Ab-initio protein structure prediction
|
|
|
- Helena Lambert
- 5 years ago
- Views:
Transcription
1 Ab-initio protein structure prediction Jaroslaw Pillardy Computational Biology Service Unit Cornell Theory Center, Cornell University Ithaca, NY USA
2 Methods for predicting protein structure 1. Homology (comparative) modeling relies on the similarity of the sequence of the protein under study to those of proteins with similar sequence and known structure. Very reliable, if sequence similarity 40%. 2. Threading (fold recognition) finding the best structure from the database, which fits the sequence studied in terms of a chosen score function. 3. Energy-based (ab initio): search of the structure as a global minimum of a designed potential-energy function, following Anfinsen s thermodynamic hypothesis.
3 Ab-initio methods: advantages They do not require existence of a similarly shaped (folded) protein in a database in order to predict a structure. Entirely new and unknown folds may be found this way. The only information necessary is a sequence, however experimental knowledge about secondary structure and other structural features may be used. They perform very well for short sequences, where other methods are not as good. In some cases they may give insight to physical properties of protein (folding pathways, statistical ensembles).
4 Ab-initio methods: limitations They are usually slower than threading and homology modeling by few orders of magnitude. They usually require big parallel computer cluster to be run, and even then it may take few days using processors to calculate something The reliability of models produced diminishes fast with the protein size the sequences shorter than amino acids work best. It is possible to treat larger proteins if some other information is used (e.g. secondary structure). The correct prediction has to be chosen from several possibilities (families) that are close in energy. Prediction based on one (lowest-energy) family only is much less reliable. Typical resolution of a model is 3-6Å.
5 Ab-initio methods Combination (R. A. Friesner, Columbia University) Uses simplified representation of protein chain with the energy function derived from protein statistics (PDB) and secondary structure elements frozen during the search (secondary structure prediction is used before the search). Monte Carlo with minimization is used as a global search Lattice folding (J. Skolnick, Danforth Plant Science Center) Uses simplified representation of protein chain on the lattice. Secondary structure prediction with multiple sequence alignment is used before search. Threading is used to derive some constraints. Monte Carlo search is used on the lattice, results are clustered, and 5 best models are refined of lattice. FRAGFOLD (D. T. Jones, Brunel University, England) Uses simplified representation of protein chain (Cα and Cβ only) with a special energy function. Fragment database is generated using standard secondary structure elements, and the energy function is optimized with simulated annealing.
6 Ab-initio methods Rosetta (D. Baker, University of Washington) Builds a database of short sequence fragments (9aa) and then uses Monte Carlo procedure to search conformational space. Resulting conformations are filtered based on backbone contacts, clustered, and re-evaluated with more complicated energy function. UNRES (H. A. Scheraga, Cornell University) Uses simplified representation of protein chain with a sophisticated, physics-based energy function. Genetic algorithm or Monte Carlo with minimization are used for global search. Structure is refined at the all-atom level with ECEPP force field.
7 Hierarchical Approach to Protein-Structure Prediction Stage 1: Global optimization of the potential-energy function in a simplified representation of the polypeptide chain. This is the key stage of the algorithm. Stage 2: Conversion of the lowest-energy structures to the all-atom representation. Stage 3: Limited energy optimization of the all-atom structures.

8 lowest in energy at the simplified level
9 What is Global Optimization? Local minimization (optimization) is an algorithm leading to a closest local minimum in the attraction basin from a given starting point in the function domain. Global minimization (optimization) is an algorithm designed to find the local minimum having the lowest (extreme) value of the function being optimized.
10 Why to do global optimization at all? Thermodynamic hypothesis (Anfinsen, 1973): The native structure corresponds to the global minimum of the free energy (F) In most cases oscillations around the most stable structure are small and the lowest energy structure well represents the minimum-free-energy ensemble. For proteins, energy functions may incorporate the solvent (all-atom models) and possibly other degrees of freedom (simplified models), but do not include the entropic free energy of backbone degrees of freedom. However, these entropic contributions are small in native states of proteins, since structural fluctuations are generally small. In this case free energy (F) may be substituted by energy (E) for optimization.
11 Global optimization: can it be done? Global optimization is a very hard NP-complete problem impossible to solve in general case as well as in most physically-interesting cases The problem lies in a huge number of local minima and the fact tha function is usually defined in very high dimensional space: For N-residue protein in an all-atom representation it is estimated to be ~10 N local minima in 6N-dimensional space (even if a lot of these minima are inaccessible due to sterical problems it is a large nuber ) For cluster of 55 identical Lennard-Jones particles the number of local minima is estimated to be ~10 10 (in 159 dimensional space not counting permutations); for 147-particle cluster this number grows to ~10 60 (in 435 dimensional space)
12 Global optimization: can it be done? General answer is: NO. It can be done in some particular cases when: Knowledge about physics of a system under study should be used to simplify the search making it plausible Global optimization method should be coupled with a potential function, i.e. they should be designed and tuned up together
13 Thermodynamic hypothesis must be supplemented by the requirement that the energy hypersurface is searchable, i.e. that the global energy minimum is preferred from the others not only by the energy at the minimum, but also by the heights of barriers
14 Hierarchical energy/free energy surface The most important feature of potential energy hypersurface is its hierachical organization helping the global optimization method to get from high to lowenergy structures. This feature may described in 2D graph through relationships between groups of minima by using: - maximum energy along the optimal trajectories connecting minima (disconnectivity tree) - temperature at which particular minima kinetically confine the system The dependence of grouping versus relation parameter (E or T) is usually graphically represented by a tree, where the root is when all the minima are equivalent, branches are groups of equivalent minima (at a given parameter value) and leaves are single minima.
15 Hierarchical energy/free energy surface energy surface disconnectivity tree
16 LJ 38 LJ 55 LJ 75 Lennard-Jones 6-12 clusters Disconnectivity graph minima diverge on plot when the energy on the Y axis is greater than he highest energy on the lowest-energy pathway connecting two minima, otherwise they are epresented together by a single vertical line.. P. K. Doye, M. A, Miller, D. J. Wales J. Chem. Phys. 1999, 111,
17 constant ε distance-dependent ε Ac-(Ala) 8 -NHMe with AMBER95 P. N. Mortenson and D. J. Wales J. Chem. Phys. 2001, 114,
18 Global Optimization: top-down versus local-minima-based Top-down method tries to explore tree-like structure of minima starting from its root and progressing to the end of longest branch (global minimum). The most important problem for such a method is how to choose an appropriate branch when a bifurcation occurs and the group of minima split. Top-down method in this meaning is equivalent to some kind of deformation. Examples: Branch-and-Bound, Diffusion Equation Method, Distance Scaling Method, Gaussian Density Annealing, Gaussian Packet Annealing, Self-Consistent Basin-to-Deformed-Basin Mapping, Simulated Annealing.
19 Global Optimization: top-down versus local-minima-based Local-minima-based method does not explore tree-like structure of minima. It starts from local minimum (or minima), progresses by finding more local minima, and builds branches of disconnectivity graph by aggregating local minima. Most important problem here is how to cover as large space as possible (usually solved by similarity classification, families) and how to obtain as low-energy trial structures as possible from current structrure(s) (solved by smart Monte Carlo moves, Genetic Algorithm, etc). Examples: Monte Carlo-Minimization, Conformational Space Annealing, Electrostatically-Driven Monte Carlo, Conformation-Family Monte Carlo. CSA and CFMC fit here as methods that try to recover hierarchy by different similarity measures (RMS, angles) and divide space into groups represented by a few (CFMC) or single (CSA) structure.
20 Global Optimization: top-down versus local-minima-based At present local-minima-based methods perform better than top-down methods, or at least they are much more cost-effective. The task of branch evaluation (predicting which branch leads to lower-energy minima) is by no means easier that designing effective algorithms for smart structure generation, but structure generation is by far more intuitive. The task of choosing a deformation that leads to a global minimum is strongly linked to a branch evaluation problem and is really very difficult as well.
21 good potential function is a necessary element of any algorithm for a ructure prediction. ood (predictive) potential function: Potential Function ) Should reproduce the experimental structure within a certain accuracy b) The structures corresponding to the lowest-energy minima found for the potential should represent plausible structures, and one of them, preferably the global minimum, should correspond to the observed experimental structure ) The lowest energy structure(s) corresponding to the native structure should be separated from other, non-physical structures by an energy gap
22 Potential Function for Proteins Fast and scaling well with the size of protein: global optimization requires large number of local minimizations to converge. Easy to parameterize: potential should be coupled with a global optimization method, what requires several Z-score/global optimization iterations Close to physical reality: this feature helps with transferability of a potential.
23 O All-atom: geometry H H O H Variables: C... N C α C 3 backbone dihedral angles average of 3 side-chain dihedral angles ϕ χ ψ... C N 6 per residue (with fixed bond lengths and bond angles) ω At least 7 centers of interaction per residue, ~15 on average Computational expense scales as ~15N*(15N-1)/2 225N 2
 Interaction centers are marked in")
24 United-residue: geometry Only 2 centers of interaction per residue Computational expense scales as ~2N*(2N-1)/2 2N 2 ariables: backbone dihedral angle backbone virtual bond angle side-chain rotation angle side-chain tilt angle per residue Side-chains are represented as ellipsoids (Gay-Berne potential) Interaction centers are marked in colors

")
25 United-residue (UNRES) representation of polypeptide chain All-atom representation of polypeptide chain in solution (explicit water)


26 UNRES: formulation X principal degrees of freedom (variables that define the C α trace of an polypeptide chain) Y secondary or less important degrees of freedom that are averaged out
27 U = + w b i< j i U U SCSC b i ( θ i j + w ) + w SCp i j rot i UNRES U U SC p rot i j ( α + SC i w el i< j 1 SC i U ) + p N i p j corr m= 2 + w - Three components (out of 13) were derived using the PDB (U SCSC, U b, U rot ) - U SCSC parametrization depends on the statistical probability of side-chain contacts in the PDB, these probabilities did not change over years (1977). Moreover, these values correlate highly (R=0.94) with the experimentally determined octanol partitioning coefficients. This suggests that these parameters reflect physical interactions. - U b and U rot are not specific and serve only to maintain reasonable values of virtual-bond angles and positions of side-chain centroids with respect to C α trace., β w ( m) corr tor U i ( m) corr U tor ( γ i ) +
 3 rd order (local and electrostatic) 5 th")
 3 rd order (local) 4 th")
28 UNRES: correlation terms 2 nd order (electrostatic) 2 nd order (local) 3 rd order (local and electrostatic) 5 th order 3 rd order (local and electrostatic) 6 th order 4 th order (local and electrostatic) 3 rd order (local) 4 th order
29 UNRES OVERVIEW : Types of residues The number of different residue types depends on the energy term considered, i.e. different numbers of different amino acid related parameters are used for different energy terms. U SCSC uses all 20 amino-acids; parameters for each interacting pair are considered independent. This energy term is the only one fully sequence-dependent. U rot and U b also use all 20 amino acids, however they represent only internal energies within the residue, not residue-residue type of interaction. All other energy terms (which are cumulant-based) use simplified set of 3 amino acids: Ala, Gly and Pro.
30 Good Global Optimization Methods: Common features Find many low-energy local minima with different topologies. Global minimum may not correspond to the native structure, therefore both global minimum and distinct low-energy conformations should be found Consider conformational space of local minima only, same as in the Monte Carlo-Minimization (MCM) Search as large part of conformational space as possible in early stages and then narrows the search to smaller region.
31
32
33
34
")
35 A road to global minimum RMSD from native Energy (kcal/mol)
36 Conformational Space Annealing Finds many low-energy local minima with different topologies. Global minimum may not correspond to the native structure, therefore both global minimum and distinct low-energy conformations should be found Considers conformational space of local minima only, same as in the Monte Carlo-Minimization (MCM) Uses genetic algorithm for structures generation Searches the whole conformational space in early stages and then narrows the search to smaller region.
37 50* random conformations Energy minimization * Arbitrary numbers Conformational Space Annealing First Bank D cut = 1 2 D ave Bank Copy Update Bank & Reduce D cut All used as a seed? No Select 20* seeds Yes Generate 50* random conformations, minimize their energies and add them to both Bank and First Bank Stop Yes No GMEC found? Generate 600 conformations (30* for each seed) by modifying seeds Energy minimization CSA is parallelized at the coarse-grain level minimizations of newly generated structures are carried out in parallel, each minimization on different processor.
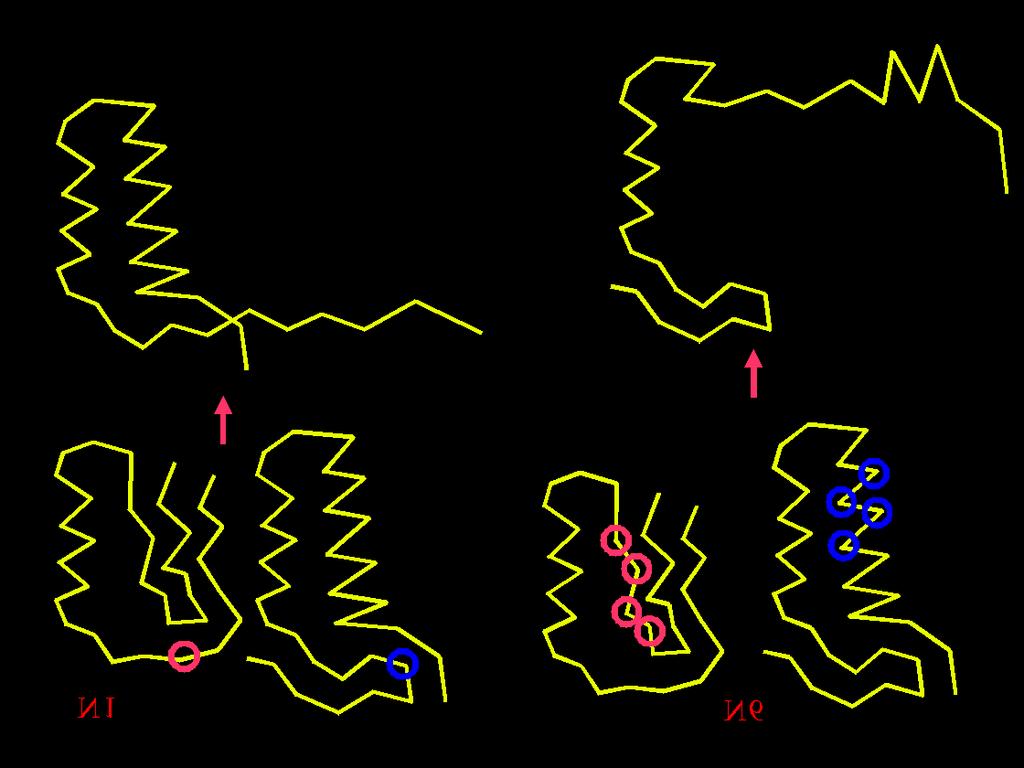
38
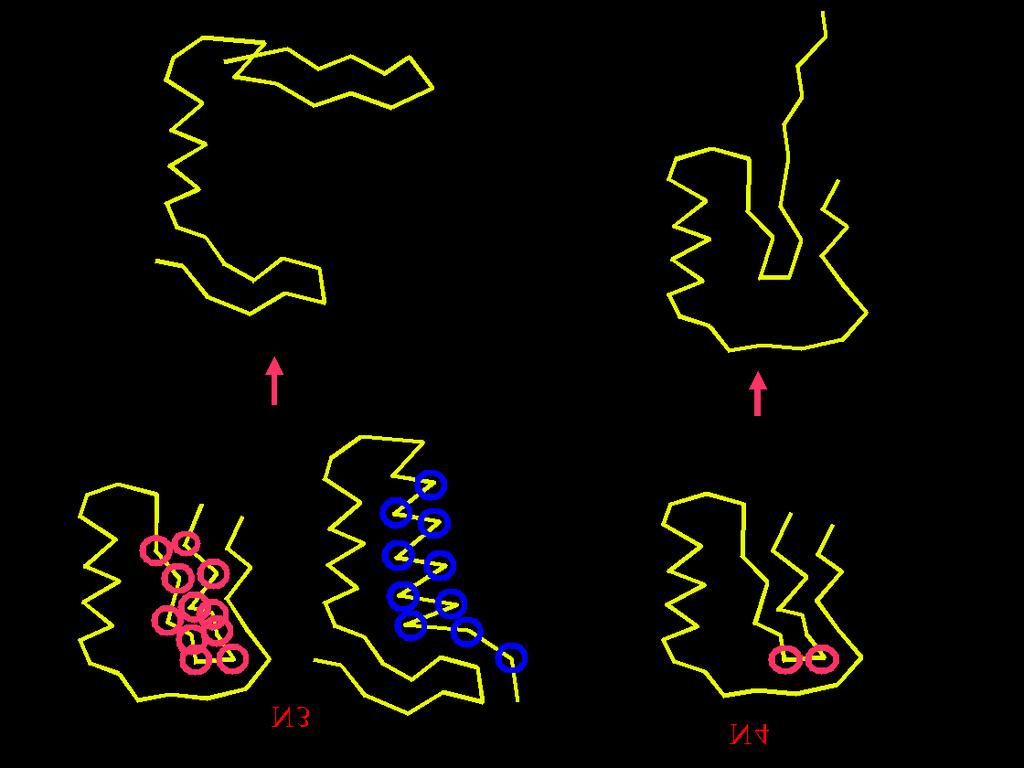
39
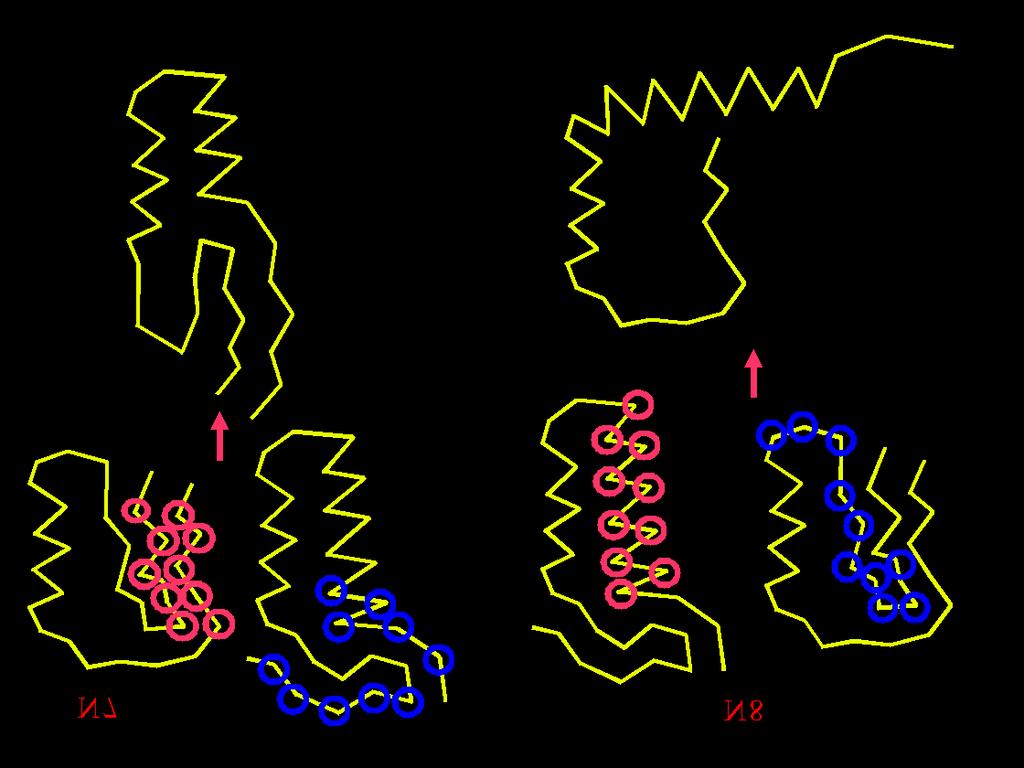
40
41
42 Monte Carlo-Minimization (MCM) Generate at random set of structures and locally minimize them. 1. Select the lowest-energy one as generative structure. 2. Carry out random change (perturbation) of generative structure to produce a new conformation. 3. Minimize the energy of the new conformation. 4. Compare its energy to energy of the generative structure by means of the Metropolis criterion [accepted with probabilty of exp(- E/kT)]. 5. If accepted in Metropolis criterion new (minimized) structure becomes the generative structure, otherwise the generative structure remains unchanged. 6. Iterate into point 3.
43 Conformation-Family Monte Carlo (CFMC) Uses the Metropolis criterion to move between families Uses the Boltzmann distribution to choose conformation from a family Does not move between structures, but between families It is equivalent to smoothed staircase deformation of a potential function Original function MCM CFMC
44 Application to UNRES polypeptide chains: Protein A Family definition was based on RMSD-based clustering (similar to minimal tree clustering method) New structures were produced by various kinds of angle perturbations (large for moves between families and small for improving a given family) and by an averaging The interfamily cut-off varied between 5Å at the beginning to 2 Å at the end of simulation The lowest-energy family was always found
 The lowest and")
 The lowest and the fourth (2.")
45 Representatives of the lowest-energy family (native) The lowest and next-to-the-lowest (3.2Å RMSD) The lowest and the fourth (2.5Å RMSD) Rotate Rotate
46 Three groups of families found by the CFMC Native Rotate Intermediate Rotate Mirror image Rotate

47 The distribution of families found by the CFMC for Protein A Energy RMSD from mirror RMSD from native Rota
 structure of 1FSD RMSD=2.")
48 Superposition of the calculated (yellow) and experimental (red) structure of 1FSD RMSD=2.8 Å
49 PROTEINS: Structure, Function, and Genetics Suppl 3: (1999) AB INITIO: ASSESSMENT Analysis and Assessment of Ab Initio Three-Dimensional Prediction, Secondary Structure, and Contacts Prediction C.A. Orengo, J.E. Bray, T. Hubbard, L. LoConte, and L. Sillitoe Protein HDEA (T61) the most impressive prediction was that of Scheraga s group, 23 using more classical ab initio methods, for which 76 residues could be superposed with an rmsd of 5.3 Å (using the Hubbard method) or 55 residues superposed to 3.81 Å (using the LCS plots of Zemla et al. 14 ) (Fig. 9). Their method uses no information from sequence alignments, secondary structure prediction, or threading.
 HDEA Segment RMSD=2.")
50 CASP3 target T006 (1BG8) HDEA RMSD=4.2 Å for 61 residues (80%, residues 25-85) HDEA Segment RMSD=2.9 Å for 27 residues (36%, residues 16-42)

51 CASP4 Target T0102, RMSD=4.2Å, native=blue, calculated=red AS48, Bacteriocin AS-48, E. faecalis, 70 amino acids, cyclical Rotate
Crystal Structure Prediction using CRYSTALG program
 Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
Crystal Structure Prediction using CRYSTALG program Yelena Arnautova Baker Laboratory of Chemistry and Chemical Biology, Cornell University Problem of crystal structure prediction: - theoretical importance
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Design of a Protein Potential Energy Landscape by Parameter Optimization
 J. Phys. Chem. B 2004, 108, 4525-4534 4525 Design of a Protein Potential Energy Landscape by Parameter Optimization Julian Lee,,, Seung-Yeon Kim, and Jooyoung Lee*, Department of Bioinformatics and Life
J. Phys. Chem. B 2004, 108, 4525-4534 4525 Design of a Protein Potential Energy Landscape by Parameter Optimization Julian Lee,,, Seung-Yeon Kim, and Jooyoung Lee*, Department of Bioinformatics and Life
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University
 Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Assignment 2 Atomic-Level Molecular Modeling
 Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
Assignment 2 Atomic-Level Molecular Modeling CS/BIOE/CME/BIOPHYS/BIOMEDIN 279 Due: November 3, 2016 at 3:00 PM The goal of this assignment is to understand the biological and computational aspects of macromolecular
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Simulating Folding of Helical Proteins with Coarse Grained Models
 366 Progress of Theoretical Physics Supplement No. 138, 2000 Simulating Folding of Helical Proteins with Coarse Grained Models Shoji Takada Department of Chemistry, Kobe University, Kobe 657-8501, Japan
366 Progress of Theoretical Physics Supplement No. 138, 2000 Simulating Folding of Helical Proteins with Coarse Grained Models Shoji Takada Department of Chemistry, Kobe University, Kobe 657-8501, Japan
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation
 Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Energy landscapes of model polyalanines
 JOURNAL OF CHEMICAL PHYSICS VOLUME 117, NUMBER 3 15 JULY 2002 Energy landscapes of model polyalanines Paul N. Mortenson, David A. Evans, and David J. Wales University Chemical Laboratories, Lensfield Road,
JOURNAL OF CHEMICAL PHYSICS VOLUME 117, NUMBER 3 15 JULY 2002 Energy landscapes of model polyalanines Paul N. Mortenson, David A. Evans, and David J. Wales University Chemical Laboratories, Lensfield Road,
Computer simulations of protein folding with a small number of distance restraints
 Vol. 49 No. 3/2002 683 692 QUARTERLY Computer simulations of protein folding with a small number of distance restraints Andrzej Sikorski 1, Andrzej Kolinski 1,2 and Jeffrey Skolnick 2 1 Department of Chemistry,
Vol. 49 No. 3/2002 683 692 QUARTERLY Computer simulations of protein folding with a small number of distance restraints Andrzej Sikorski 1, Andrzej Kolinski 1,2 and Jeffrey Skolnick 2 1 Department of Chemistry,
Universal Similarity Measure for Comparing Protein Structures
 Marcos R. Betancourt Jeffrey Skolnick Laboratory of Computational Genomics, The Donald Danforth Plant Science Center, 893. Warson Rd., Creve Coeur, MO 63141 Universal Similarity Measure for Comparing Protein
Marcos R. Betancourt Jeffrey Skolnick Laboratory of Computational Genomics, The Donald Danforth Plant Science Center, 893. Warson Rd., Creve Coeur, MO 63141 Universal Similarity Measure for Comparing Protein
JOOYOUNG LEE*, ADAM LIWO*, AND HAROLD A. SCHERAGA*
 Proc. Natl. Acad. Sci. USA Vol. 96, pp. 2025 2030, March 1999 Biophysics Energy-based de novo protein folding by conformational space annealing and an off-lattice united-residue force field: Application
Proc. Natl. Acad. Sci. USA Vol. 96, pp. 2025 2030, March 1999 Biophysics Energy-based de novo protein folding by conformational space annealing and an off-lattice united-residue force field: Application
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009
 114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
CS 273 Prof. Serafim Batzoglou Prof. Jean-Claude Latombe Spring Lecture 12 : Energy maintenance (1) Lecturer: Prof. J.C.
 Lecturer: Prof. J.C.") CS 273 Prof. Serafim Batzoglou Prof. Jean-Claude Latombe Spring 2006 Lecture 12 : Energy maintenance (1) Lecturer: Prof. J.C. Latombe Scribe: Neda Nategh How do you update the energy function during the
CS 273 Prof. Serafim Batzoglou Prof. Jean-Claude Latombe Spring 2006 Lecture 12 : Energy maintenance (1) Lecturer: Prof. J.C. Latombe Scribe: Neda Nategh How do you update the energy function during the
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Structure Determination from Pseudocontact Shifts Using ROSETTA
 Supporting Information Protein Structure Determination from Pseudocontact Shifts Using ROSETTA Christophe Schmitz, Robert Vernon, Gottfried Otting, David Baker and Thomas Huber Table S0. Biological Magnetic
Supporting Information Protein Structure Determination from Pseudocontact Shifts Using ROSETTA Christophe Schmitz, Robert Vernon, Gottfried Otting, David Baker and Thomas Huber Table S0. Biological Magnetic
Protein Structure Prediction
 Protein Structure Prediction Michael Feig MMTSB/CTBP 2006 Summer Workshop From Sequence to Structure SEALGDTIVKNA Ab initio Structure Prediction Protocol Amino Acid Sequence Conformational Sampling to
Protein Structure Prediction Michael Feig MMTSB/CTBP 2006 Summer Workshop From Sequence to Structure SEALGDTIVKNA Ab initio Structure Prediction Protocol Amino Acid Sequence Conformational Sampling to
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Programme Last week s quiz results + Summary Fold recognition Break Exercise: Modelling remote homologues
 Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Protein Structure Prediction
 Protein Structure Prediction Michael Feig MMTSB/CTBP 2009 Summer Workshop From Sequence to Structure SEALGDTIVKNA Folding with All-Atom Models AAQAAAAQAAAAQAA All-atom MD in general not succesful for real
Protein Structure Prediction Michael Feig MMTSB/CTBP 2009 Summer Workshop From Sequence to Structure SEALGDTIVKNA Folding with All-Atom Models AAQAAAAQAAAAQAA All-atom MD in general not succesful for real
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Protein Structures. 11/19/2002 Lecture 24 1
 Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Structure Determination
 Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Folding of small proteins using a single continuous potential
 JOURNAL OF CHEMICAL PHYSICS VOLUME 120, NUMBER 17 1 MAY 2004 Folding of small proteins using a single continuous potential Seung-Yeon Kim School of Computational Sciences, Korea Institute for Advanced
JOURNAL OF CHEMICAL PHYSICS VOLUME 120, NUMBER 17 1 MAY 2004 Folding of small proteins using a single continuous potential Seung-Yeon Kim School of Computational Sciences, Korea Institute for Advanced
Protein structure analysis. Risto Laakso 10th January 2005
 Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Exploring the energy landscape
 Exploring the energy landscape ChE210D Today's lecture: what are general features of the potential energy surface and how can we locate and characterize minima on it Derivatives of the potential energy
Exploring the energy landscape ChE210D Today's lecture: what are general features of the potential energy surface and how can we locate and characterize minima on it Derivatives of the potential energy
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
09/06/25. Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Non-uniform distribution of folds. Scheme of protein structure predicition
 Non-uniform distribution of folds. Scheme of protein structure predicition") Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Chemical Shift Restraints Tools and Methods. Andrea Cavalli
 Chemical Shift Restraints Tools and Methods Andrea Cavalli Overview Methods Overview Methods Details Overview Methods Details Results/Discussion Overview Methods Methods Cheshire base solid-state Methods
Chemical Shift Restraints Tools and Methods Andrea Cavalli Overview Methods Overview Methods Details Overview Methods Details Results/Discussion Overview Methods Methods Cheshire base solid-state Methods
A new combination of replica exchange Monte Carlo and histogram analysis for protein folding and thermodynamics
 JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 3 15 JULY 2001 A new combination of replica exchange Monte Carlo and histogram analysis for protein folding and thermodynamics Dominik Gront Department of
JOURNAL OF CHEMICAL PHYSICS VOLUME 115, NUMBER 3 15 JULY 2001 A new combination of replica exchange Monte Carlo and histogram analysis for protein folding and thermodynamics Dominik Gront Department of
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence
 Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Protein dynamics. Folding/unfolding dynamics. Fluctuations near the folded state
 Protein dynamics Folding/unfolding dynamics Passage over one or more energy barriers Transitions between infinitely many conformations B. Ozkan, K.A. Dill & I. Bahar, Protein Sci. 11, 1958-1970, 2002 Fluctuations
Protein dynamics Folding/unfolding dynamics Passage over one or more energy barriers Transitions between infinitely many conformations B. Ozkan, K.A. Dill & I. Bahar, Protein Sci. 11, 1958-1970, 2002 Fluctuations
Protein Modeling. Generating, Evaluating and Refining Protein Homology Models
 Protein Modeling Generating, Evaluating and Refining Protein Homology Models Troy Wymore and Kristen Messinger Biomedical Initiatives Group Pittsburgh Supercomputing Center Homology Modeling of Proteins
Protein Modeling Generating, Evaluating and Refining Protein Homology Models Troy Wymore and Kristen Messinger Biomedical Initiatives Group Pittsburgh Supercomputing Center Homology Modeling of Proteins
AbInitioProteinStructurePredictionviaaCombinationof Threading,LatticeFolding,Clustering,andStructure Refinement
 PROTEINS: Structure, Function, and Genetics Suppl 5:149 156 (2001) DOI 10.1002/prot.1172 AbInitioProteinStructurePredictionviaaCombinationof Threading,LatticeFolding,Clustering,andStructure Refinement
PROTEINS: Structure, Function, and Genetics Suppl 5:149 156 (2001) DOI 10.1002/prot.1172 AbInitioProteinStructurePredictionviaaCombinationof Threading,LatticeFolding,Clustering,andStructure Refinement
Orientational degeneracy in the presence of one alignment tensor.
 Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Contact map guided ab initio structure prediction
 Contact map guided ab initio structure prediction S M Golam Mortuza Postdoctoral Research Fellow I-TASSER Workshop 2017 North Carolina A&T State University, Greensboro, NC Outline Ab initio structure prediction:
Contact map guided ab initio structure prediction S M Golam Mortuza Postdoctoral Research Fellow I-TASSER Workshop 2017 North Carolina A&T State University, Greensboro, NC Outline Ab initio structure prediction:
Introduction to Computational Structural Biology
 Introduction to Computational Structural Biology Part I 1. Introduction The disciplinary character of Computational Structural Biology The mathematical background required and the topics covered Bibliography
Introduction to Computational Structural Biology Part I 1. Introduction The disciplinary character of Computational Structural Biology The mathematical background required and the topics covered Bibliography
Many proteins spontaneously refold into native form in vitro with high fidelity and high speed.
 Macromolecular Processes 20. Protein Folding Composed of 50 500 amino acids linked in 1D sequence by the polypeptide backbone The amino acid physical and chemical properties of the 20 amino acids dictate
Macromolecular Processes 20. Protein Folding Composed of 50 500 amino acids linked in 1D sequence by the polypeptide backbone The amino acid physical and chemical properties of the 20 amino acids dictate
Multi-Scale Hierarchical Structure Prediction of Helical Transmembrane Proteins
 Multi-Scale Hierarchical Structure Prediction of Helical Transmembrane Proteins Zhong Chen Dept. of Biochemistry and Molecular Biology University of Georgia, Athens, GA 30602 Email: zc@csbl.bmb.uga.edu
Multi-Scale Hierarchical Structure Prediction of Helical Transmembrane Proteins Zhong Chen Dept. of Biochemistry and Molecular Biology University of Georgia, Athens, GA 30602 Email: zc@csbl.bmb.uga.edu
Presenter: She Zhang
 Presenter: She Zhang Introduction Dr. David Baker Introduction Why design proteins de novo? It is not clear how non-covalent interactions favor one specific native structure over many other non-native
Presenter: She Zhang Introduction Dr. David Baker Introduction Why design proteins de novo? It is not clear how non-covalent interactions favor one specific native structure over many other non-native
Soot - Developing anisotropic potentials from first principles for PAH molecules. Tim Totton, Alston Misquitta and Markus Kraft 12/11/2009
 Soot - Developing anisotropic potentials from first principles for PAH molecules. Tim Totton, Alston Misquitta and 12/11/2009 HRTEM images of soot Some evidence for different soot structures based on different
Soot - Developing anisotropic potentials from first principles for PAH molecules. Tim Totton, Alston Misquitta and 12/11/2009 HRTEM images of soot Some evidence for different soot structures based on different
Improving De novo Protein Structure Prediction using Contact Maps Information
 CIBCB 2017 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology Improving De novo Protein Structure Prediction using Contact Maps Information Karina Baptista dos Santos
CIBCB 2017 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology Improving De novo Protein Structure Prediction using Contact Maps Information Karina Baptista dos Santos
Protein Folding & Stability. Lecture 11: Margaret A. Daugherty. Fall How do we go from an unfolded polypeptide chain to a
 Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2004 How do we go from an unfolded polypeptide chain to a compact folded protein? (Folding of thioredoxin, F. Richards) Structure - Function
Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2004 How do we go from an unfolded polypeptide chain to a compact folded protein? (Folding of thioredoxin, F. Richards) Structure - Function
Molecular Mechanics. I. Quantum mechanical treatment of molecular systems
 Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Molecular Mechanics I. Quantum mechanical treatment of molecular systems The first principle approach for describing the properties of molecules, including proteins, involves quantum mechanics. For example,
Supplemental Materials for. Structural Diversity of Protein Segments Follows a Power-law Distribution
 Supplemental Materials for Structural Diversity of Protein Segments Follows a Power-law Distribution Yoshito SAWADA and Shinya HONDA* National Institute of Advanced Industrial Science and Technology (AIST),
Supplemental Materials for Structural Diversity of Protein Segments Follows a Power-law Distribution Yoshito SAWADA and Shinya HONDA* National Institute of Advanced Industrial Science and Technology (AIST),
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Procheck output. Bond angles (Procheck) Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.
 Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.") Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Supplementary Figures:
 Supplementary Figures: Supplementary Figure 1: The two strings converge to two qualitatively different pathways. A) Models of active (red) and inactive (blue) states used as end points for the string calculations
Supplementary Figures: Supplementary Figure 1: The two strings converge to two qualitatively different pathways. A) Models of active (red) and inactive (blue) states used as end points for the string calculations
arxiv: v1 [cond-mat.soft] 22 Oct 2007
![arxiv: v1 [cond-mat.soft] 22 Oct 2007](/thumbs/78/78401876.jpg "arxiv: v1 [cond-mat.soft] 22 Oct 2007") Conformational Transitions of Heteropolymers arxiv:0710.4095v1 [cond-mat.soft] 22 Oct 2007 Michael Bachmann and Wolfhard Janke Institut für Theoretische Physik, Universität Leipzig, Augustusplatz 10/11,
Conformational Transitions of Heteropolymers arxiv:0710.4095v1 [cond-mat.soft] 22 Oct 2007 Michael Bachmann and Wolfhard Janke Institut für Theoretische Physik, Universität Leipzig, Augustusplatz 10/11,
PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS
 TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
Molecular Interactions F14NMI. Lecture 4: worked answers to practice questions
 Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Molecular Interactions F14NMI Lecture 4: worked answers to practice questions http://comp.chem.nottingham.ac.uk/teaching/f14nmi jonathan.hirst@nottingham.ac.uk (1) (a) Describe the Monte Carlo algorithm
Conformational Sampling in Template-Free Protein Loop Structure Modeling: An Overview
 # of Loops, http://dx.doi.org/10.5936/csbj.201302003 CSBJ Conformational Sampling in Template-Free Protein Loop Structure Modeling: An Overview Yaohang Li a,* Abstract: Accurately modeling protein loops
# of Loops, http://dx.doi.org/10.5936/csbj.201302003 CSBJ Conformational Sampling in Template-Free Protein Loop Structure Modeling: An Overview Yaohang Li a,* Abstract: Accurately modeling protein loops
ICCP Project 2 - Advanced Monte Carlo Methods Choose one of the three options below
 ICCP Project 2 - Advanced Monte Carlo Methods Choose one of the three options below Introduction In statistical physics Monte Carlo methods are considered to have started in the Manhattan project (1940
ICCP Project 2 - Advanced Monte Carlo Methods Choose one of the three options below Introduction In statistical physics Monte Carlo methods are considered to have started in the Manhattan project (1940
Lecture 11: Protein Folding & Stability
 Structure - Function Protein Folding: What we know Lecture 11: Protein Folding & Stability 1). Amino acid sequence dictates structure. 2). The native structure represents the lowest energy state for a
Structure - Function Protein Folding: What we know Lecture 11: Protein Folding & Stability 1). Amino acid sequence dictates structure. 2). The native structure represents the lowest energy state for a
Protein Folding & Stability. Lecture 11: Margaret A. Daugherty. Fall Protein Folding: What we know. Protein Folding
 Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2003 Structure - Function Protein Folding: What we know 1). Amino acid sequence dictates structure. 2). The native structure represents
Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2003 Structure - Function Protein Folding: What we know 1). Amino acid sequence dictates structure. 2). The native structure represents
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Course Notes: Topics in Computational. Structural Biology.
 Course Notes: Topics in Computational Structural Biology. Bruce R. Donald June, 2010 Copyright c 2012 Contents 11 Computational Protein Design 1 11.1 Introduction.........................................
Course Notes: Topics in Computational Structural Biology. Bruce R. Donald June, 2010 Copyright c 2012 Contents 11 Computational Protein Design 1 11.1 Introduction.........................................
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling lecture 2
 Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
Limitations of temperature replica exchange (T-REMD) for protein folding simulations
 for protein folding simulations") Limitations of temperature replica exchange (T-REMD) for protein folding simulations Jed W. Pitera, William C. Swope IBM Research pitera@us.ibm.com Anomalies in protein folding kinetic thermodynamic 322K
Limitations of temperature replica exchange (T-REMD) for protein folding simulations Jed W. Pitera, William C. Swope IBM Research pitera@us.ibm.com Anomalies in protein folding kinetic thermodynamic 322K
Molecular Structure Prediction by Global Optimization
 Molecular Structure Prediction by Global Optimization K.A. DILL Department of Pharmaceutical Chemistry, University of California at San Francisco, San Francisco, CA 94118 A.T. PHILLIPS Computer Science
Molecular Structure Prediction by Global Optimization K.A. DILL Department of Pharmaceutical Chemistry, University of California at San Francisco, San Francisco, CA 94118 A.T. PHILLIPS Computer Science
LOCAL MINIMA HOPPING ALONG THE PROTEIN ENERGY SURFACE
 LOCAL MINIMA HOPPING ALONG THE PROTEIN ENERGY SURFACE by Brian Olson A Thesis Submitted to the Graduate Faculty of George Mason University In Partial Fulfillment of The Requirements for the Degree of Master
LOCAL MINIMA HOPPING ALONG THE PROTEIN ENERGY SURFACE by Brian Olson A Thesis Submitted to the Graduate Faculty of George Mason University In Partial Fulfillment of The Requirements for the Degree of Master
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
 Title Theory of solutions in the energy r of the molecular flexibility Author(s) Matubayasi, N; Nakahara, M Citation JOURNAL OF CHEMICAL PHYSICS (2003), 9702 Issue Date 2003-11-08 URL http://hdl.handle.net/2433/50354
Title Theory of solutions in the energy r of the molecular flexibility Author(s) Matubayasi, N; Nakahara, M Citation JOURNAL OF CHEMICAL PHYSICS (2003), 9702 Issue Date 2003-11-08 URL http://hdl.handle.net/2433/50354
Protein Folding Prof. Eugene Shakhnovich
 Protein Folding Eugene Shakhnovich Department of Chemistry and Chemical Biology Harvard University 1 Proteins are folded on various scales As of now we know hundreds of thousands of sequences (Swissprot)
Protein Folding Eugene Shakhnovich Department of Chemistry and Chemical Biology Harvard University 1 Proteins are folded on various scales As of now we know hundreds of thousands of sequences (Swissprot)
Crystal structure prediction is one of the most challenging and
 Conformation-family Monte Carlo: A new method for crystal structure prediction Jaroslaw Pillardy, Yelena A. Arnautova, Cezary Czaplewski, Kenneth D. Gibson, and Harold A. Scheraga Baker Laboratory of Chemistry
Conformation-family Monte Carlo: A new method for crystal structure prediction Jaroslaw Pillardy, Yelena A. Arnautova, Cezary Czaplewski, Kenneth D. Gibson, and Harold A. Scheraga Baker Laboratory of Chemistry
Prediction and refinement of NMR structures from sparse experimental data
 Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
Lecture 11: Potential Energy Functions
 Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Lecture 11: Potential Energy Functions Dr. Ronald M. Levy ronlevy@temple.edu Originally contributed by Lauren Wickstrom (2011) Microscopic/Macroscopic Connection The connection between microscopic interactions
Advanced Molecular Dynamics
 Advanced Molecular Dynamics Introduction May 2, 2017 Who am I? I am an associate professor at Theoretical Physics Topics I work on: Algorithms for (parallel) molecular simulations including GPU acceleration
Advanced Molecular Dynamics Introduction May 2, 2017 Who am I? I am an associate professor at Theoretical Physics Topics I work on: Algorithms for (parallel) molecular simulations including GPU acceleration
SUPPLEMENTAL MATERIAL
 SUPPLEMENTAL MATERIAL Systematic Coarse-Grained Modeling of Complexation between Small Interfering RNA and Polycations Zonghui Wei 1 and Erik Luijten 1,2,3,4,a) 1 Graduate Program in Applied Physics, Northwestern
SUPPLEMENTAL MATERIAL Systematic Coarse-Grained Modeling of Complexation between Small Interfering RNA and Polycations Zonghui Wei 1 and Erik Luijten 1,2,3,4,a) 1 Graduate Program in Applied Physics, Northwestern
Template-Based Modeling of Protein Structure
 Template-Based Modeling of Protein Structure David Constant Biochemistry 218 December 11, 2011 Introduction. Much can be learned about the biology of a protein from its structure. Simply put, structure
Template-Based Modeling of Protein Structure David Constant Biochemistry 218 December 11, 2011 Introduction. Much can be learned about the biology of a protein from its structure. Simply put, structure
Molecular dynamics simulations of anti-aggregation effect of ibuprofen. Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov
 Biophysical Journal, Volume 98 Supporting Material Molecular dynamics simulations of anti-aggregation effect of ibuprofen Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov Supplemental
Biophysical Journal, Volume 98 Supporting Material Molecular dynamics simulations of anti-aggregation effect of ibuprofen Wenling E. Chang, Takako Takeda, E. Prabhu Raman, and Dmitri Klimov Supplemental
Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization
 Biophysical Journal Volume 101 November 2011 2525 2534 2525 Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization Dong Xu and Yang Zhang
Biophysical Journal Volume 101 November 2011 2525 2534 2525 Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization Dong Xu and Yang Zhang
Monte Carlo (MC) Simulation Methods. Elisa Fadda
 Simulation Methods. Elisa Fadda") Monte Carlo (MC) Simulation Methods Elisa Fadda 1011-CH328, Molecular Modelling & Drug Design 2011 Experimental Observables A system observable is a property of the system state. The system state i is
Monte Carlo (MC) Simulation Methods Elisa Fadda 1011-CH328, Molecular Modelling & Drug Design 2011 Experimental Observables A system observable is a property of the system state. The system state i is
Molecular Modelling. part of Bioinformatik von RNA- und Proteinstrukturen. Sonja Prohaska. Leipzig, SS Computational EvoDevo University Leipzig
 part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
Simulation of mutation: Influence of a side group on global minimum structure and dynamics of a protein model
 JOURNAL OF CHEMICAL PHYSICS VOLUME 111, NUMBER 8 22 AUGUST 1999 Simulation of mutation: Influence of a side group on global minimum structure and dynamics of a protein model Benjamin Vekhter and R. Stephen
JOURNAL OF CHEMICAL PHYSICS VOLUME 111, NUMBER 8 22 AUGUST 1999 Simulation of mutation: Influence of a side group on global minimum structure and dynamics of a protein model Benjamin Vekhter and R. Stephen
TASSER: An Automated Method for the Prediction of Protein Tertiary Structures in CASP6
 PROTEINS: Structure, Function, and Bioinformatics Suppl 7:91 98 (2005) TASSER: An Automated Method for the Prediction of Protein Tertiary Structures in CASP6 Yang Zhang, Adrian K. Arakaki, and Jeffrey
PROTEINS: Structure, Function, and Bioinformatics Suppl 7:91 98 (2005) TASSER: An Automated Method for the Prediction of Protein Tertiary Structures in CASP6 Yang Zhang, Adrian K. Arakaki, and Jeffrey
All-atom Molecular Mechanics. Trent E. Balius AMS 535 / CHE /27/2010
 All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
All-atom Molecular Mechanics Trent E. Balius AMS 535 / CHE 535 09/27/2010 Outline Molecular models Molecular mechanics Force Fields Potential energy function functional form parameters and parameterization
Protein Structure Analysis with Sequential Monte Carlo Method. Jinfeng Zhang Computational Biology Lab Department of Statistics Harvard University
 Protein Structure Analysis with Sequential Monte Carlo Method Jinfeng Zhang Computational Biology Lab Department of Statistics Harvard University Introduction Structure Function & Interaction Protein structure
Protein Structure Analysis with Sequential Monte Carlo Method Jinfeng Zhang Computational Biology Lab Department of Statistics Harvard University Introduction Structure Function & Interaction Protein structure
StructuralBiology. October 18, 2018
 StructuralBiology October 18, 2018 1 Lecture 18: Computational Structural Biology CBIO (CSCI) 4835/6835: Introduction to Computational Biology 1.1 Overview and Objectives Even though we re officially moving
StructuralBiology October 18, 2018 1 Lecture 18: Computational Structural Biology CBIO (CSCI) 4835/6835: Introduction to Computational Biology 1.1 Overview and Objectives Even though we re officially moving
Energy Minimization of Protein Tertiary Structure by Parallel Simulated Annealing using Genetic Crossover
 Minimization of Protein Tertiary Structure by Parallel Simulated Annealing using Genetic Crossover Tomoyuki Hiroyasu, Mitsunori Miki, Shinya Ogura, Keiko Aoi, Takeshi Yoshida, Yuko Okamoto Jack Dongarra
Minimization of Protein Tertiary Structure by Parallel Simulated Annealing using Genetic Crossover Tomoyuki Hiroyasu, Mitsunori Miki, Shinya Ogura, Keiko Aoi, Takeshi Yoshida, Yuko Okamoto Jack Dongarra
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations
 Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations Alexandre V. Morozov, Tanja Kortemme, Kiril Tsemekhman, David Baker
Protein structure forces, and folding
 Harvard-MIT Division of Health Sciences and Technology HST.508: Quantitative Genomics, Fall 2005 Instructors: Leonid Mirny, Robert Berwick, Alvin Kho, Isaac Kohane Protein structure forces, and folding
Harvard-MIT Division of Health Sciences and Technology HST.508: Quantitative Genomics, Fall 2005 Instructors: Leonid Mirny, Robert Berwick, Alvin Kho, Isaac Kohane Protein structure forces, and folding