CS612 - Algorithms in Bioinformatics
|
|
|
- Godfrey Austin
- 5 years ago
- Views:
Transcription
1 Fall 2017 Protein Structure Detection Methods October 30, 2017
2 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing the structure s x assumed by a sequence x of amino acids: Structure is a function of sequence: So, s x = f (x)? The function f encodes how the sequence x determines the structure s x Given another protein of sequence y and known structure s y, we can infer: IF x y THEN s x s y It is important that x and y be similar enough An important question: how similar?
3 Comparative Modeling Some Terminology The protein of unknown structure is the query or the target The protein of known structure whose sequence is similar to that of the target is the template The process of inferring the coordinates for the target is called model building Comparative modeling builds the model, completes it, refines it, and then evaluates it
4 Why Use Comparative Modeling? Structures of proteins in a given functional family are more conserved than their sequences About a third of all sequences assume known structures The number of unique protein folds is limited If not applicable to yield a high-resolution structure, comparative modeling can at least yield the fold for a sequence Currently, comparative modeling is both faster and more accurate (as long as the sequence identity is high) than ab initio or de novo methods for structure prediction

5 When to Use Comparative Modeling? How similar do x and y have to be to infer that the structure assumed by the sequence x is similar to that assumed by the sequence y? Statistical analysis of sequences with known structure reveals: Sequences with no less than 50% sequence identity assume very similar structures Minimum sequence identity for structural similarity: 25-30% Higher than 30% sequence identity often results in very similar structures
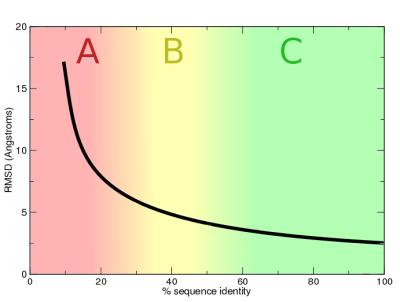
6 Sequence-structure Relationship
7 A Simplistic View of Comparative Modeling Target sequence Select target(s) from PDB Align target sequence with template structure(s) Build model, refine, evaluate Alignment is the most critical step. comparative modeling cannot recover from a bad alignment.
8 Basic Steps of Homology Modeling Crucial step! Raw model Loop modeling Side-chain placement Refinement Figure from M. A. Marti-Renom and A. Sali Modeling Protein Structure from Its Sequence Current Prototocols in Bioinformatics (2003)
9 Basic Steps of Homology Modeling 1 Query a database of protein sequences with known structures with the target sequence, focusing on those with 30% seq. identity to the target sequence 2 Align obtained sequences to target to choose templates 3 Identify structurally conserved (SC) and variable (SV) regions 4 Generate coordinates for the core region of the target 5 Complete the structure of the target generate coordinates for loop regions generate coordinates for side-chains 6 Refine the completed structure using energy minimization 7 Validate/evaluate completed structure

10 Step 1 Query PDB PRTEINSEQENCEPRTEINSEQUENC EPRTEINSEQNCEQWERYTRASDFHG TREWQIYPASDFGHKLMCNASQERWW PRETWQLKHGFDSADAMNCVCNQWER GFDHSDASFWERQWK Query Sequence PDB
11 Step 1 Query PDB PRTEINSEQENCEPRTEINSEQUENC EPRTEINSEQNCEQWERYTRASDFHG TREWQIYPASDFGHKLMCNASQERWW PRETWQLKHGFDSADAMNCVCNQWER GFDHSDASFWERQWK Query Sequence PRTEINSEQENCEPRTEINSEQUENC EPRTEINSEQNCEQWERYTRASDFHG TREWQIYPASDFGHKLMCNASQERWW PRETWQLKHGFDSADAMNCVCNQWER GFDHSDASFWERQWK PRTEINSEQENCEPRTEINSEQUENC EPRTEINSEQQWEWEWQWEWEQWEWE WQRYEYEWQWNCEQWERYTRASDFHG TREWQIYPASDWERWEREWRFDSFG PRTEINSEQENCEPRTEINSEQUENC EPRTEINSEQNCEQWERYTRASDFHG TREWQIYPASDFGHKLMCNASQERWW PRETWQLKHGFDSADAMNCVCNQWER GFDHSDASFWERQWK Hit #1 PRTEINSEQENCEPRTEINSEQUENC EPRTEINSEQQWEWEWQWEWEQWEWE WQRYEYEWQWNCEQWERYTRASDFHG TR PRTEINSEQENCEPRTEINSEQUENC EPRTEINSEQNCEQWERYTRASDFHG TREWQIYPASDFG Hit #2 PRTEINSEQENCEPRTEINSEQUENC EPRTEINSEQNCEQWERYTRASDFHG TREWQIYPASDFGPRTEINSEQENCE PRTEINSEQUENCEPRTEINSEQNCE QWERYTRASDFHGTREWQIYPASDFG TREWQIYPASDFGPRTEINSEQENCE PRTEINSEQUENCEPRTEINSEQNCE QWERYTRASDFHGTREWQ PRTEINSEQENCEPRTEINSEQUENC EPRTEINSEQNCEQWERYTRASDFHG TREWQIYPASDFG PRTEINSEQENCEPRTEINSEQUENC EPRTEINSEQNCEQWERYTRASDFHG TREWQIYPASDFGPRTEINSEQENC PDB
12 Step 2 Alignment G E N E T I C S G E N E S I S G E 40 G E N E T I C S N E S I S Dynamic programming 10
13 Step 2 Alignment Goal: Find a template or templates pairwise sequence alignment finds high homology sequences BLAST Improved multiple sequence alignment methods improves sensitivity remote homologs PSI-BLAST, CLUSTAL
14 Step 2 Alignment Pairwise sequence alignment: BLAST, FASTA, WU-BLAST, SSEARCH, and more Available as web servers and standalone software Basic functionality needed: compare target sequence with sequences in the PDB (or any other comprehensive structural database)? BLAST scans the sequence for 3-letter words (wmers, where w = 3) and expands alignments from 3-mers Statistically significant alignments are hits Templates are hits with no lower than 30% sequence identity
15 Step 2 Alignment Query ACDEFGHIKLMNPQRST--FGHQWERT-----TYREWYEG Hit #1 ASDEYAHLRILDPQRSTVAYAYE--KSFAPPGSFKWEYEA Hit #2 MCDEYAHIRLMNPERSTVAGGHQWERT----GSFKEWYAA Hit #1 Hit #2
16 Step 2 Alignment Global (Needleman-Wunsch) alignment can be used Alignment is the most crucial step, as comparative modeling can never recover from a bad alignment A small error in the alignment can translate to a significant error in the reconstructed model Multiple sequence alignments (that also align the templates to one another) is often better than pairwise alignment
17 Step 2 Alignment A good template is closest to the target in terms of subfamilies This means that high overall sequence similarity is needed The template environment like ph, ligands, etc., should be the same as that of the target The quality of the experimentally-available template structure - the resolution, R-factor, etc. - should be high When choosing a template for a protein-ligand model, it is preferred that the template have the same ligand When modeling an active site a high resolution template structure with ligand is important
18 Step 2 Get Template A good template is closest to the target in terms of subfamilies This means that high overall sequence similarity is needed The template environment like ph, ligands, etc., should be the same as that of the target The quality of the experimentally-available template structure - the resolution, R-factor, etc. - should be high When choosing a template for a protein-ligand model, it is preferred that the template have the same ligand When modeling an active site a high resolution template structure with ligand is important
19 Step 3 Detect Structurally Conserved Regions (SCRs) Query Hit #1 Hit #2 ACDEFGHIKLMNPQRST--FGHQWERT-----TYREWYEG ASDEYAHLRILDPQRSTVAYAYE--KSFAPPGSFKWEYEA MCDEYAHIRLMNPERSTVAGGHQWERT----GSFKEWYAA HHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCBBBBBBBBB SCR #1 SCR #2 Hit #1 Hit #2
20 Step 3 Detect SCRs SCRs correspond to the most stable structures or regions (usually in the interior/core) of the protein SCRs also often correspond to sequence regions with the lowest level of gapping and highest level of sequence conservation SCRs are often the secondary structures
21 Step 3 Detect Structurally Variable Regions (SVRs) Query Hit #1 Hit #2 ACDEFGHIKLMNPQRST--FGHQWERT-----TYREWYEG ASDEYAHLRILDPQRSTVAYAYE--KSFAPPGSFKWEYEA MCDEYAHIRLMNPERSTVAGGHQWERT----GSFKEWYAA HHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCBBBBBBBBB SVR (loop) Hit #1 Hit #2
22 Step 3 Detect SVRs SVRs correspond to the least stable or the most flexible regions (usually in the exterior/surface) of the protein SVRs correspond to sequence regions with the highest level of gapping and lowest level of sequence conservation SVRs are usually loops and turns
23 Step 4 Threading Core conserved regions Protein fold Variable loop regions Side chains Calculate the framework from average of all template structures Multiple templates Generate model for each template and evaluate
24 Step 4 Threading For identical amino acids, just transfer all atom coordinates (x, y, z) to the query protein (both backbone and side-chain atoms are identical)? For similar amino acids, transfer the backbone coordinates and replace side-chain atoms while respecting χ angles For different amino acids, one can only transfer the backbone coordinates (x, y, z) to query sequence The side chains of different amino acids have to be built at a later stage, when completing the model






25 Step 5 Loop Modeling Look up a fragment database Query Hit #1 FGHQWERT YAYE--KS
26 Step 5 Loop Modeling Loops result from substitutions and ins/dels in same family Mini protein folding problem loops can be very long in membrane proteins Ab-initio methods generate various random loop conformations and evaluate/score Compare the loop sequence string to PDB, get hits, and evaluate/score Some comparative modeling methods have fewer loops to be added because of extensive multiple sequence alignment of profiles
27 Step 5 Loop Modeling Ab-initio loop modeling Monte Carlo, Monte Carlo with simulated annealing, MD, main chain dihedral angle search biased with the data from PDB, inverse kinematics-based, etc. Energy functions used: physics-based (CHARMM, AMBER, etc.) or knowledge-based (built with statistics obtained from PDB)? Ab-initio methods allow simultaneous addition of several loops, which yields a conformational ensemble view for the loop

28 Step 5 Side Chain Modeling

29 Step 5 Side Chain Modeling Side-chain packing is critical to studying ligand binding to proteins Side chain builder Predicted from similar structures Built from steric and energetic considerations with robust conformational search algorithms Combination of rotamer library and energy evaluations
30 Step 5 Side Chain Modeling Rotamer libraries have been created (statistical analysis of torsion angles of side chains of amino acids) from structures in the PDB Two main effects in predicting side chains How it sits on top of the main chain(very critical)? Continuous variation of side chain torsions - only 6% varies +/- 40 degrees from the rotamer libraries Current techniques predict side chains up to 1.5Å accuracy for a fixed backbone for the core residues Solvation and H-bond terms are very important in modeling exposed side chains
31 Step 5 Side Chain Modeling Methods available SCWRL, SCAP, MODELLER, Insight II, WhatIf, SCREAM etc. Evaluation of all three methods for backbone < 4Å lrmsd to native all work equally 50% of χ1 and 35% of χ2 and χ3 SCWRL Decomposition of protein to non-interacting parts, collision free energy function. Fast, works quite well SCREAM works well accurate energy analysis computationally intensive
32 Step 6 Refinement Completed model may undergo a short energy minimization Physics-based or knowledge-based functions may be used The minimization may help remove steric clashes and improve favorable interactions in the completed model prior to the final evaluation of the built model for the target

33 Comparative Modeling Example Yellow M. Jann. Predicted Blue Bac. Thermophillus 4ts1 Predicted Structure of M.Jann TyrRS, Zhang et al PNAS
34 Evaluation of Model Given a predicted structure: Ramachandran plot allowed regions for backbone torsions Calculate the Hydrogen-bond network - use Quanta or WhatIf or MolProbity normally calculated for heteroatoms with distance cutoff Identify hydrophobic residues on the surface Identify hydrophilic residues in the core satisfied with salt bridges? Voids in the core are typically small two water cluster?
35 Comparative Modeling on the WWW Prior to 1998, comparative modeling could only be done with commercial software or command-line freeware The process was time-consuming and labor-intensive The past few years has seen an explosion in automated web-based comparative modeling servers Now anyone can! (but you still have to know what you re doing...)
36 What are Folds Anyway? Family: clear evolutionary relationship Sequence identity 30%, but similar functions and structures indicate common descent even on low sequence identity Globins family has members with sequence identities of only 15% Superfamily: probable common evolutionary origin Low sequence identities, but structural and functional features suggest a common evolutionary origin Actin, ATPase domain of heat shock protein, and hexakinase together form a superfamily Fold : major structural similarity (possibly no common ancestor) Proteins have a common fold if they have the same major secondary structures in the same arrangement and with the same topological connections, though length of regions can change Structural similarities can arise just from the physics and chemistry of proteins favoring certain packing arrangements and chain topologies
37 Many Sequences Map to Limited Folds A large number of protein sequences adopt similar folds Even when sequence identity is lower than 25% Such sequences are known as remote homologues Sequence universe Fold universe
38 Threading: Towards Ab-initio Methods Remote homologues necessitate a novel computational approach Between comparative modeling and ab initio: Threading Threading may detect structural similarities that are not accompanied by detectable or significant sequence similarities The environment of a particular amino acid is expected to be more conserved than the actual sequence identity of the amino acid

39 When to Resort to Threading? A pair of structures within the same family that have sequence identity of 32%: Maybe comparative modeling can be applied here

40 When to Resort to Threading? A pair of structures within the same super-family that have sequence identity 16%: beyond the applicability of comparative modeling
41 A Simplistic View of Threading Target sequence G-A-L-T-E-S-Q-V-P-... Fold library Build model Calculate energetic score Rank and evaluate model
42 An Algorithmic View of Threading Step 1: Construction of template library from known folds Step 2: Design of (energetic) scoring function Step 3: Sequence-template alignment Step 4: Template selection and model construction Step 5: Model completion, refinement, and evaluation



?")
43 Threading: Problem Statement Threading essentially approaches the following problem: Given a query sequence, find which fold fits best from a library of known folds This essential component of threading is often known as fold recognition (recognizing the best fitting fold among the library of available folds)? MTYKLILNGKTKGETTTEAVDAA TAEKVFQYANDNGVDGEWTYTE Query sequence Fold Library
44 Threading: Pictorial Presentation
45 Threading: Sequence-structure Alignment Finding the best fold for a query sequence entails addressing the sequence-template alignment problem, which consists of two sub-problems: Design of an effective scoring function to determine the goodness of a fit that results from an alignment Known as the threading energy function or potential Rapid alignment algorithm so that the sequence can be efficiently threaded to many folds during the search for the best one Different methods spanning from hard to soft threading consider aligning sequence to sequence (like comparative modeling), sequence to structure, sequence to contact environment etc.
46 Threading Energy Function Sequence-template alignments are scored using a threading energy (objective) function The function scores the compatibility between the query sequence of amino acids and their corresponding positions in a given template The objective function essentially scores compatibility using specifically-chosen parameters such as: Amino-acid preferences for solvent accessibility Amino-acid preferences for particular secondary structures Interactions between neighboring amino acids Inexpensive physics-based terms are also incorporated
47 Threading Energy Function how preferable to put two particular residues nearby: E p (Pairwise potential) alignment gap penalty: E g? (gap score)? how well a residue fits a structural environment: E s (Fitness score)? sequence similarity between query and template proteins: E m (Mutation score)?? Consistency with secondary structures: E ss E = E p + E s + E m + E g + E ss Minimize E to find the best sequence-template alignment
48 Threading Energy Function There are three main approaches to the alignment sub-problem: 1 Sequence-sequence alignment (1D-1D) Align query sequence with template sequences This alignment guides the threading of sequence into structure 2 Consider structural environment in addition to sequence (3D-1D) Align query sequence to a string of descriptors that describe the 3D environment of the considered folds 3 Consider pairwise contacts in folds Contact graph guides the alignment of the query sequence Most successful threading methods fall in this category
49 Sequence - Sequence Alignment to Template Essentially similar to the process used in comparative modeling Advantage: Simple dynamic programming methods can used to align the query sequence to the sequences of the templates Disadvantages: Templates may have low sequence similarity to query sequence Fails to consider interactions between neighboring amino acids There is no rigorous boundary between comparative modeling and threading in terms of methodology: rule of thumb is that when the alignment takes into consideration structural aspects (besides sequence aspects) and the templates are remote homologues, then we talk about threading rather than comparative modeling.
50 Sequence - Sequence Alignment to Template 1 Sequence-sequence alignment (1D-1D)?Align query sequence with template sequences This alignment guides the threading of sequence into structure 2 Consider structural environment in addition to sequence (3D-1D)?Align query sequence to a string of descriptors that describe the 3D environment of the considered folds 3 Consider pairwise contacts in folds Contact graph guides the alignment of the query sequence Most successful threading methods fall in this category
51 Sequence - Sequence Alignment to Template Instead of aligning the query sequence to a template sequence, the query sequence is aligned to a string of descriptors that capture the 3D environment of the template structure For each amino-acid position in a template structure, one determines: How buried it is (buried, partly buried or exposed)? The fraction of surrounding environment that is polar (or apolar)? The local secondary structure (helix, sheet, or other)? This information is encoded in a scoring matrix that (similar to the scoring matrices used for sequence alignment) is used to guide the alignment in a dynamic programming framework
52 Amino Acid Environments Each environment is further divided into three sub-classes according to the secondary structure of the amino acid (α-helix, β-strand, or other)? B1: buried and hydrophobic environment B2: buried and moderately polar environment B3: and buried and polar environment P1: partially buried and moderately polar environment P2: partially buried and polar environment E: exposed to solvent Bowie, J.U., Luthy, R., and Eisenberg, D A method to identify protein sequences that fold into a known three-dimensional structure. Science 253:
53 Tabulating Amino Acid Environments Each position in the template structure is mapped into one of the 18 possible environment classes (a vector of 18 descriptors) Different amino-acids prefer different environments This preference is captured by compiling descriptors for each amino acid over structures in the PDB The number of times an amino acid appears in a specific environment class is tabulated to obtain frequencies These frequencies are normalized for each amino acid to obtain probabilities of the form P(x, y)? P(x,y) = probability of finding amino acid x in environment class y
54 Amino-acid Environment Matrix 20 amino acids Environment class W F Y... B1α B1β classes Score = ln Pr(residue j in environment i Pr(residue j in any environment)
55 From Environment Matrix to Profile Each template structure is mapped to an environmental profile Each position in the template is mapped by the sequence identity of the amino acid (find column) and the actual environment of that amino acid in the structure (find row)? Each position is scored in this way? total profile score of the template is additive, so obtained by summing over the scores of the amino acids Environment class W F Y... B1α B1β
56 Environment and Sequence Alignment Aligning the query sequence to the profile of a specific template is similar to the traditional sequence-sequence alignment with DP: For each amino acid s i in the query sequence, the cost of modeling it through amino-acid tj in the template is either: that of mutation : environment cost provides this that of insertion: gap penalty incurred s 1 s 2... s m new sequence e 1 e 2... e n environment classes
57 Sequence - 3D Profile Alignment Advantages: Provides good-quality models It considers not only sequence, but structural considerations encoded in the environment of an amino acid Disadvantages: Amino acids are considered/threaded independently of one another This is inherent in the additive score used in the dynamic programming formulation of the problem
58 Sequence - Sequence Alignment to Template 1 Sequence-sequence alignment (1D-1D) Align query sequence with template sequences This alignment guides the threading of sequence into structure 2 Consider structural environment in addition to sequence (3D-1D) Align query sequence to a string of descriptors that describe the 3D environment of the considered folds 3 Consider pairwise contacts in folds Contact graph guides the alignment of the query sequence Most successful threading methods fall in this category
?")
59 Contact Graph Template Each residue as a vertex One edge between two residues if in contact (their spatial distance is within a given cutoff)? Cores are the most conserved segments in the template: alpha-helices and beta-sheets
60 Simplified Contact Graph
61 Contact Graph and Alignment Diagram
62 Contact Graph and Alignment Diagram
63 Variables in the Alignment x (i,l) denotes core i is aligned to sequence position l y (i,l,j,k) denotes that core i is aligned to position l and core j is aligned to position k at the same time D[i]: all the valid query sequence positions that c i could be aligned to. R[i, j, l]: All the valid alignment positions of c j given that c i is aligned to s l
64 Alignment as a Linear Programming Problem Minimize: E = a i,l x i,l + b (i,l),(j,k) y (i,l),(j,k) s.t. x i,l, y (i,l),(j,k) {0, 1} x i,l + x i+1,k 1 k D[i + 1] R[i, i + 1, l] y (i,l),(j,k) = x i,l x j,k x i,l = 1 l D(i) E encodes the scoring function a i,l encodes the individual energy terms (E s,e ss,e m ) b (i,l),(j,k) encodes the pairwise terms (E g,e p )
65 Alignment as a Linear Programming Problem Minimize: E = a i,l x i,l + b (i,l),(j,k) y (i,l),(j,k) (1) s.t. (2) x i,l, y (i,l),(j,k) {0, 1} (3) x i,l + x i+1,k 1 k D[i + 1] R[i, i + 1, l] (4) y (i,l),(j,k) = x i,l x j,k (5) x i,l = 1 (6) l D(i) 1 The objective function 2 Subject to the following constraints: 3 Binary variables (either put an amino acid in a position or not etc.) 4 No conflicts in the alignment 5 Pairwise and individual terms have to be compatible (not linear but can be made such quite easily) 6 Only one position is selected for each residue
66 Alignment with RAPTOR Minimize: E = a i,l x i,l + b (i,l),(j,k) y (i,l),(j,k) s.t. x i,l = y (i,l),(j,k), l D[i] x j,k = k R[i,j,l] l R[j,k,i] x i,l, y (i,l),(j,k) {0, 1} x i,l = 1 l D(i) y (i,l),(j,k), k D[j]
, 95-118. J. Xu, M.")
67 Alignment with RAPTOR Target ID: T Target Size:144 Superimposable size within 5Å: 118 RMSD:1.9Å J. Xu, M. Li, D. Kim, Y. Xu, Journal of Bioinformatics and Computational Biology, 1:1(2003), J. Xu, M. Li, PROTEINS: Structure, Function, and Genetics, CASP5 special issue Red: Experimental Structure Blue/Green: Raptor model
68 Final Words on Threading More than 99% threading instances can be solved directly by linear programming The rest can be solved by branch-and-bound with only several branch nodes Less memory consumption Less computational time Easy to extend to incorporate other constraints
69 Ab Initio Folding State of the art methods use fragment assembly, deep learning, HMM. Search is done using optimization techniques (Monte Carlo, MD etc.). Example Rosetta (also expanded to using distributed computing), TASSER, I-TASSER etc.
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Procheck output. Bond angles (Procheck) Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.
 Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.") Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Programme Last week s quiz results + Summary Fold recognition Break Exercise: Modelling remote homologues
 Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Modeling for 3D structure prediction
 Modeling for 3D structure prediction What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing
Modeling for 3D structure prediction What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Building 3D models of proteins
 Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University
 Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
Template-Based Modeling of Protein Structure
 Template-Based Modeling of Protein Structure David Constant Biochemistry 218 December 11, 2011 Introduction. Much can be learned about the biology of a protein from its structure. Simply put, structure
Template-Based Modeling of Protein Structure David Constant Biochemistry 218 December 11, 2011 Introduction. Much can be learned about the biology of a protein from its structure. Simply put, structure
Protein Structure Determination
 Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Protein Modeling. Generating, Evaluating and Refining Protein Homology Models
 Protein Modeling Generating, Evaluating and Refining Protein Homology Models Troy Wymore and Kristen Messinger Biomedical Initiatives Group Pittsburgh Supercomputing Center Homology Modeling of Proteins
Protein Modeling Generating, Evaluating and Refining Protein Homology Models Troy Wymore and Kristen Messinger Biomedical Initiatives Group Pittsburgh Supercomputing Center Homology Modeling of Proteins
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
09/06/25. Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Non-uniform distribution of folds. Scheme of protein structure predicition
 Non-uniform distribution of folds. Scheme of protein structure predicition") Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
Can protein model accuracy be. identified? NO! CBS, BioCentrum, Morten Nielsen, DTU
 Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
FlexPepDock In a nutshell
 FlexPepDock In a nutshell All Tutorial files are located in http://bit.ly/mxtakv FlexPepdock refinement Step 1 Step 3 - Refinement Step 4 - Selection of models Measure of fit FlexPepdock Ab-initio Step
FlexPepDock In a nutshell All Tutorial files are located in http://bit.ly/mxtakv FlexPepdock refinement Step 1 Step 3 - Refinement Step 4 - Selection of models Measure of fit FlexPepdock Ab-initio Step
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics
 Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
HMM applications. Applications of HMMs. Gene finding with HMMs. Using the gene finder
 HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Prediction and refinement of NMR structures from sparse experimental data
 Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Contact map guided ab initio structure prediction
 Contact map guided ab initio structure prediction S M Golam Mortuza Postdoctoral Research Fellow I-TASSER Workshop 2017 North Carolina A&T State University, Greensboro, NC Outline Ab initio structure prediction:
Contact map guided ab initio structure prediction S M Golam Mortuza Postdoctoral Research Fellow I-TASSER Workshop 2017 North Carolina A&T State University, Greensboro, NC Outline Ab initio structure prediction:
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5
 Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Bioinformatics. Macromolecular structure
 Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence
 Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
Molecular Modeling Lecture 7. Homology modeling insertions/deletions manual realignment
 Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
HOMOLOGY MODELING. The sequence alignment and template structure are then used to produce a structural model of the target.
 HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009
 114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
Basics of protein structure
 Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Protein Structures. 11/19/2002 Lecture 24 1
 Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
THEORY. Based on sequence Length According to the length of sequence being compared it is of following two types
 Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Exp 11- THEORY Sequence Alignment is a process of aligning two sequences to achieve maximum levels of identity between them. This help to derive functional, structural and evolutionary relationships between
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
7.91 Amy Keating. Solving structures using X-ray crystallography & NMR spectroscopy
 7.91 Amy Keating Solving structures using X-ray crystallography & NMR spectroscopy How are X-ray crystal structures determined? 1. Grow crystals - structure determination by X-ray crystallography relies
7.91 Amy Keating Solving structures using X-ray crystallography & NMR spectroscopy How are X-ray crystal structures determined? 1. Grow crystals - structure determination by X-ray crystallography relies
Tiffany Samaroo MB&B 452a December 8, Take Home Final. Topic 1
 Tiffany Samaroo MB&B 452a December 8, 2003 Take Home Final Topic 1 Prior to 1970, protein and DNA sequence alignment was limited to visual comparison. This was a very tedious process; even proteins with
Tiffany Samaroo MB&B 452a December 8, 2003 Take Home Final Topic 1 Prior to 1970, protein and DNA sequence alignment was limited to visual comparison. This was a very tedious process; even proteins with
proteins Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field Dong Xu1 and Yang Zhang1,2* 1 Department
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field Dong Xu1 and Yang Zhang1,2* 1 Department
Physiochemical Properties of Residues
 Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Steps in protein modelling. Structure prediction, fold recognition and homology modelling. Basic principles of protein structure
 Structure prediction, fold recognition and homology modelling Marjolein Thunnissen Lund September 2012 Steps in protein modelling 3-D structure known Comparative Modelling Sequence of interest Similarity
Structure prediction, fold recognition and homology modelling Marjolein Thunnissen Lund September 2012 Steps in protein modelling 3-D structure known Comparative Modelling Sequence of interest Similarity
Neural Networks for Protein Structure Prediction Brown, JMB CS 466 Saurabh Sinha
 Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Conformational Sampling in Template-Free Protein Loop Structure Modeling: An Overview
 # of Loops, http://dx.doi.org/10.5936/csbj.201302003 CSBJ Conformational Sampling in Template-Free Protein Loop Structure Modeling: An Overview Yaohang Li a,* Abstract: Accurately modeling protein loops
# of Loops, http://dx.doi.org/10.5936/csbj.201302003 CSBJ Conformational Sampling in Template-Free Protein Loop Structure Modeling: An Overview Yaohang Li a,* Abstract: Accurately modeling protein loops
InDel 3-5. InDel 8-9. InDel 3-5. InDel 8-9. InDel InDel 8-9
 Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Lecture 5 Alignment I. Introduction. For sequence data, the process of generating an alignment establishes positional homologies; that is, alignment provides the identification of homologous phylogenetic
Protein Structures: Experiments and Modeling. Patrice Koehl
 Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Protein Modeling Methods. Knowledge. Protein Modeling Methods. Fold Recognition. Knowledge-based methods. Introduction to Bioinformatics
 Protein Modeling Methods Introduction to Bioinformatics Iosif Vaisman Ab initio methods Energy-based methods Knowledge-based methods Email: ivaisman@gmu.edu Protein Modeling Methods Ab initio methods:
Protein Modeling Methods Introduction to Bioinformatics Iosif Vaisman Ab initio methods Energy-based methods Knowledge-based methods Email: ivaisman@gmu.edu Protein Modeling Methods Ab initio methods:
Introduction to" Protein Structure
 Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Computational protein design
 Computational protein design There are astronomically large number of amino acid sequences that needs to be considered for a protein of moderate size e.g. if mutating 10 residues, 20^10 = 10 trillion sequences
Computational protein design There are astronomically large number of amino acid sequences that needs to be considered for a protein of moderate size e.g. if mutating 10 residues, 20^10 = 10 trillion sequences
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Large-Scale Genomic Surveys
 Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Bioinformatics Subtopics Fold Recognition Secondary Structure Prediction Docking & Drug Design Protein Geometry Protein Flexibility Homology Modeling Sequence Alignment Structure Classification Gene Prediction
Analysis and Prediction of Protein Structure (I)
") Analysis and Prediction of Protein Structure (I) Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 2006 Free for academic use. Copyright @ Jianlin Cheng
Analysis and Prediction of Protein Structure (I) Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 2006 Free for academic use. Copyright @ Jianlin Cheng
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Homology Modeling I. Growth of the Protein Data Bank PDB. Basel, September 30, EMBnet course: Introduction to Protein Structure Bioinformatics
 Swiss Institute of Bioinformatics EMBnet course: Introduction to Protein Structure Bioinformatics Homology Modeling I Basel, September 30, 2004 Torsten Schwede Biozentrum - Universität Basel Swiss Institute
Swiss Institute of Bioinformatics EMBnet course: Introduction to Protein Structure Bioinformatics Homology Modeling I Basel, September 30, 2004 Torsten Schwede Biozentrum - Universität Basel Swiss Institute
Protein Structure Prediction
 Protein Structure Prediction Michael Feig MMTSB/CTBP 2009 Summer Workshop From Sequence to Structure SEALGDTIVKNA Folding with All-Atom Models AAQAAAAQAAAAQAA All-atom MD in general not succesful for real
Protein Structure Prediction Michael Feig MMTSB/CTBP 2009 Summer Workshop From Sequence to Structure SEALGDTIVKNA Folding with All-Atom Models AAQAAAAQAAAAQAA All-atom MD in general not succesful for real
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT
 3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
3. SEQUENCE ANALYSIS BIOINFORMATICS COURSE MTAT.03.239 25.09.2012 SEQUENCE ANALYSIS IS IMPORTANT FOR... Prediction of function Gene finding the process of identifying the regions of genomic DNA that encode
Protein structure alignments
 Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
SCOP. all-β class. all-α class, 3 different folds. T4 endonuclease V. 4-helical cytokines. Globin-like
 SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
Finding Similar Protein Structures Efficiently and Effectively
 Finding Similar Protein Structures Efficiently and Effectively by Xuefeng Cui A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Doctor of Philosophy
Finding Similar Protein Structures Efficiently and Effectively by Xuefeng Cui A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Doctor of Philosophy
Tools and Algorithms in Bioinformatics
 Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Tools and Algorithms in Bioinformatics GCBA815, Fall 2015 Week-4 BLAST Algorithm Continued Multiple Sequence Alignment Babu Guda, Ph.D. Department of Genetics, Cell Biology & Anatomy Bioinformatics and
Orientational degeneracy in the presence of one alignment tensor.
 Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Secondary Structure. Bioch/BIMS 503 Lecture 2. Structure and Function of Proteins. Further Reading. Φ, Ψ angles alone determine protein structure
 Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Computational Molecular Biology. Protein Structure and Homology Modeling
 Computational Molecular Biology Protein Structure and Homology Modeling Prof. Alejandro Giorge1 Dr. Francesco Musiani Sequence, function and structure relationships v Life is the ability to metabolize
Computational Molecular Biology Protein Structure and Homology Modeling Prof. Alejandro Giorge1 Dr. Francesco Musiani Sequence, function and structure relationships v Life is the ability to metabolize
2MHR. Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity.
 Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein quality assessment
 Protein quality assessment Speaker: Renzhi Cao Advisor: Dr. Jianlin Cheng Major: Computer Science May 17 th, 2013 1 Outline Introduction Paper1 Paper2 Paper3 Discussion and research plan Acknowledgement
Protein quality assessment Speaker: Renzhi Cao Advisor: Dr. Jianlin Cheng Major: Computer Science May 17 th, 2013 1 Outline Introduction Paper1 Paper2 Paper3 Discussion and research plan Acknowledgement
Ch. 9 Multiple Sequence Alignment (MSA)
") Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
Ch. 9 Multiple Sequence Alignment (MSA) - gather seqs. to make MSA - doing MSA with ClustalW - doing MSA with Tcoffee - comparing seqs. that cannot align Introduction - from pairwise alignment to MSA -
Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization
 Biophysical Journal Volume 101 November 2011 2525 2534 2525 Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization Dong Xu and Yang Zhang
Biophysical Journal Volume 101 November 2011 2525 2534 2525 Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization Dong Xu and Yang Zhang
Ab-initio protein structure prediction
 Ab-initio protein structure prediction Jaroslaw Pillardy Computational Biology Service Unit Cornell Theory Center, Cornell University Ithaca, NY USA Methods for predicting protein structure 1. Homology
Ab-initio protein structure prediction Jaroslaw Pillardy Computational Biology Service Unit Cornell Theory Center, Cornell University Ithaca, NY USA Methods for predicting protein structure 1. Homology
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools. Giri Narasimhan
 CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinff18.html Proteins and Protein Structure
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinff18.html Proteins and Protein Structure
In-Depth Assessment of Local Sequence Alignment
 2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
2012 International Conference on Environment Science and Engieering IPCBEE vol.3 2(2012) (2012)IACSIT Press, Singapoore In-Depth Assessment of Local Sequence Alignment Atoosa Ghahremani and Mahmood A.
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Assignment 2 Atomic-Level Molecular Modeling
 Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
Structure to Function. Molecular Bioinformatics, X3, 2006
 Structure to Function Molecular Bioinformatics, X3, 2006 Structural GeNOMICS Structural Genomics project aims at determination of 3D structures of all proteins: - organize known proteins into families
Structure to Function Molecular Bioinformatics, X3, 2006 Structural GeNOMICS Structural Genomics project aims at determination of 3D structures of all proteins: - organize known proteins into families
Protein Structure Prediction
 Protein Structure Prediction Michael Feig MMTSB/CTBP 2006 Summer Workshop From Sequence to Structure SEALGDTIVKNA Ab initio Structure Prediction Protocol Amino Acid Sequence Conformational Sampling to
Protein Structure Prediction Michael Feig MMTSB/CTBP 2006 Summer Workshop From Sequence to Structure SEALGDTIVKNA Ab initio Structure Prediction Protocol Amino Acid Sequence Conformational Sampling to
3D Structure. Prediction & Assessment Pt. 2. David Wishart 3-41 Athabasca Hall
 3D Structure Prediction & Assessment Pt. 2 David Wishart 3-41 Athabasca Hall david.wishart@ualberta.ca Objectives Become familiar with methods and algorithms for secondary Structure Prediction Become familiar
3D Structure Prediction & Assessment Pt. 2 David Wishart 3-41 Athabasca Hall david.wishart@ualberta.ca Objectives Become familiar with methods and algorithms for secondary Structure Prediction Become familiar
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
CSCE555 Bioinformatics. Protein Function Annotation
 CSCE555 Bioinformatics Protein Function Annotation Why we need to do function annotation? Fig from: Network-based prediction of protein function. Molecular Systems Biology 3:88. 2007 What s function? The
CSCE555 Bioinformatics Protein Function Annotation Why we need to do function annotation? Fig from: Network-based prediction of protein function. Molecular Systems Biology 3:88. 2007 What s function? The
Motif Prediction in Amino Acid Interaction Networks
 Motif Prediction in Amino Acid Interaction Networks Omar GACI and Stefan BALEV Abstract In this paper we represent a protein as a graph where the vertices are amino acids and the edges are interactions
Motif Prediction in Amino Acid Interaction Networks Omar GACI and Stefan BALEV Abstract In this paper we represent a protein as a graph where the vertices are amino acids and the edges are interactions
Protein Secondary Structure Assignment and Prediction
 1 Protein Secondary Structure Assignment and Prediction Defining SS features - Dihedral angles, alpha helix, beta stand (Hydrogen bonds) Assigned manually by crystallographers or Automatic DSSP (Kabsch
1 Protein Secondary Structure Assignment and Prediction Defining SS features - Dihedral angles, alpha helix, beta stand (Hydrogen bonds) Assigned manually by crystallographers or Automatic DSSP (Kabsch
proteins Estimating quality of template-based protein models by alignment stability Hao Chen 1 and Daisuke Kihara 1,2,3,4 * INTRODUCTION
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS Estimating quality of template-based protein models by alignment stability Hao Chen 1 and Daisuke Kihara 1,2,3,4 * 1 Department of Biological Sciences, College
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Estimating quality of template-based protein models by alignment stability Hao Chen 1 and Daisuke Kihara 1,2,3,4 * 1 Department of Biological Sciences, College
1. Protein Data Bank (PDB) 1. Protein Data Bank (PDB)
 1. Protein Data Bank (PDB)") Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
Protein Structure. Hierarchy of Protein Structure. Tertiary structure. independently stable structural unit. includes disulfide bonds
 Protein Structure Hierarchy of Protein Structure 2 3 Structural element Primary structure Secondary structure Super-secondary structure Domain Tertiary structure Quaternary structure Description amino
Protein Structure Hierarchy of Protein Structure 2 3 Structural element Primary structure Secondary structure Super-secondary structure Domain Tertiary structure Quaternary structure Description amino
COMP 598 Advanced Computational Biology Methods & Research. Introduction. Jérôme Waldispühl School of Computer Science McGill University
 COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
Sequence Alignment Techniques and Their Uses
 Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Sequence Alignment Techniques and Their Uses Sarah Fiorentino Since rapid sequencing technology and whole genomes sequencing, the amount of sequence information has grown exponentially. With all of this
Supersecondary Structures (structural motifs)
") Supersecondary Structures (structural motifs) Various Sources Slide 1 Supersecondary Structures (Motifs) Supersecondary Structures (Motifs): : Combinations of secondary structures in specific geometric
Supersecondary Structures (structural motifs) Various Sources Slide 1 Supersecondary Structures (Motifs) Supersecondary Structures (Motifs): : Combinations of secondary structures in specific geometric