristic algorithms FASTA BLAST
CONCEPT OF SEQUENCE COMPARISON. Natapol Pornputtapong 18 January 2018
 CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
CONCEPT OF SEQUENCE COMPARISON Natapol Pornputtapong 18 January 2018 SEQUENCE ANALYSIS - A ROSETTA STONE OF LIFE Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT
 5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
5. MULTIPLE SEQUENCE ALIGNMENT BIOINFORMATICS COURSE MTAT.03.239 03.10.2012 ALIGNMENT Alignment is the task of locating equivalent regions of two or more sequences to maximize their similarity. Homology:
Pairwise & Multiple sequence alignments
 Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Pairwise & Multiple sequence alignments Urmila Kulkarni-Kale Bioinformatics Centre 411 007 urmila@bioinfo.ernet.in Basis for Sequence comparison Theory of evolution: gene sequences have evolved/derived
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment
 Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Module: Sequence Alignment Theory and Applications Session: Introduction to Searching and Sequence Alignment Introduction to Bioinformatics online course : IBT Jonathan Kayondo Learning Objectives Understand
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
In-Depth Assessment of Local Sequence Alignment
 2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
Practical considerations of working with sequencing data
 Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Practical considerations of working with sequencing data File Types Fastq ->aligner -> reference(genome) coordinates Coordinate files SAM/BAM most complete, contains all of the info in fastq and more!
Basic Local Alignment Search Tool
 Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Basic Local Alignment Search Tool Alignments used to uncover homologies between sequences combined with phylogenetic studies o can determine orthologous and paralogous relationships Local Alignment uses
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Bioinformatics and BLAST
 Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Computational Biology
 Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Computational Biology Lecture 6 31 October 2004 1 Overview Scoring matrices (Thanks to Shannon McWeeney) BLAST algorithm Start sequence alignment 2 1 What is a homologous sequence? A homologous sequence,
Collected Works of Charles Dickens
 Collected Works of Charles Dickens A Random Dickens Quote If there were no bad people, there would be no good lawyers. Original Sentence It was a dark and stormy night; the night was dark except at sunny
Collected Works of Charles Dickens A Random Dickens Quote If there were no bad people, there would be no good lawyers. Original Sentence It was a dark and stormy night; the night was dark except at sunny
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Tools and Algorithms in Bioinformatics GCBA815, Fall 2013 Week3: Blast Algorithm, theory and practice Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and Systems Biology
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Pairwise Sequence Alignment
 Introduction to Bioinformatics Pairwise Sequence Alignment Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Outline Introduction to sequence alignment pair wise sequence alignment The Dot Matrix Scoring
Introduction to Bioinformatics Pairwise Sequence Alignment Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Outline Introduction to sequence alignment pair wise sequence alignment The Dot Matrix Scoring
Sequence Alignment (chapter 6)
") Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Sequence lignment (chapter 6) he biological problem lobal alignment Local alignment Multiple alignment Introduction to bioinformatics, utumn 6 Background: comparative genomics Basic question in biology:
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Quantifying sequence similarity
 Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Quantifying sequence similarity Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, February 16 th 2016 After this lecture, you can define homology, similarity, and identity
Pairwise sequence alignments
 Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Pairwise sequence alignments Volker Flegel VI, October 2003 Page 1 Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs VI, October
Introduction to Bioinformatics
 Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Introduction to Bioinformatics Jianlin Cheng, PhD Department of Computer Science Informatics Institute 2011 Topics Introduction Biological Sequence Alignment and Database Search Analysis of gene expression
Sequence Comparison. mouse human
 Sequence Comparison Sequence Comparison mouse human Why Compare Sequences? The first fact of biological sequence analysis In biomolecular sequences (DNA, RNA, or amino acid sequences), high sequence similarity
Sequence Comparison Sequence Comparison mouse human Why Compare Sequences? The first fact of biological sequence analysis In biomolecular sequences (DNA, RNA, or amino acid sequences), high sequence similarity
Bioinformatics for Biologists
 Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Bioinformatics for Biologists Sequence Analysis: Part I. Pairwise alignment and database searching Fran Lewitter, Ph.D. Head, Biocomputing Whitehead Institute Bioinformatics Definitions The use of computational
Pairwise Alignment. Guan-Shieng Huang. Dept. of CSIE, NCNU. Pairwise Alignment p.1/55
 Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
Pairwise Alignment Guan-Shieng Huang shieng@ncnu.edu.tw Dept. of CSIE, NCNU Pairwise Alignment p.1/55 Approach 1. Problem definition 2. Computational method (algorithms) 3. Complexity and performance Pairwise
Pairwise sequence alignment
 Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Department of Evolutionary Biology Example Alignment between very similar human alpha- and beta globins: GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKL G+ +VK+HGKKV A+++++AH+D++ +++++LS+LH KL GNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKL
Pairwise sequence alignments. Vassilios Ioannidis (From Volker Flegel )
") Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
Pairwise sequence alignments Vassilios Ioannidis (From Volker Flegel ) Outline Introduction Definitions Biological context of pairwise alignments Computing of pairwise alignments Some programs Importance
B I O I N F O R M A T I C S
 B I O I N F O R M A T I C S Kristel Van Steen, PhD 2 Montefiore Institute - Systems and Modeling GIGA - Bioinformatics ULg kristel.vansteen@ulg.ac.be K Van Steen CH4 : 1 CHAPTER 4: SEQUENCE COMPARISON
B I O I N F O R M A T I C S Kristel Van Steen, PhD 2 Montefiore Institute - Systems and Modeling GIGA - Bioinformatics ULg kristel.vansteen@ulg.ac.be K Van Steen CH4 : 1 CHAPTER 4: SEQUENCE COMPARISON
Sequence Alignments. Dynamic programming approaches, scoring, and significance. Lucy Skrabanek ICB, WMC January 31, 2013
 Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Sequence Alignments Dynamic programming approaches, scoring, and significance Lucy Skrabanek ICB, WMC January 31, 213 Sequence alignment Compare two (or more) sequences to: Find regions of conservation
Alignment principles and homology searching using (PSI-)BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)
BLAST. Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU)") Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
Alignment principles and homology searching using (PSI-)BLAST Jaap Heringa Centre for Integrative Bioinformatics VU (IBIVU) http://ibivu.cs.vu.nl Bioinformatics Nothing in Biology makes sense except in
20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, Global and local alignment of two sequences using dynamic programming
 20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, 2008 4 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance 4. Global and local alignment
20 Grundlagen der Bioinformatik, SS 08, D. Huson, May 27, 2008 4 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance 4. Global and local alignment
Sequence Database Search Techniques I: Blast and PatternHunter tools
 Sequence Database Search Techniques I: Blast and PatternHunter tools Zhang Louxin National University of Singapore Outline. Database search 2. BLAST (and filtration technique) 3. PatternHunter (empowered
Sequence Database Search Techniques I: Blast and PatternHunter tools Zhang Louxin National University of Singapore Outline. Database search 2. BLAST (and filtration technique) 3. PatternHunter (empowered
Scoring Matrices. Shifra Ben-Dor Irit Orr
 Scoring Matrices Shifra Ben-Dor Irit Orr Scoring matrices Sequence alignment and database searching programs compare sequences to each other as a series of characters. All algorithms (programs) for comparison
Scoring Matrices Shifra Ben-Dor Irit Orr Scoring matrices Sequence alignment and database searching programs compare sequences to each other as a series of characters. All algorithms (programs) for comparison
Alignment & BLAST. By: Hadi Mozafari KUMS
 Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
Alignment & BLAST By: Hadi Mozafari KUMS SIMILARITY - ALIGNMENT Comparison of primary DNA or protein sequences to other primary or secondary sequences Expecting that the function of the similar sequence
Similarity or Identity? When are molecules similar?
 Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Similarity or Identity? When are molecules similar? Mapping Identity A -> A T -> T G -> G C -> C or Leu -> Leu Pro -> Pro Arg -> Arg Phe -> Phe etc If we map similarity using identity, how similar are
Introduction to Computation & Pairwise Alignment
 Introduction to Computation & Pairwise Alignment Eunok Paek eunokpaek@hanyang.ac.kr Algorithm what you already know about programming Pan-Fried Fish with Spicy Dipping Sauce This spicy fish dish is quick
Introduction to Computation & Pairwise Alignment Eunok Paek eunokpaek@hanyang.ac.kr Algorithm what you already know about programming Pan-Fried Fish with Spicy Dipping Sauce This spicy fish dish is quick
An Introduction to Sequence Similarity ( Homology ) Searching
 Searching") An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
An Introduction to Sequence Similarity ( Homology ) Searching Gary D. Stormo 1 UNIT 3.1 1 Washington University, School of Medicine, St. Louis, Missouri ABSTRACT Homologous sequences usually have the same,
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Sepp Hochreiter
 Sepp Hochreiter") Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Bioinformatics for Computer Scientists (Part 2 Sequence Alignment) Institute of Bioinformatics Johannes Kepler University, Linz, Austria Sequence Alignment 2. Sequence Alignment Sequence Alignment 2.1
Motivating the need for optimal sequence alignments...
 1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
1 Motivating the need for optimal sequence alignments... 2 3 Note that this actually combines two objectives of optimal sequence alignments: (i) use the score of the alignment o infer homology; (ii) use
Administration. ndrew Torda April /04/2008 [ 1 ]
![Administration. ndrew Torda April /04/2008 [ 1 ]](/thumbs/73/68891262.jpg "Administration. ndrew Torda April /04/2008 [ 1 ]") ndrew Torda April 2008 Administration 22/04/2008 [ 1 ] Sprache? zu verhandeln (Englisch, Hochdeutsch, Bayerisch) Selection of topics Proteins / DNA / RNA Two halves to course week 1-7 Prof Torda (larger
ndrew Torda April 2008 Administration 22/04/2008 [ 1 ] Sprache? zu verhandeln (Englisch, Hochdeutsch, Bayerisch) Selection of topics Proteins / DNA / RNA Two halves to course week 1-7 Prof Torda (larger
Sequence Alignment: Scoring Schemes. COMP 571 Luay Nakhleh, Rice University
 Sequence Alignment: Scoring Schemes COMP 571 Luay Nakhleh, Rice University Scoring Schemes Recall that an alignment score is aimed at providing a scale to measure the degree of similarity (or difference)
Sequence Alignment: Scoring Schemes COMP 571 Luay Nakhleh, Rice University Scoring Schemes Recall that an alignment score is aimed at providing a scale to measure the degree of similarity (or difference)
BLAST. Varieties of BLAST
 BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
BLAST Basic Local Alignment Search Tool (1990) Altschul, Gish, Miller, Myers, & Lipman Uses short-cuts or heuristics to improve search speed Like speed-reading, does not examine every nucleotide of database
Introduction to sequence alignment. Local alignment the Smith-Waterman algorithm
 Lecture 2, 12/3/2003: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties Local alignment the Smith-Waterman algorithm 1 Computational
Lecture 2, 12/3/2003: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties Local alignment the Smith-Waterman algorithm 1 Computational
Background: comparative genomics. Sequence similarity. Homologs. Similarity vs homology (2) Similarity vs homology. Sequence Alignment (chapter 6)
 Similarity vs homology. Sequence Alignment (chapter 6)") Sequence lignment (chapter ) he biological problem lobal alignment Local alignment Multiple alignment Background: comparative genomics Basic question in biology: what properties are shared among organisms?
Sequence lignment (chapter ) he biological problem lobal alignment Local alignment Multiple alignment Background: comparative genomics Basic question in biology: what properties are shared among organisms?
Chapter 7: Rapid alignment methods: FASTA and BLAST
 Chapter 7: Rapid alignment methods: FASTA and BLAST The biological problem Search strategies FASTA BLAST Introduction to bioinformatics, Autumn 2007 117 BLAST: Basic Local Alignment Search Tool BLAST (Altschul
Chapter 7: Rapid alignment methods: FASTA and BLAST The biological problem Search strategies FASTA BLAST Introduction to bioinformatics, Autumn 2007 117 BLAST: Basic Local Alignment Search Tool BLAST (Altschul
Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM)
") Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
Bioinformatics II Probability and Statistics Universität Zürich and ETH Zürich Spring Semester 2009 Lecture 4: Evolutionary Models and Substitution Matrices (PAM and BLOSUM) Dr Fraser Daly adapted from
BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University
 BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University Measures of Sequence Similarity Alignment with dot
BIO 285/CSCI 285/MATH 285 Bioinformatics Programming Lecture 8 Pairwise Sequence Alignment 2 And Python Function Instructor: Lei Qian Fisk University Measures of Sequence Similarity Alignment with dot
Local Alignment Statistics
 Local Alignment Statistics Stephen Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Bethesda, MD Central Issues in Biological Sequence Comparison
Local Alignment Statistics Stephen Altschul National Center for Biotechnology Information National Library of Medicine National Institutes of Health Bethesda, MD Central Issues in Biological Sequence Comparison
Sequence comparison: Score matrices
 Sequence comparison: Score matrices http://facultywashingtonedu/jht/gs559_2013/ Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best
Sequence comparison: Score matrices http://facultywashingtonedu/jht/gs559_2013/ Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best
Bioinformatics. Part 8. Sequence Analysis An introduction. Mahdi Vasighi
 Bioinformatics Sequence Analysis An introduction Part 8 Mahdi Vasighi Sequence analysis Some of the earliest problems in genomics concerned how to measure similarity of DNA and protein sequences, either
Bioinformatics Sequence Analysis An introduction Part 8 Mahdi Vasighi Sequence analysis Some of the earliest problems in genomics concerned how to measure similarity of DNA and protein sequences, either
BIOINFORMATICS: An Introduction
 BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
BIOINFORMATICS: An Introduction What is Bioinformatics? The term was first coined in 1988 by Dr. Hwa Lim The original definition was : a collective term for data compilation, organisation, analysis and
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences
 Analysis of Biological Sequences") First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
First generation sequencing and pairwise alignment (High-tech, not high throughput) Analysis of Biological Sequences 140.638 where do sequences come from? DNA is not hard to extract (getting DNA from a
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best alignment path onsider the last step in
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas FYI - informal inductive proof of best alignment path onsider the last step in
Sequence Analysis 17: lecture 5. Substitution matrices Multiple sequence alignment
 Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Sequence Analysis 17: lecture 5 Substitution matrices Multiple sequence alignment Substitution matrices Used to score aligned positions, usually of amino acids. Expressed as the log-likelihood ratio of
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Lecture 1, 31/10/2001: Introduction to sequence alignment. The Needleman-Wunsch algorithm for global sequence alignment: description and properties
 Lecture 1, 31/10/2001: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties 1 Computational sequence-analysis The major goal of computational
Lecture 1, 31/10/2001: Introduction to sequence alignment The Needleman-Wunsch algorithm for global sequence alignment: description and properties 1 Computational sequence-analysis The major goal of computational
Bioinformatics. Scoring Matrices. David Gilbert Bioinformatics Research Centre
 Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Bioinformatics Scoring Matrices David Gilbert Bioinformatics Research Centre www.brc.dcs.gla.ac.uk Department of Computing Science, University of Glasgow Learning Objectives To explain the requirement
Chapter 5. Proteomics and the analysis of protein sequence Ⅱ
 Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
Proteomics Chapter 5. Proteomics and the analysis of protein sequence Ⅱ 1 Pairwise similarity searching (1) Figure 5.5: manual alignment One of the amino acids in the top sequence has no equivalent and
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
C E N T R. Introduction to bioinformatics 2007 E B I O I N F O R M A T I C S V U F O R I N T. Lecture 5 G R A T I V. Pair-wise Sequence Alignment
 C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 5 Pair-wise Sequence Alignment Bioinformatics Nothing in Biology makes sense except in
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Introduction to bioinformatics 2007 Lecture 5 Pair-wise Sequence Alignment Bioinformatics Nothing in Biology makes sense except in
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm. Alignment scoring schemes and theory: substitution matrices and gap models
 Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Lecture 2, 5/12/2001: Local alignment the Smith-Waterman algorithm Alignment scoring schemes and theory: substitution matrices and gap models 1 Local sequence alignments Local sequence alignments are necessary
Lecture 5,6 Local sequence alignment
 Lecture 5,6 Local sequence alignment Chapter 6 in Jones and Pevzner Fall 2018 September 4,6, 2018 Evolution as a tool for biological insight Nothing in biology makes sense except in the light of evolution
Lecture 5,6 Local sequence alignment Chapter 6 in Jones and Pevzner Fall 2018 September 4,6, 2018 Evolution as a tool for biological insight Nothing in biology makes sense except in the light of evolution
... and searches for related sequences probably make up the vast bulk of bioinformatics activities.
 1 2 ... and searches for related sequences probably make up the vast bulk of bioinformatics activities. 3 The terms homology and similarity are often confused and used incorrectly. Homology is a quality.
1 2 ... and searches for related sequences probably make up the vast bulk of bioinformatics activities. 3 The terms homology and similarity are often confused and used incorrectly. Homology is a quality.
Grundlagen der Bioinformatik, SS 08, D. Huson, May 2,
 Grundlagen der Bioinformatik, SS 08, D. Huson, May 2, 2008 39 5 Blast This lecture is based on the following, which are all recommended reading: R. Merkl, S. Waack: Bioinformatik Interaktiv. Chapter 11.4-11.7
Grundlagen der Bioinformatik, SS 08, D. Huson, May 2, 2008 39 5 Blast This lecture is based on the following, which are all recommended reading: R. Merkl, S. Waack: Bioinformatik Interaktiv. Chapter 11.4-11.7
Comparative genomics: Overview & Tools + MUMmer algorithm
 Comparative genomics: Overview & Tools + MUMmer algorithm Urmila Kulkarni-Kale Bioinformatics Centre University of Pune, Pune 411 007. urmila@bioinfo.ernet.in Genome sequence: Fact file 1995: The first
Comparative genomics: Overview & Tools + MUMmer algorithm Urmila Kulkarni-Kale Bioinformatics Centre University of Pune, Pune 411 007. urmila@bioinfo.ernet.in Genome sequence: Fact file 1995: The first
Bioinformatics Exercises
 Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
Bioinformatics Exercises AP Biology Teachers Workshop Susan Cates, Ph.D. Evolution of Species Phylogenetic Trees show the relatedness of organisms Common Ancestor (Root of the tree) 1 Rooted vs. Unrooted
8 Grundlagen der Bioinformatik, SoSe 11, D. Huson, April 18, 2011
 8 Grundlagen der Bioinformatik, SoSe 11, D. Huson, April 18, 2011 2 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance and alignment 4. The number
8 Grundlagen der Bioinformatik, SoSe 11, D. Huson, April 18, 2011 2 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance and alignment 4. The number
Study and Implementation of Various Techniques Involved in DNA and Protein Sequence Analysis
 Study and Implementation of Various Techniques Involved in DNA and Protein Sequence Analysis Kumud Joseph Kujur, Sumit Pal Singh, O.P. Vyas, Ruchir Bhatia, Varun Singh* Indian Institute of Information
Study and Implementation of Various Techniques Involved in DNA and Protein Sequence Analysis Kumud Joseph Kujur, Sumit Pal Singh, O.P. Vyas, Ruchir Bhatia, Varun Singh* Indian Institute of Information
BLAST: Basic Local Alignment Search Tool
 .. CSC 448 Bioinformatics Algorithms Alexander Dekhtyar.. (Rapid) Local Sequence Alignment BLAST BLAST: Basic Local Alignment Search Tool BLAST is a family of rapid approximate local alignment algorithms[2].
.. CSC 448 Bioinformatics Algorithms Alexander Dekhtyar.. (Rapid) Local Sequence Alignment BLAST BLAST: Basic Local Alignment Search Tool BLAST is a family of rapid approximate local alignment algorithms[2].
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
BLAST Database Searching. BME 110: CompBio Tools Todd Lowe April 8, 2010
 BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
BLAST Database Searching BME 110: CompBio Tools Todd Lowe April 8, 2010 Admin Reading: Read chapter 7, and the NCBI Blast Guide and tutorial http://www.ncbi.nlm.nih.gov/blast/why.shtml Read Chapter 8 for
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Tiffany Samaroo MB&B 452a December 8, Take Home Final. Topic 1
 Tiffany Samaroo MB&B 452a December 8, 2003 Take Home Final Topic 1 Prior to 1970, protein and DNA sequence alignment was limited to visual comparison. This was a very tedious process; even proteins with
Tiffany Samaroo MB&B 452a December 8, 2003 Take Home Final Topic 1 Prior to 1970, protein and DNA sequence alignment was limited to visual comparison. This was a very tedious process; even proteins with
1.5 Sequence alignment
 1.5 Sequence alignment The dramatic increase in the number of sequenced genomes and proteomes has lead to development of various bioinformatic methods and algorithms for extracting information (data mining)
1.5 Sequence alignment The dramatic increase in the number of sequenced genomes and proteomes has lead to development of various bioinformatic methods and algorithms for extracting information (data mining)
Sequence comparison: Score matrices. Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas
 Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
Sequence comparison: Score matrices Genome 559: Introduction to Statistical and omputational Genomics Prof James H Thomas Informal inductive proof of best alignment path onsider the last step in the best
8 Grundlagen der Bioinformatik, SS 09, D. Huson, April 28, 2009
 8 Grundlagen der Bioinformatik, SS 09, D. Huson, April 28, 2009 2 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance and alignment 4. The number
8 Grundlagen der Bioinformatik, SS 09, D. Huson, April 28, 2009 2 Pairwise alignment We will discuss: 1. Strings 2. Dot matrix method for comparing sequences 3. Edit distance and alignment 4. The number
Introduction to Bioinformatics
 Introduction to Bioinformatics Lecture : p he biological problem p lobal alignment p Local alignment p Multiple alignment 6 Background: comparative genomics p Basic question in biology: what properties
Introduction to Bioinformatics Lecture : p he biological problem p lobal alignment p Local alignment p Multiple alignment 6 Background: comparative genomics p Basic question in biology: what properties
Global alignments - review
 Global alignments - review Take two sequences: X[j] and Y[j] M[i-1, j-1] ± 1 M[i, j] = max M[i, j-1] 2 M[i-1, j] 2 The best alignment for X[1 i] and Y[1 j] is called M[i, j] X[j] Initiation: M[,]= pply
Global alignments - review Take two sequences: X[j] and Y[j] M[i-1, j-1] ± 1 M[i, j] = max M[i, j-1] 2 M[i-1, j] 2 The best alignment for X[1 i] and Y[1 j] is called M[i, j] X[j] Initiation: M[,]= pply
Comparing whole genomes
 BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
Lecture 2: Pairwise Alignment. CG Ron Shamir
 Lecture 2: Pairwise Alignment 1 Main source 2 Why compare sequences? Human hexosaminidase A vs Mouse hexosaminidase A 3 www.mathworks.com/.../jan04/bio_genome.html Sequence Alignment עימוד רצפים The problem:
Lecture 2: Pairwise Alignment 1 Main source 2 Why compare sequences? Human hexosaminidase A vs Mouse hexosaminidase A 3 www.mathworks.com/.../jan04/bio_genome.html Sequence Alignment עימוד רצפים The problem:
Introduction to Sequence Alignment. Manpreet S. Katari
 Introduction to Sequence Alignment Manpreet S. Katari 1 Outline 1. Global vs. local approaches to aligning sequences 1. Dot Plots 2. BLAST 1. Dynamic Programming 3. Hash Tables 1. BLAT 4. BWT (Burrow Wheeler
Introduction to Sequence Alignment Manpreet S. Katari 1 Outline 1. Global vs. local approaches to aligning sequences 1. Dot Plots 2. BLAST 1. Dynamic Programming 3. Hash Tables 1. BLAT 4. BWT (Burrow Wheeler
Copyright 2000 N. AYDIN. All rights reserved. 1
 Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Introduction to Bioinformatics Prof. Dr. Nizamettin AYDIN naydin@yildiz.edu.tr Multiple Sequence Alignment Outline Multiple sequence alignment introduction to msa methods of msa progressive global alignment
Introduction to protein alignments
 Introduction to protein alignments Comparative Analysis of Proteins Experimental evidence from one or more proteins can be used to infer function of related protein(s). Gene A Gene X Protein A compare
Introduction to protein alignments Comparative Analysis of Proteins Experimental evidence from one or more proteins can be used to infer function of related protein(s). Gene A Gene X Protein A compare
Substitution matrices
 Introduction to Bioinformatics Substitution matrices Jacques van Helden Jacques.van-Helden@univ-amu.fr Université d Aix-Marseille, France Lab. Technological Advances for Genomics and Clinics (TAGC, INSERM
Introduction to Bioinformatics Substitution matrices Jacques van Helden Jacques.van-Helden@univ-amu.fr Université d Aix-Marseille, France Lab. Technological Advances for Genomics and Clinics (TAGC, INSERM
MATHEMATICAL MODELS - Vol. III - Mathematical Modeling and the Human Genome - Hilary S. Booth MATHEMATICAL MODELING AND THE HUMAN GENOME
 MATHEMATICAL MODELING AND THE HUMAN GENOME Hilary S. Booth Australian National University, Australia Keywords: Human genome, DNA, bioinformatics, sequence analysis, evolution. Contents 1. Introduction:
MATHEMATICAL MODELING AND THE HUMAN GENOME Hilary S. Booth Australian National University, Australia Keywords: Human genome, DNA, bioinformatics, sequence analysis, evolution. Contents 1. Introduction:
Phylogenetic inference
 Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
Phylogenetic inference Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, March 7 th 016 After this lecture, you can discuss (dis-) advantages of different information types
CSE : Computational Issues in Molecular Biology. Lecture 6. Spring 2004
 CSE 397-497: Computational Issues in Molecular Biology Lecture 6 Spring 2004-1 - Topics for today Based on premise that algorithms we've studied are too slow: Faster method for global comparison when sequences
CSE 397-497: Computational Issues in Molecular Biology Lecture 6 Spring 2004-1 - Topics for today Based on premise that algorithms we've studied are too slow: Faster method for global comparison when sequences
Pairwise alignment. 2.1 Introduction GSAQVKGHGKKVADALTNAVAHVDDMPNALSALSD----LHAHKL
 2 Pairwise alignment 2.1 Introduction The most basic sequence analysis task is to ask if two sequences are related. This is usually done by first aligning the sequences (or parts of them) and then deciding
2 Pairwise alignment 2.1 Introduction The most basic sequence analysis task is to ask if two sequences are related. This is usually done by first aligning the sequences (or parts of them) and then deciding
COPIA: A New Software for Finding Consensus Patterns. Chengzhi Liang. A thesis. presented to the University ofwaterloo. in fulfilment of the
 COPIA: A New Software for Finding Consensus Patterns in Unaligned Protein Sequences by Chengzhi Liang A thesis presented to the University ofwaterloo in fulfilment of the thesis requirement for the degree
COPIA: A New Software for Finding Consensus Patterns in Unaligned Protein Sequences by Chengzhi Liang A thesis presented to the University ofwaterloo in fulfilment of the thesis requirement for the degree
Introduction to Bioinformatics Online Course: IBT
 Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
Introduction to Bioinformatics Online Course: IBT Multiple Sequence Alignment Building Multiple Sequence Alignment Lec1 Building a Multiple Sequence Alignment Learning Outcomes 1- Understanding Why multiple
SEQUENCE ALIGNMENT BACKGROUND: BIOINFORMATICS. Prokaryotes and Eukaryotes. DNA and RNA
 SEQUENCE ALIGNMENT BACKGROUND: BIOINFORMATICS 1 Prokaryotes and Eukaryotes 2 DNA and RNA 3 4 Double helix structure Codons Codons are triplets of bases from the RNA sequence. Each triplet defines an amino-acid.
SEQUENCE ALIGNMENT BACKGROUND: BIOINFORMATICS 1 Prokaryotes and Eukaryotes 2 DNA and RNA 3 4 Double helix structure Codons Codons are triplets of bases from the RNA sequence. Each triplet defines an amino-acid.
Local Alignment: Smith-Waterman algorithm
 Local Alignment: Smith-Waterman algorithm Example: a shared common domain of two protein sequences; extended sections of genomic DNA sequence. Sensitive to detect similarity in highly diverged sequences.
Local Alignment: Smith-Waterman algorithm Example: a shared common domain of two protein sequences; extended sections of genomic DNA sequence. Sensitive to detect similarity in highly diverged sequences.