Protein Secondary Structure Assignment and Prediction
|
|
|
- Aldous Elvin Craig
- 5 years ago
- Views:
Transcription
 STRIDE (Frishman & Argos, 1995) Continuum - PCE: Protein Continuum Electrostatics (Andersen et al.")
1 1 Protein Secondary Structure Assignment and Prediction Defining SS features - Dihedral angles, alpha helix, beta stand (Hydrogen bonds) Assigned manually by crystallographers or Automatic DSSP (Kabsch & Sander,1983) STRIDE (Frishman & Argos, 1995) Continuum - PCE: Protein Continuum Electrostatics (Andersen et al.) Structure Prediction methods Dihedral Angles From ϕ phi - dihedral angle about the N-Cα bond ψ psi - dihedral angle about the Cα-C bond ω - dihedral angle about the C-N (peptide) bond
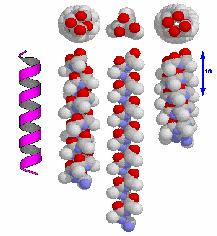
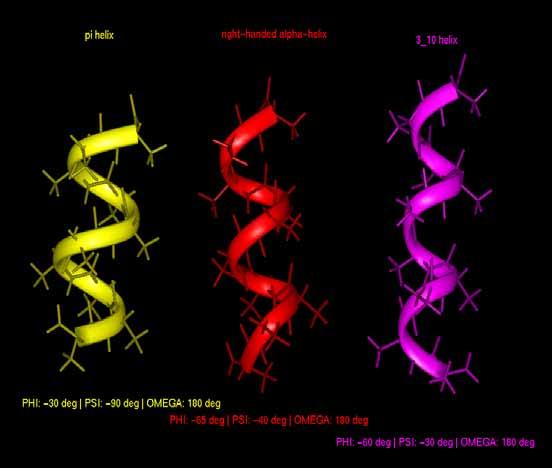
2 α Helix phi(deg) psi(deg) H-bond pattern right-handed alpha-helix i+4 π-helix i helix i+3 (ω is 180 deg in all cases) 2 π-helix α-helix 3/10helix α-helix 3/10helix π-helix

 and green lines (parallel strands) connecting the")

3 3 Beta Strand phi(deg) psi(deg) omega (deg) beta strand Hydrogen bond patterns in beta sheets. Here a four-stranded beta sheet is drawn schematically which contains three anti-parallel and one parallel strand. Hydrogen bonds are indicated with red lines (anti-parallel strands) and green lines (parallel strands) connecting the hydrogen and receptor oxygen. Secondary Structure Types * H = alpha helix * B = residue in isolated beta-bridge * E = extended strand, participates in beta ladder * G = 3-helix (3/10 helix) * I = 5 helix (p helix) * T = hydrogen bonded turn * S = bend Automatic assignment programs
 Neural Networks (different inputs) Raw Sequence (late 80 s) Blosum matrix (e.g., PhD, early 90 s) Position specific alignment profiles (e.")
4 DSSP ( ) STRIDE ( 4 Secondary Structure Prediction What to predict? All the 8 types of structures Simple alignments Heuristic Methods (e.g., Chou-Fasman, 1974) Neural Networks (different inputs) Raw Sequence (late 80 s) Blosum matrix (e.g., PhD, early 90 s) Position specific alignment profiles (e.g., PsiPred, late 90 s) Multiple networks balloting, probability conversion, output expansion Improvement of accuracy 1974 Chou & Fasman ~50-53% 1978 Garnier 63% 1987 Zvelebil 66% 1988 Quian & Sejnowski 64.3% 1993 Levine 69% 1994 Rost & Sander % 1997 Frishman & Argos <75% 1999 Cuff & Barton 72.9% 1999 Jones 76.5% 2000 Petersen et al. 77.9% Simple Alignments Solved structures homologous to query needed
5 Homologous proteins have ~88% identical (3 state) secondary structure If no homologue can be identified alignment will give almost random results 5 Chou-Fasman based on the propensities of amino acids to adopt secondary structures based on the observation of their location in 15 protein structures determined by X-ray diffraction - these statistics derive from the particular stereochemical and physicochemical properties of the amino acids. See for example, glycine and proline. These statistics have been refined over the years by a number of authors (including Chou and Fasman themselves) using a larger set of proteins. Rather than a position by position analysis the propensity of a position is calculated using an average over 5 or 6 residues surrounding each position. On a larger set of 62 proteins the base method reports a success rate of 50%. Propensity is defined by where i the 20 aa types, fiϕ Pi ϕ = [1] f ϕ ϕ conformations - α helix, β strand and coiled coil f ϕ denotes the fraction of all the aa (20 types) belong to the ϕ th type SS, and f iϕ denotes the fraction of the ith type aa belongs to the ϕth type SS, that is ni ϕ Nϕ f iϕ = and fϕ =, N i N T N i, N ϕ = the number of type i and ϕ th aa, n iϕ = the number of type i aa belonging to the ϕ th type SS, N T = the total number of aa.

6 6 H strong, h intermediate, I weak, I insensitive, b break, B strongly breaking
, f(i+1), f(i+2) and f(i+3), which corresponding to the frequency with which the aa was observed in the first, second, third and fourth")
7 Chou-Fasman parameters for the 20 amino acids 7 - each aa is assigned several conformational parameters, P(a), P(b), and P(turn) - represent the propensity of each aa to participate in alpha, beta sheet and turns - each aa is assign 4 turn parameters, f(i), f(i+1), f(i+2) and f(i+3), which corresponding to the frequency with which the aa was observed in the first, second, third and fourth position of a hairpin turn - The algorithm for assigning secondary structure proceeds as follows: - Identify alpha - Identify beta - Identify turns (a) for each residue i, calculate the turn propensity, P(t) = f(i)*f(i+1)*f(i+2)*f(i+3) (b) predict a hairpin turn starting at each position i that satisfies the following criteria: (i) P(t) > 7.5 e-5 (ii) The average P(turn) value for the four aa at positions i through i+3 > 100 (iii) a) P( turn) > P ( < P( b) over the four aa in position i through i+3 Example Given a protein seq. CAENKLDHVADCC, use the Chou-Fashman method to predict the turn. aa P(t) Average P(turn) Average P(a) Average P(b) Is it a turn? C = 0.149*0.076*0.077* No = 7.9e-5 A = 0.06*0.06*0.191* No = 6.5e-5 E N K
8 8 GOR method 1978 Garnier, Osguthorpe and Robson (GOR) introduce a more sophisticate method and defined an information measure Eq.[2] is same as Eq.[1], Bayes Rule I(S;a) = log[p(s a)/p(s)] [2] I(S;a) = log[p(s,a)/(p(s)p(a))] [3] where S is secondary structure, a is amino acid, P(S a) is the conditional probability of conformation S given residue a, and P(a) is the probability of a.a. a, and P(S) is the probability of conformation S. I( S; a) niϕ Nϕ = log( / ) same as Eq.[1] N N i GOR define information gain, I( S;a), in order to avoid data size and sampling variation; Substitute Eq. [3] into [4], one obtained T I( S;a) = I(S;a) - I(not S;a) [4] P( S, a) 1 P( S) I( S; a) = log[ ] + log[ ] [5] 1 P( S, a) P( S) where the probability of finding a.a. a not in conformation S is 1-P(S,a), and of not finding any aa in conformation S is 1-P(S). GOR method considered a sliding window of size 17 with a.a. a X in the center, and considered the effects of the eight a.a. on the left and right hand sides of a.a. a X, hence values for I( S ϕ ;a X ) in Eq.[5] is calculated according to I ( Sϕ, a X ) I( Sϕ, a X + i ) 8 i= 8 where ϕ is the conformations, and predict state with highest gain. Amino acid preferences in α-helix Amino acid preferences in β-strand
9 9 Amino acid preferences in coil Garnier improved the method by using statistically significant pair-wise interactions as a determinant of the statistical significance. This improved the success rate to 62% Levin improved the prediction level by using MSA. The reasoning is as follows. Conserved regions in a MSA provides a strong evolutionary indicator of a role in the function of the protein. Those regions are also likely to have conserved structure, including secondary structure and strengthen the prediction by their joint propensities. This improved the success rate to 69% , 1994 Rost and Sander combined neural networks with MSA. The idea of a neural net is to create a complex network of interconnected nodes, where progress from one node to the next depends on satisfying a weighted function that has been derived by training the net with data of known results, in this case protein sequences with known secondary structures. The success rate is 72%. Neural Network Methods such as PHD, Pred2ary PHD combine MSA and the optimization strength of the Neural Network formulism


10 10 Consensus Approach Jpred at EMBL - it runs prediction methods such as PHD, PREDATOR, DSC, NNSSP, ZPRED and MULPRED - if sufficient methods predict an identical SSE for the position, that structure is taken as the consensus prediction for the position - if no consensus reached, the PHD predication is taken
11 11 Summary and Current Statue of SS prediction - Various different induction techniques over same data, give modest improvements Linear discriminant Analysis (LDA) Decision trees Neural networks - GOR method improves 8-9% points (to about 64% correct residue by residue). - Similar improvement for NNs (to ~ 68%) - Prediction quality has not improved much even with huge growth of training data. - Secondary structure is not completely determined by local forces Long distance interactions do not appear in sliding window Secondary Structure Prediction Tools ProfPHD - PSIPRED - BCM PSSP - PedictProtein - o PHDsec, PHDacc, PHDhtm, PHDtopology, PHDthreader, MaxHom, EvalSec from Columbia University HNN - o Hierarchical Neural Network method (Guermeur, 1997 Jpred - o A consensus method for protein secondary structure prediction at EBI GORIV - o GOR Secondary Structure Prediction (Garnier et.al.,1996)
12 Threading (sequence-structure alignment approach) 12 based on the observation that many protein structures in the PDB are very similar. For example, there are many 4-helical bundles, TIM barrels, globins, etc. in the set of solved structures it is conjectured there are only a limited number of unique protein folds in nature Estimates that are < 1000 different protein folds Methodology ii- instead of aligning a sequence to a sequence, we align a sequence to a string of descriptors that describe the 3D environment of the target structure - for each residue position in the structure, we determine: - how buried it is - the fraction of surrounding environment that is polar - the local secondary structure (α-helix, β-sheet or other) - For each position in the structure, we categorize it into one of 18 environment classes using these characteristics (see Figure 15.3) Area Buried Very buried, Highly polar Exposed Fraction of polar - the key observation is that different amino acids prefer different environments
13 13 - For all proteins, we can tabulate the number of times we see a particular residue in a particular environment class, and use this to compute a score for each environment class and each amino acid pair. In particular, we compute a log-odds score of This gives us an 18x20 table as follows: - build a 3D profile for a particular structure using this table. Namely, for each position in our structure, we determine its environment class, and the score of a particular amino acid in this position depends on the table we built above. Thus, for example, if the first position in our structure has environment class B 1 β, the score of having a tyrosine (Y) in that position is Thus, for example, if there are n positions in our structure, we build a table as follows (the gap penalties are chosen to discourage gaps in positions within α-helices and β-sheets); Then to align a sequence s with a structure, we align the sequence with the descriptors of the 3D environment of the target structure. To find the best alignment, we use a 2D dynamic programming matrix as for regular sequence alignment: Thus, to use the 3D profile method for fold recognition, for a particular sequence we calculate its score (using dynamic programming) for all structures. Significance of a score for a particular structure is given by scoring a large sequence database against the structure and calculating where µ is the mean score for that structure, and s is the standard deviation of the scores. The advantages of the 3D profile method over regular sequence alignment is that environmental tendencies may be more informative than simple amino acid similarity, and that structural information is actually used. Additionally, this is a fast method with reasonably good performance. The major disadvantage of this method is that it assumes independence between all positions in the structure.
14 14 My research proposal Knowledge based fold recognition method relies on the extraction of statistical parameters from an experimental determined protein structure database and it has demonstrated some successes. Protein Structure Learning Data Set and Database Construction A set of protein structures will be selected from the DSSP database by referring to the SCOP classes as our input data set. In the SCOP database, proteins are classified in a hierarchy according to their evolutionary origin and structural similarity. There are >25000 PDB entries and >55000 domains. According to the SCOP release 1.65, the number of protein folds, superfamilies and families for the all-α, all-β, and alpha and beta proteins (α/β, α+β) are given by the table below; We will extract the environment information, for example, the residue solvent accessible surface areas (buried (B), partly buried (PB) and exposed (E)), number of hydrogen bondings and secondary structures (such as α-helix, β-sheet and coil) data from the DSSP database. For instance, one can use the following nine environment classes, that is (B, PB, E) α,β, where c stands for the coil region. The DSSP database utilizes the DSSP program to define secondary structure, geometrical features and solvent accessible area of proteins given atomic coordinates in PDB. Figure 1 is part of the secondary structure definition by the program DSSP for deoxyhemoglobin protein (1A00).



15 15 Fig. 1 Part of the secondary structure definition by the program DSSP for deoxyhemoglobin protein (1A00). We use the CGI language, PHP, to extract the environment information from DSSP. Embedding the PHP results in HTML, we have developed a web base interface for data retrieval, and using the molecular graphic tool, Rasmol, for protein 3D structure demonstration (Figure 2). Fig 2 Part of environment information for protein, 1A00, from DSSP and the 3D structure view. 3D structure view of protein, 1A00, using molecule graphics tool RasMol.

16 16 Fig. 3 Environment data for protein, 1A00, where res_sum, aa, rsas, sec_summary, three_turns, four_turns and five_turns represent the residue number, amino acid type, residue solvent accessible area surface, secondary structure summary, three helix, four helix and five helix respectively. Structure profile method (a) 3D profile method and residue environment The 3D profile method uses structural information. Instead of doing sequences alignment, the 3D profile method align a sequence to a string of descriptors that describe the 3D environment of the target structure. That is for each residue position in the structure we determine: solvent accessible surface area (buried, partly buried or exposed), the local secondary structure (α-helix, β-sheet and coil), and the fraction of surrounding environment that is polar (O and N) The basic assumption of this method is that the environment of a particular residue is expected to be more conserve than the actual residue itself, and so the method is able to detect more distant sequence-structure relationship than purely sequence-based method. (b) Residues environment score matrix The probability, P(i,m), associated with residue i in an environment m (in our study it is the solvent accessible area A) is given by P ( i, m) = n( i, m) / N( i) where n(i,m) is the number of residue i with solvent accessible area A, and N(i) is the total number of residue i. For instance, one can compute the probability of having residue i s solvent accessible surface area in a buried, partly buried or exposed environment with one of the three secondary structures. The cutoffs used in defining buried (B), partly buried (PB) and exposed (E) are taken to be 0-10%, 11-40% and >40% of the maximal accessibility for the residues. Thus, each position i the 3D protein structure is assigned to one of nine environment classes. Our study will consider four classes of proteins: all-α protein, all-β protein, α+β protein and α/β
17 17 protein (multi-domain proteins, membrane and cell surface proteins and small proteins will also be considered in case of need), which belong to the SCOP database. The residue solvent accessible surface area and secondary structures data are retrieved from the DSSP database. The matrix score element of the 3D structure profile, M ij, for environment class i and residue j is given by; M ij = ln P P( residue j in environment i) ( residue j in any environment) The denominator is obtained from the residues frequency in the DSSP database, where j is one of the nine environment classes; (B, PB, E) α,β,c. Given the scoring matrix for a class of protein family, we can build a 3D profile for a particular structure using this matrix. That is, for each position in the known protein structure, one can determine its environment class, and the score of a particular residue in this position is given by the score matrix value. As an illustration, we had computed the 3D profile for sperm whale myoglobin sequences, which belong to the globins protein family according to SCOP classification scheme. We consider three environment classes in our preliminary study and more classes will be consider in order to model the residue environment more precisely. This result is tabular in Table 1. Table 1. Environment classes (E, PB, E) and score value, S ij, of the sperm whale myoglobin sequences in comparison with the hydrophobicity KD scale. Residue Buried (B) Partly buried (PB) Exposed (E) KD scale I V L F C M A G T S W x -0.9 Y P x H N x D x E x Q K R
18 18 A large negative score value indicates a strong preference for the particular environment whereas large positive score value indicates an aversion. It is evident from the Table 1 that residues P, N, D, and E were not found at the buried state. Furthermore, residues Q, K and R all have a positive score, which is an indication of aversion, hence, these are the polar residues. Similarly, residues I, V, L, F and M are found to prefer reside in the buried state by examining the exposed column score values (a large positive or small negative value) in Table 1, these are the hydrophobic residues. These two conclusions are well consistent with the experimental determined hydrophobicity KD scale results given in the last column in Table 1. Figure 4. Environment classes (E, PB, E) and score value, S ij, of the sperm whale myoglobin sequences Given the score matrix (Table 1) one can build a 3D profile for a particular structure using this score matrix. For each position in the structure we determine its environment class and the score value of a particular residue in this position depends on the score matrix we built. For instance, if the first position in our structure has the environment class buried, the score of having residue K in that position is Thus, if there are n residues in the structure, we can build a profile for the known protein structure. For example, one can construct the following chart, Position in fold Environment class.. Q K R 1 B 0.7 (5.07) PB


19 19 To align a sequence with a structure, one aligns the sequence with the descriptors of the 3D environment of the known protein structure using the dynamic programming algorithm in order to find the optimal alignment. SSE prediction servers Network Protein Sequence Analysis Meta-servers Integrate predictions from several other servers Significantly better predictions than any individual approach provides access to various fold recognition and local structure prediction methods Fold Recognition and Structure Predication Tools UCLA-DOE Protein 3D-Structure Prediction Server



20 20 3D-PSSM o Protein fold recognition using 1D and 3D sequence profiles coupled with secondary structure information (Foldfit) 123D o combines sequence profiles, secondary structure prediction, and contact capacity potentials to thread a protein sequence through the set of structures FFAS o Fold assignment method is based on the profile-profile matching algorithm
21 Online Hypertext Book A Guide to Structure Prediction (ICRF) Protein Secondary Structure Prediction with Neural Networks: A Tutorial (UCL) BioComputing Hypertext Coursebook (VSNS-BCD, Germany) Coils-(Lupa s method) o Paircoil- (Berger's method) o Multicoil - Prediction of two- and three-stranded coiled coils o 21 References Mona Singh, Lecture 15 The Threading Approach to Tertiary Structure Prediction.
22 22 Transmembrane Segments - form a distinct topological class due to the presence of one or more transmembrane seq. segments - the transmembrane segments are subject to severe restrictions imposed by the lipid bilayer of the cell membrane Coiled-coil structures
23 23 Protein Secondary Structure Assignment and Prediction - Assignment (1) Proof Equation 5. (2) Given a protein seq. CAENKLDHVADCC, use the Chou-Fashman method to predict the turn. Fill in the boxes for the residues E, N and K. aa P(t) Average P(turn) Average P(a) Average P(b) Is it a turn? C = 0.149*0.076*0.077* No = 7.9e-5 A = 0.06*0.06*0.191* No = 6.5e-5 E N K
Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction. Sepp Hochreiter
 Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction Institute of Bioinformatics Johannes Kepler University, Linz, Austria Chapter 4 Protein Secondary
Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction Institute of Bioinformatics Johannes Kepler University, Linz, Austria Chapter 4 Protein Secondary
Protein Secondary Structure Prediction
 Protein Secondary Structure Prediction Doug Brutlag & Scott C. Schmidler Overview Goals and problem definition Existing approaches Classic methods Recent successful approaches Evaluating prediction algorithms
Protein Secondary Structure Prediction Doug Brutlag & Scott C. Schmidler Overview Goals and problem definition Existing approaches Classic methods Recent successful approaches Evaluating prediction algorithms
Protein Secondary Structure Prediction
 part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 the goal is the prediction of the secondary structure conformation which is local each amino
part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 the goal is the prediction of the secondary structure conformation which is local each amino
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Protein structure. Protein structure. Amino acid residue. Cell communication channel. Bioinformatics Methods
 Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Protein Structure Prediction and Display
 Protein Structure Prediction and Display Goal Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each
Protein Structure Prediction and Display Goal Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Getting To Know Your Protein
 Getting To Know Your Protein Comparative Protein Analysis: Part III. Protein Structure Prediction and Comparison Robert Latek, PhD Sr. Bioinformatics Scientist Whitehead Institute for Biomedical Research
Getting To Know Your Protein Comparative Protein Analysis: Part III. Protein Structure Prediction and Comparison Robert Latek, PhD Sr. Bioinformatics Scientist Whitehead Institute for Biomedical Research
SUPPLEMENTARY MATERIALS
 SUPPLEMENTARY MATERIALS Enhanced Recognition of Transmembrane Protein Domains with Prediction-based Structural Profiles Baoqiang Cao, Aleksey Porollo, Rafal Adamczak, Mark Jarrell and Jaroslaw Meller Contact:
SUPPLEMENTARY MATERIALS Enhanced Recognition of Transmembrane Protein Domains with Prediction-based Structural Profiles Baoqiang Cao, Aleksey Porollo, Rafal Adamczak, Mark Jarrell and Jaroslaw Meller Contact:
BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer Protein Structure Prediction I
 BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 9. Protein Structure Prediction I Structure Prediction Overview Overview of problem variants Secondary structure prediction
BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 9. Protein Structure Prediction I Structure Prediction Overview Overview of problem variants Secondary structure prediction
Basics of protein structure
 Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Protein Structures: Experiments and Modeling. Patrice Koehl
 Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Analysis and Prediction of Protein Structure (I)
") Analysis and Prediction of Protein Structure (I) Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 2006 Free for academic use. Copyright @ Jianlin Cheng
Analysis and Prediction of Protein Structure (I) Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 2006 Free for academic use. Copyright @ Jianlin Cheng
Physiochemical Properties of Residues
 Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein structure alignments
 Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
IT og Sundhed 2010/11
 IT og Sundhed 2010/11 Sequence based predictors. Secondary structure and surface accessibility Bent Petersen 13 January 2011 1 NetSurfP Real Value Solvent Accessibility predictions with amino acid associated
IT og Sundhed 2010/11 Sequence based predictors. Secondary structure and surface accessibility Bent Petersen 13 January 2011 1 NetSurfP Real Value Solvent Accessibility predictions with amino acid associated
1-D Predictions. Prediction of local features: Secondary structure & surface exposure
 1-D Predictions Prediction of local features: Secondary structure & surface exposure 1 Learning Objectives After today s session you should be able to: Explain the meaning and usage of the following local
1-D Predictions Prediction of local features: Secondary structure & surface exposure 1 Learning Objectives After today s session you should be able to: Explain the meaning and usage of the following local
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics
 Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
SCOP. all-β class. all-α class, 3 different folds. T4 endonuclease V. 4-helical cytokines. Globin-like
 SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
Bioinformatics: Secondary Structure Prediction
 Bioinformatics: Secondary Structure Prediction Prof. David Jones d.jones@cs.ucl.ac.uk LMLSTQNPALLKRNIIYWNNVALLWEAGSD The greatest unsolved problem in molecular biology:the Protein Folding Problem? Entries
Bioinformatics: Secondary Structure Prediction Prof. David Jones d.jones@cs.ucl.ac.uk LMLSTQNPALLKRNIIYWNNVALLWEAGSD The greatest unsolved problem in molecular biology:the Protein Folding Problem? Entries
PROTEIN SECONDARY STRUCTURE PREDICTION: AN APPLICATION OF CHOU-FASMAN ALGORITHM IN A HYPOTHETICAL PROTEIN OF SARS VIRUS
 Int. J. LifeSc. Bt & Pharm. Res. 2012 Kaladhar, 2012 Research Paper ISSN 2250-3137 www.ijlbpr.com Vol.1, Issue. 1, January 2012 2012 IJLBPR. All Rights Reserved PROTEIN SECONDARY STRUCTURE PREDICTION:
Int. J. LifeSc. Bt & Pharm. Res. 2012 Kaladhar, 2012 Research Paper ISSN 2250-3137 www.ijlbpr.com Vol.1, Issue. 1, January 2012 2012 IJLBPR. All Rights Reserved PROTEIN SECONDARY STRUCTURE PREDICTION:
Neural Networks for Protein Structure Prediction Brown, JMB CS 466 Saurabh Sinha
 Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Bioinformatics: Secondary Structure Prediction
 Bioinformatics: Secondary Structure Prediction Prof. David Jones d.t.jones@ucl.ac.uk Possibly the greatest unsolved problem in molecular biology: The Protein Folding Problem MWMPPRPEEVARK LRRLGFVERMAKG
Bioinformatics: Secondary Structure Prediction Prof. David Jones d.t.jones@ucl.ac.uk Possibly the greatest unsolved problem in molecular biology: The Protein Folding Problem MWMPPRPEEVARK LRRLGFVERMAKG
Steps in protein modelling. Structure prediction, fold recognition and homology modelling. Basic principles of protein structure
 Structure prediction, fold recognition and homology modelling Marjolein Thunnissen Lund September 2012 Steps in protein modelling 3-D structure known Comparative Modelling Sequence of interest Similarity
Structure prediction, fold recognition and homology modelling Marjolein Thunnissen Lund September 2012 Steps in protein modelling 3-D structure known Comparative Modelling Sequence of interest Similarity
3D Structure. Prediction & Assessment Pt. 2. David Wishart 3-41 Athabasca Hall
 3D Structure Prediction & Assessment Pt. 2 David Wishart 3-41 Athabasca Hall david.wishart@ualberta.ca Objectives Become familiar with methods and algorithms for secondary Structure Prediction Become familiar
3D Structure Prediction & Assessment Pt. 2 David Wishart 3-41 Athabasca Hall david.wishart@ualberta.ca Objectives Become familiar with methods and algorithms for secondary Structure Prediction Become familiar
BCB 444/544 Fall 07 Dobbs 1
 BCB 444/544 Lecture 21 Protein Structure Visualization, Classification & Comparison Secondary Structure #21_Oct10 Required Reading (before lecture) Mon Oct 8 - Lecture 20 Protein Secondary Structure Chp
BCB 444/544 Lecture 21 Protein Structure Visualization, Classification & Comparison Secondary Structure #21_Oct10 Required Reading (before lecture) Mon Oct 8 - Lecture 20 Protein Secondary Structure Chp
Lecture 7. Protein Secondary Structure Prediction. Secondary Structure DSSP. Master Course DNA/Protein Structurefunction.
 C N T R F O R N T G R A T V B O N F O R M A T C S V U Master Course DNA/Protein Structurefunction Analysis and Prediction Lecture 7 Protein Secondary Structure Prediction Protein primary structure 20 amino
C N T R F O R N T G R A T V B O N F O R M A T C S V U Master Course DNA/Protein Structurefunction Analysis and Prediction Lecture 7 Protein Secondary Structure Prediction Protein primary structure 20 amino
Improved Protein Secondary Structure Prediction
 Improved Protein Secondary Structure Prediction Secondary Structure Prediction! Given a protein sequence a 1 a 2 a N, secondary structure prediction aims at defining the state of each amino acid ai as
Improved Protein Secondary Structure Prediction Secondary Structure Prediction! Given a protein sequence a 1 a 2 a N, secondary structure prediction aims at defining the state of each amino acid ai as
Amino Acid Structures from Klug & Cummings. 10/7/2003 CAP/CGS 5991: Lecture 7 1
 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 1 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 2 Amino Acid Structures from Klug & Cummings
Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 1 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 2 Amino Acid Structures from Klug & Cummings
HMM applications. Applications of HMMs. Gene finding with HMMs. Using the gene finder
 HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
Protein Secondary Structure Prediction
 C E N T R F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U E Master Course DNA/Protein Structurefunction Analysis and Prediction Lecture 7 Protein Secondary Structure Prediction Protein primary
C E N T R F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U E Master Course DNA/Protein Structurefunction Analysis and Prediction Lecture 7 Protein Secondary Structure Prediction Protein primary
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools. Giri Narasimhan
 CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinff18.html Proteins and Protein Structure
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinff18.html Proteins and Protein Structure
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
PROTEIN SECONDARY STRUCTURE PREDICTION USING NEURAL NETWORKS AND SUPPORT VECTOR MACHINES
 PROTEIN SECONDARY STRUCTURE PREDICTION USING NEURAL NETWORKS AND SUPPORT VECTOR MACHINES by Lipontseng Cecilia Tsilo A thesis submitted to Rhodes University in partial fulfillment of the requirements for
PROTEIN SECONDARY STRUCTURE PREDICTION USING NEURAL NETWORKS AND SUPPORT VECTOR MACHINES by Lipontseng Cecilia Tsilo A thesis submitted to Rhodes University in partial fulfillment of the requirements for
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence
 Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
HIV protease inhibitor. Certain level of function can be found without structure. But a structure is a key to understand the detailed mechanism.
 Proteins are linear polypeptide chains (one or more) Building blocks: 20 types of amino acids. Range from a few 10s-1000s They fold into varying three-dimensional shapes structure medicine Certain level
Proteins are linear polypeptide chains (one or more) Building blocks: 20 types of amino acids. Range from a few 10s-1000s They fold into varying three-dimensional shapes structure medicine Certain level
2MHR. Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity.
 Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Protein Secondary Structure Prediction using Feed-Forward Neural Network
 COPYRIGHT 2010 JCIT, ISSN 2078-5828 (PRINT), ISSN 2218-5224 (ONLINE), VOLUME 01, ISSUE 01, MANUSCRIPT CODE: 100713 Protein Secondary Structure Prediction using Feed-Forward Neural Network M. A. Mottalib,
COPYRIGHT 2010 JCIT, ISSN 2078-5828 (PRINT), ISSN 2218-5224 (ONLINE), VOLUME 01, ISSUE 01, MANUSCRIPT CODE: 100713 Protein Secondary Structure Prediction using Feed-Forward Neural Network M. A. Mottalib,
Useful background reading
 Overview of lecture * General comment on peptide bond * Discussion of backbone dihedral angles * Discussion of Ramachandran plots * Description of helix types. * Description of structures * NMR patterns
Overview of lecture * General comment on peptide bond * Discussion of backbone dihedral angles * Discussion of Ramachandran plots * Description of helix types. * Description of structures * NMR patterns
Research Article Extracting Physicochemical Features to Predict Protein Secondary Structure
 The Scientific World Journal Volume 2013, Article ID 347106, 8 pages http://dx.doi.org/10.1155/2013/347106 Research Article Extracting Physicochemical Features to Predict Protein Secondary Structure Yin-Fu
The Scientific World Journal Volume 2013, Article ID 347106, 8 pages http://dx.doi.org/10.1155/2013/347106 Research Article Extracting Physicochemical Features to Predict Protein Secondary Structure Yin-Fu
Identification of Representative Protein Sequence and Secondary Structure Prediction Using SVM Approach
 Identification of Representative Protein Sequence and Secondary Structure Prediction Using SVM Approach Prof. Dr. M. A. Mottalib, Md. Rahat Hossain Department of Computer Science and Information Technology
Identification of Representative Protein Sequence and Secondary Structure Prediction Using SVM Approach Prof. Dr. M. A. Mottalib, Md. Rahat Hossain Department of Computer Science and Information Technology
7 Protein secondary structure
 78 Grundlagen der Bioinformatik, SS 1, D. Huson, June 17, 21 7 Protein secondary structure Sources for this chapter, which are all recommended reading: Introduction to Protein Structure, Branden & Tooze,
78 Grundlagen der Bioinformatik, SS 1, D. Huson, June 17, 21 7 Protein secondary structure Sources for this chapter, which are all recommended reading: Introduction to Protein Structure, Branden & Tooze,
Presentation Outline. Prediction of Protein Secondary Structure using Neural Networks at Better than 70% Accuracy
 Prediction of Protein Secondary Structure using Neural Networks at Better than 70% Accuracy Burkhard Rost and Chris Sander By Kalyan C. Gopavarapu 1 Presentation Outline Major Terminology Problem Method
Prediction of Protein Secondary Structure using Neural Networks at Better than 70% Accuracy Burkhard Rost and Chris Sander By Kalyan C. Gopavarapu 1 Presentation Outline Major Terminology Problem Method
Intro Secondary structure Transmembrane proteins Function End. Last time. Domains Hidden Markov Models
 Last time Domains Hidden Markov Models Today Secondary structure Transmembrane proteins Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL
Last time Domains Hidden Markov Models Today Secondary structure Transmembrane proteins Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL
Today. Last time. Secondary structure Transmembrane proteins. Domains Hidden Markov Models. Structure prediction. Secondary structure
 Last time Today Domains Hidden Markov Models Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL SSLGPVVDAHPEYEEVALLERMVIPERVIE FRVPWEDDNGKVHVNTGYRVQFNGAIGPYK
Last time Today Domains Hidden Markov Models Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL SSLGPVVDAHPEYEEVALLERMVIPERVIE FRVPWEDDNGKVHVNTGYRVQFNGAIGPYK
Software and Databases for Protein Structure Classification. Some slides are modified from Kun Huang (OSU) and Doug Brutlag (Stanford)
 and Doug Brutlag (Stanford)") Software and Databases for Protein Structure Classification Some slides are modified from Kun Huang (OSU) and Doug Brutlag (Stanford) Proteins If there is a job to be done in the molecular world of our
Software and Databases for Protein Structure Classification Some slides are modified from Kun Huang (OSU) and Doug Brutlag (Stanford) Proteins If there is a job to be done in the molecular world of our
Protein Secondary Structure Prediction using Pattern Recognition Neural Network
 Protein Secondary Structure Prediction using Pattern Recognition Neural Network P.V. Nageswara Rao 1 (nagesh@gitam.edu), T. Uma Devi 1, DSVGK Kaladhar 1, G.R. Sridhar 2, Allam Appa Rao 3 1 GITAM University,
Protein Secondary Structure Prediction using Pattern Recognition Neural Network P.V. Nageswara Rao 1 (nagesh@gitam.edu), T. Uma Devi 1, DSVGK Kaladhar 1, G.R. Sridhar 2, Allam Appa Rao 3 1 GITAM University,
Lecture 14 Secondary Structure Prediction
 C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Lecture 14 Secondary Structure Prediction Bioinformatics Center IBIVU Protein structure Linus Pauling (1951) Atomic Coordinates and
C E N T R E F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U Lecture 14 Secondary Structure Prediction Bioinformatics Center IBIVU Protein structure Linus Pauling (1951) Atomic Coordinates and
Lecture 14 Secondary Structure Prediction
 C N T R F O R I N T G R A T I V Protein structure B I O I N F O R M A T I C S V U Lecture 14 Secondary Structure Prediction Bioinformatics Center IBIVU James Watson & Francis Crick (1953) Linus Pauling
C N T R F O R I N T G R A T I V Protein structure B I O I N F O R M A T I C S V U Lecture 14 Secondary Structure Prediction Bioinformatics Center IBIVU James Watson & Francis Crick (1953) Linus Pauling
Bioinformatics. Macromolecular structure
 Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
8 Protein secondary structure
 Grundlagen der Bioinformatik, SoSe 11, D. Huson, June 6, 211 13 8 Protein secondary structure Sources for this chapter, which are all recommended reading: Introduction to Protein Structure, Branden & Tooze,
Grundlagen der Bioinformatik, SoSe 11, D. Huson, June 6, 211 13 8 Protein secondary structure Sources for this chapter, which are all recommended reading: Introduction to Protein Structure, Branden & Tooze,
HOMOLOGY MODELING. The sequence alignment and template structure are then used to produce a structural model of the target.
 HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
Protein Structure. Hierarchy of Protein Structure. Tertiary structure. independently stable structural unit. includes disulfide bonds
 Protein Structure Hierarchy of Protein Structure 2 3 Structural element Primary structure Secondary structure Super-secondary structure Domain Tertiary structure Quaternary structure Description amino
Protein Structure Hierarchy of Protein Structure 2 3 Structural element Primary structure Secondary structure Super-secondary structure Domain Tertiary structure Quaternary structure Description amino
1. Protein Data Bank (PDB) 1. Protein Data Bank (PDB)
 1. Protein Data Bank (PDB)") Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
12 Protein secondary structure
 Grundlagen der Bioinformatik, SoSe 14, D. Huson, July 2, 214 147 12 Protein secondary structure Sources for this chapter, which are all recommended reading: Introduction to Protein Structure, Branden &
Grundlagen der Bioinformatik, SoSe 14, D. Huson, July 2, 214 147 12 Protein secondary structure Sources for this chapter, which are all recommended reading: Introduction to Protein Structure, Branden &
Biochemistry Prof. S. DasGupta Department of Chemistry Indian Institute of Technology Kharagpur. Lecture - 06 Protein Structure IV
 Biochemistry Prof. S. DasGupta Department of Chemistry Indian Institute of Technology Kharagpur Lecture - 06 Protein Structure IV We complete our discussion on Protein Structures today. And just to recap
Biochemistry Prof. S. DasGupta Department of Chemistry Indian Institute of Technology Kharagpur Lecture - 06 Protein Structure IV We complete our discussion on Protein Structures today. And just to recap
COMP 598 Advanced Computational Biology Methods & Research. Introduction. Jérôme Waldispühl School of Computer Science McGill University
 COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Introduction to Bioinformatics Lecture 13. Protein Secondary Structure Elements and their Prediction. Protein Secondary Structure
 Introduction to Bioinformatics Lecture 13 Protein Secondary Structure C E N T R F O R I N T B I O I N F E Protein Secondary Structure Elements and their Prediction IBIVU Centre E G R A T I V E O R M A
Introduction to Bioinformatics Lecture 13 Protein Secondary Structure C E N T R F O R I N T B I O I N F E Protein Secondary Structure Elements and their Prediction IBIVU Centre E G R A T I V E O R M A
Optimization of the Sliding Window Size for Protein Structure Prediction
 Optimization of the Sliding Window Size for Protein Structure Prediction Ke Chen* 1, Lukasz Kurgan 1 and Jishou Ruan 2 1 University of Alberta, Department of Electrical and Computer Engineering, Edmonton,
Optimization of the Sliding Window Size for Protein Structure Prediction Ke Chen* 1, Lukasz Kurgan 1 and Jishou Ruan 2 1 University of Alberta, Department of Electrical and Computer Engineering, Edmonton,
Procheck output. Bond angles (Procheck) Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.
 Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.") Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Bioinformatics. Proteins II. - Pattern, Profile, & Structure Database Searching. Robert Latek, Ph.D. Bioinformatics, Biocomputing
 Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Bioinformatics Proteins II. - Pattern, Profile, & Structure Database Searching Robert Latek, Ph.D. Bioinformatics, Biocomputing WIBR Bioinformatics Course, Whitehead Institute, 2002 1 Proteins I.-III.
Prediction of protein secondary structure by mining structural fragment database
 Polymer 46 (2005) 4314 4321 www.elsevier.com/locate/polymer Prediction of protein secondary structure by mining structural fragment database Haitao Cheng a, Taner Z. Sen a, Andrzej Kloczkowski a, Dimitris
Polymer 46 (2005) 4314 4321 www.elsevier.com/locate/polymer Prediction of protein secondary structure by mining structural fragment database Haitao Cheng a, Taner Z. Sen a, Andrzej Kloczkowski a, Dimitris
Bayesian Network Multi-classifiers for Protein Secondary Structure Prediction
 Bayesian Network Multi-classifiers for Protein Secondary Structure Prediction Víctor Robles a, Pedro Larrañaga b,josém.peña a, Ernestina Menasalvas a,maría S. Pérez a, Vanessa Herves a and Anita Wasilewska
Bayesian Network Multi-classifiers for Protein Secondary Structure Prediction Víctor Robles a, Pedro Larrañaga b,josém.peña a, Ernestina Menasalvas a,maría S. Pérez a, Vanessa Herves a and Anita Wasilewska
Predicting Secondary Structures of Proteins
 CHALLENGES IN PROTEOMICS BACKGROUND PHOTODISC, FOREGROUND IMAGE: U.S. DEPARTMENT OF ENERGY GENOMICS: GTL PROGRAM, HTTP://WWW.ORNL.GOV.HGMIS BY JACEK BLAŻEWICZ, PETER L. HAMMER, AND PIOTR LUKASIAK Predicting
CHALLENGES IN PROTEOMICS BACKGROUND PHOTODISC, FOREGROUND IMAGE: U.S. DEPARTMENT OF ENERGY GENOMICS: GTL PROGRAM, HTTP://WWW.ORNL.GOV.HGMIS BY JACEK BLAŻEWICZ, PETER L. HAMMER, AND PIOTR LUKASIAK Predicting
Protein Structure & Motifs
 & Motifs Biochemistry 201 Molecular Biology January 12, 2000 Doug Brutlag Introduction Proteins are more flexible than nucleic acids in structure because of both the larger number of types of residues
& Motifs Biochemistry 201 Molecular Biology January 12, 2000 Doug Brutlag Introduction Proteins are more flexible than nucleic acids in structure because of both the larger number of types of residues
Protein structure analysis. Risto Laakso 10th January 2005
 Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Bayesian Network Multi-classifiers for Protein Secondary Structure Prediction
 Bayesian Network Multi-classifiers for Protein Secondary Structure Prediction Víctor Robles a, Pedro Larrañaga b,josém.peña a, Ernestina Menasalvas a,maría S. Pérez a, Vanessa Herves a and Anita Wasilewska
Bayesian Network Multi-classifiers for Protein Secondary Structure Prediction Víctor Robles a, Pedro Larrañaga b,josém.peña a, Ernestina Menasalvas a,maría S. Pérez a, Vanessa Herves a and Anita Wasilewska
Secondary Structure. Bioch/BIMS 503 Lecture 2. Structure and Function of Proteins. Further Reading. Φ, Ψ angles alone determine protein structure
 Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Protein Structure: Data Bases and Classification Ingo Ruczinski
 Protein Structure: Data Bases and Classification Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Reference Bourne and Weissig Structural Bioinformatics Wiley, 2003 More References
Protein Structure: Data Bases and Classification Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Reference Bourne and Weissig Structural Bioinformatics Wiley, 2003 More References
Conformational Geometry of Peptides and Proteins:
 Conformational Geometry of Peptides and Proteins: Before discussing secondary structure, it is important to appreciate the conformational plasticity of proteins. Each residue in a polypeptide has three
Conformational Geometry of Peptides and Proteins: Before discussing secondary structure, it is important to appreciate the conformational plasticity of proteins. Each residue in a polypeptide has three
Molecular Modelling. part of Bioinformatik von RNA- und Proteinstrukturen. Sonja Prohaska. Leipzig, SS Computational EvoDevo University Leipzig
 part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 Protein Structure levels or organization Primary structure: sequence of amino acids (from
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Two-Stage Multi-Class Support Vector Machines to Protein Secondary Structure Prediction. M.N. Nguyen and J.C. Rajapakse
 Two-Stage Multi-Class Support Vector Machines to Protein Secondary Structure Prediction M.N. Nguyen and J.C. Rajapakse Pacific Symposium on Biocomputing 10:346-357(2005) TWO-STAGE MULTI-CLASS SUPPORT VECTOR
Two-Stage Multi-Class Support Vector Machines to Protein Secondary Structure Prediction M.N. Nguyen and J.C. Rajapakse Pacific Symposium on Biocomputing 10:346-357(2005) TWO-STAGE MULTI-CLASS SUPPORT VECTOR
Proteins: Structure & Function. Ulf Leser
 Proteins: Structure & Function Ulf Leser This Lecture Proteins Structure Function Databases Predicting Protein Secondary Structure Many figures from Zvelebil, M. and Baum, J. O. (2008). "Understanding
Proteins: Structure & Function Ulf Leser This Lecture Proteins Structure Function Databases Predicting Protein Secondary Structure Many figures from Zvelebil, M. and Baum, J. O. (2008). "Understanding
Conditional Graphical Models
 PhD Thesis Proposal Conditional Graphical Models for Protein Structure Prediction Yan Liu Language Technologies Institute University Thesis Committee Jaime Carbonell (Chair) John Lafferty Eric P. Xing
PhD Thesis Proposal Conditional Graphical Models for Protein Structure Prediction Yan Liu Language Technologies Institute University Thesis Committee Jaime Carbonell (Chair) John Lafferty Eric P. Xing
Protein Structure Prediction Using Neural Networks
 Protein Structure Prediction Using Neural Networks Martha Mercaldi Kasia Wilamowska Literature Review December 16, 2003 The Protein Folding Problem Evolution of Neural Networks Neural networks originally
Protein Structure Prediction Using Neural Networks Martha Mercaldi Kasia Wilamowska Literature Review December 16, 2003 The Protein Folding Problem Evolution of Neural Networks Neural networks originally
The Structure and Functions of Proteins
 Wright State University CORE Scholar Computer Science and Engineering Faculty Publications Computer Science and Engineering 2003 The Structure and Functions of Proteins Dan E. Krane Wright State University
Wright State University CORE Scholar Computer Science and Engineering Faculty Publications Computer Science and Engineering 2003 The Structure and Functions of Proteins Dan E. Krane Wright State University
Protein structure prediction and conformational transitions
 Graduate Theses and Dissertations Graduate College 2009 Protein structure prediction and conformational transitions Haitao Cheng Iowa State University Follow this and additional works at: http://lib.dr.iastate.edu/etd
Graduate Theses and Dissertations Graduate College 2009 Protein structure prediction and conformational transitions Haitao Cheng Iowa State University Follow this and additional works at: http://lib.dr.iastate.edu/etd
Computational Molecular Modeling
 Computational Molecular Modeling Lecture 1: Structure Models, Properties Chandrajit Bajaj Today s Outline Intro to atoms, bonds, structure, biomolecules, Geometry of Proteins, Nucleic Acids, Ribosomes,
Computational Molecular Modeling Lecture 1: Structure Models, Properties Chandrajit Bajaj Today s Outline Intro to atoms, bonds, structure, biomolecules, Geometry of Proteins, Nucleic Acids, Ribosomes,
DATE A DAtabase of TIM Barrel Enzymes
 DATE A DAtabase of TIM Barrel Enzymes 2 2.1 Introduction.. 2.2 Objective and salient features of the database 2.2.1 Choice of the dataset.. 2.3 Statistical information on the database.. 2.4 Features....
DATE A DAtabase of TIM Barrel Enzymes 2 2.1 Introduction.. 2.2 Objective and salient features of the database 2.2.1 Choice of the dataset.. 2.3 Statistical information on the database.. 2.4 Features....
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009
 114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5
 Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Sequence and Structure Alignment Z. Luthey-Schulten, UIUC Pittsburgh, 2006 VMD 1.8.5 Why Look at More Than One Sequence? 1. Multiple Sequence Alignment shows patterns of conservation 2. What and how many
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Bayesian Models and Algorithms for Protein Beta-Sheet Prediction
 0 Bayesian Models and Algorithms for Protein Beta-Sheet Prediction Zafer Aydin, Student Member, IEEE, Yucel Altunbasak, Senior Member, IEEE, and Hakan Erdogan, Member, IEEE Abstract Prediction of the three-dimensional
0 Bayesian Models and Algorithms for Protein Beta-Sheet Prediction Zafer Aydin, Student Member, IEEE, Yucel Altunbasak, Senior Member, IEEE, and Hakan Erdogan, Member, IEEE Abstract Prediction of the three-dimensional
Protein Structure Prediction using String Kernels. Technical Report
 Protein Structure Prediction using String Kernels Technical Report Department of Computer Science and Engineering University of Minnesota 4-192 EECS Building 200 Union Street SE Minneapolis, MN 55455-0159
Protein Structure Prediction using String Kernels Technical Report Department of Computer Science and Engineering University of Minnesota 4-192 EECS Building 200 Union Street SE Minneapolis, MN 55455-0159
Orientational degeneracy in the presence of one alignment tensor.
 Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Protein 8-class Secondary Structure Prediction Using Conditional Neural Fields
 2010 IEEE International Conference on Bioinformatics and Biomedicine Protein 8-class Secondary Structure Prediction Using Conditional Neural Fields Zhiyong Wang, Feng Zhao, Jian Peng, Jinbo Xu* Toyota
2010 IEEE International Conference on Bioinformatics and Biomedicine Protein 8-class Secondary Structure Prediction Using Conditional Neural Fields Zhiyong Wang, Feng Zhao, Jian Peng, Jinbo Xu* Toyota
Building 3D models of proteins
 Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Sequence Analysis, '18 -- lecture 9. Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene.
 Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
Sequence Analysis, '18 -- lecture 9 Families and superfamilies. Sequence weights. Profiles. Logos. Building a representative model for a gene. How can I represent thousands of homolog sequences in a compact
CS612 - Algorithms in Bioinformatics
 Fall 2017 Protein Structure Detection Methods October 30, 2017 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing
Fall 2017 Protein Structure Detection Methods October 30, 2017 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural