SUPPLEMENTARY MATERIALS
|
|
|
- Junior Jeremy Collins
- 6 years ago
- Views:
Transcription
1 SUPPLEMENTARY MATERIALS Enhanced Recognition of Transmembrane Protein Domains with Prediction-based Structural Profiles Baoqiang Cao, Aleksey Porollo, Rafal Adamczak, Mark Jarrell and Jaroslaw Meller Contact: Server available at SP1. Multistage protocol for transmembrane helix prediction in MINNOU In addition to a simple NN-based classifier developed and assessed in the cross-validation study (see the Systems and Methods section of the main body of the paper), we also developed a multistage protocol for enhanced prediction of transmembrane (TM) helices. For the final predictor we do not consider the MAbased representation, which is shown using cross-validation to yield a lower accuracy compared with the proposed compact prediction-based structural profiles. We also excluded hydropathy scales from the representation of an amino acid residue, as it was observed to lead to a higher level of confusion with globular proteins and signal peptides. Moreover, statistical propensities of amino acids (e.g. to lipid aqueous phase interfaces) that could be used, in principle, to improve the prediction of TM segments, are not taken into account at this stage. Consequently, each residue is initially represented by five numbers: the predicted real valued RSA, confidence of RSA prediction and probabilities of each of the three secondary structures (as predicted by SABLE, SABLE predictions are derived from the multiple alignment, hydropathy scales and other attributes, which are commonly used by other state-of-theart methods. Therefore, it is expected that other accurate methods for RSA and SS prediction will be useful in that regard as well. This is a subject of a future investigation. Following in the footsteps of other studies (Rost et al., 1995), we use a two-stage prediction system, with the second layer (structure-to-structure) NNs allowing one to average and smooth over the initial classification obtained using the first (sequence-to-structure) layer predictor. The architecture of the first and second layer NNs is similar to that used for the cross-validation study. Namely, a simple feedforward topology with one hidden layer, fully interconnected with the input and output layers, is employed. The choice of the sliding window size, the number of nodes in the hidden layer, training protocols and other characteristics of these NNs are discussed below. In order to reduce the danger of overfitting and achieve regularization as well as improvement in accuracy, a consensus of twenty different networks was used to generate predictions at each stage. These different networks were trained on different subsets of the training set that were also used for the crossvalidation study. Multiple NNs were trained on each subset of the data, with different number of nodes in the hidden layer and with different size of the sliding window. The number of nodes in the hidden layer was again varied between 8 and 18 and the size of the sliding window was varied between 11 and 31. The stopping criteria were defined in terms of improvement (or lack thereof) on the corresponding validation set. From multiple networks trained on each subset of the data, the one providing the highest per residue accuracy on the corresponding validation set was then selected for the final consensus predictor. In that sense, the whole training set of 73 protein chains is used here to optimize the parameters of each of the networks. However, using different subsets of the data provides a better sampling of local minima. The first ten networks were trained on different subsets of the data using the representation with five attributes per amino acid residue, as described above. The other ten networks included in the consensus were trained with only four attributes per residue. Namely, the two nodes related to relative solvent accessibility prediction are replaced by one node, which represents a discretized RSA assignment. In other words, such a binary node indicates if a given residue is predicted to be buried or exposed (with a threshold of 25% RSA used to project the real valued SABLE prediction into two classes). Adding these additional networks to the consensus was found to improve somewhat the per segment classification accuracy. The consensus predictor is based on a simple majority voting. The source code for the MINNOU package, with the definition of all the NNs used for the consensus prediction (including a detailed description of their topology and parameters) can be downloaded from ftp://ftp.chmcc.org/pdi/jmeller/minnou/. To further improve the performance of the first layer classifier, we introduced a second layer prediction system. The output of the first layer is used at this stage as input. In addition, the SABLE predictions used in 1 st layer are included in the input as well. After some experimentation we found that the best results are obtained when the output of the first layer system is combined as follows to be included in the input of the 2 nd layer. For each residue, two additional nodes are used to represent averaged (over the
2 networks included in the consensus) excitation of the two output nodes corresponding to two different classes. Another pair of nodes is used to represent the maximum (again over the different networks) excitation for each class. Thus, the number of nodes per residue in the 2 nd layer is either nine or eight, depending on the number of attributes used in the first layer. The choice of optimal topology and other settings for the neural networks and the training procedures follows that for the 1 st layers (see also the MINNOU package for further details). While the second layer NNs lead to significant smoothing of the prediction and improves the overall accuracy in terms of both: sensitivity and specificity, some long or short helices are still occasionally predicted. We estimated the probability density distribution for the length of TM helices and used it as a guideline in the design of a filter, applied to the second layer prediction in order to avoid such unphysical predictions. Similar filters have been used before by other groups (Rost et al., 1995). Basically, the final filter is applied to either split predicted long TM helices or delete too short ones, which are observed with very low frequency in the known sample of helical TM domains. The presence of relatively short (e.g., horizontally oriented) membrane embedded helices as well as relatively long ( skewed ) helices that occur in some ion channels, for instance, influences the choice of the length thresholds applied here. Specifically, if only a single membrane segment is predicted in a protein, then it is deleted if its length is shorter than 14 residues; if more than one membrane segment is predicted, then it is deleted if shorter than 8 residues. On the other hand, if there is a continuous membrane segment of length greater than 44, it is split into two segments in the middle (by introducing an artificial loop of length one); and if the predicted segment is longer than 66 residues (the latter happened seven times in the set of 2247 segments predicted for the TMH Benchmarks test), then it is split into three segments. Even though this final post-processing step does not affect significantly the per residue accuracy, it does help to smooth the predictions and to improve the per segment accuracy, Q OK, as defined in (Chen et al., 2002; Kernytsky and Rost, 2003). It also helps reducing further the observed level of confusion with signal peptides and globular proteins by filtering out unphysical short TM helices (see Table 6 and also the Results section of the main body of the paper for further discussion). However, this final prediction stage is likely to be improved further, for example, by considering explicitly the overall topology of membrane proteins and characteristics of loops connecting TM segments. This is a subject of a future work.
3 SP2. Tables and figures Table 5 Per residue classification accuracy (Q 3 ) of secondary structure predictions obtained using SABLE (Adamczak et al., 2005) and PSIPRED servers (Jones, 1999). The results on non-redundant sets of 73 alpha-helical and 15 beta-barrel membrane proteins, respectively, are shown. Test set SABLE PSIPRED Alpha-helical: all 78.2% 76.6% Alpha-helical: TM segments only 83.3% 80.6% Beta-barrel: all 68.7% 75.7% Beta-barrel: TM segments only 64.9% 77.6% Table 6 Improvements due to 1 st layer, 2 nd layer and filter (F) based predictors according to TMH Benchmark evaluation. Per segment (Q OK ) and per residue (Q 2 ) classification accuracies and confusion levels with signal peptides (csp) and globular proteins (cgp) are given in the units of per cent. Five different prediction systems are compared. The first two involve the first layer predictions: (a) training with the standard training set containing membrane proteins only; and (b) training with the augmented training set that includes signal peptides and some false positives from the first iteration. The next two include the corresponding results for the second layer predictors that use the results of the first layer classifiers as part of their input. The last row contains results of the second layer prediction with filtering of short, unphysical segments, which improves the prediction in some respects. High resolution Low resolution Q OK Q 2 Q OK Q 2 1 st layer (a) st layer (b) nd layer (a) nd layer (b) nd layer (F) Figure 1 Examples of MINNOU transmembrane helix predictions for an ion channel, PDB code 1OTS, (panel A), and a glutamate transporter protein, PDB code 1XFH (panel B). The amino acid sequence is included in the first row, with the actual and predicted membrane segments highlighted using bold and yellow boxes, respectively. The secondary structures and relative solvent accessibilities, predicted using the SABLE server, are shown in the second and third rows, respectively. The alpha-helices are represented by red ribbons, the beta-strands by green arrows and coils by blue lines, respectively. The level of predicted aqueous solvent exposure is represented by shaded boxes, with fully buried and fully exposed residues represented by black and white boxes, respectively. The secondary structures observed in the experimentally resolved structures are shown in the last row for comparison. See text for further details.
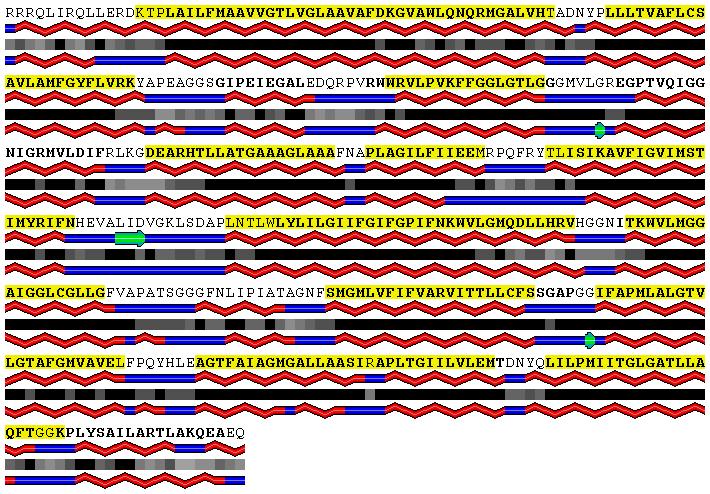

4 A. B.
5 Figure 2 The distribution of lengths for TM segments included in the MPtopo (Jayasinghe et al., 2001) database of membrane proteins. 3D_helix and 1D_helix refer to helical membrane proteins with or without resolved 3D structure, respectively (see text for details). Thus, in case of 1D_helix set only low resolution information derived from various experimental studies is available. Consequently, the delineation of TM segments is laden with additional uncertainty. Moreover, it was suggested that prediction methods may have been used to derive the actual boundaries of TM segments for some of the 1D_helix proteins (Chen et al., 2002). As can be seen from the figure, the distribution of lengths for low resolution structures is indeed qualitatively different, compared to high resolution (3D_helix) set. In particular, a sharp peak is observed around the canonical length of an alpha-helical TM segment (20-22 residues) for low resolution structures. This is in stark contrast to a much wider distribution for high resolution structures. Because of these differences and higher uncertainty in the annotation of low resolution structures, we used only 3D_helix set for training of MINNOU. The 3D_other set includes beta barrel membrane proteins with known 3D structures and is characterized by much shorter membrane spanning segments dhelix 1dhelix 3dother Number of TM segments TM length
6 Figure 3 Dependence of prediction accuracy on the size of the sliding window, using 10-fold crossvalidation on the non-redundant training set of 73 alpha-helical membrane protein chains (see text for details). The results of a simple NN, with one hidden layer consisting of 10 nodes, trained for 1000 cycles with the default learning parameter and the Rprop delta parameter set to 0.1, are compared in terms of Matthews Correlation Coefficients (MCC). Fixed topology and meta-parameters of the networks allow one to compare the results directly and assess the extent of overfitting due to increased windows size. Each amino acid residue in the sliding window is represented here by five numbers, using the prediction-based structural profiles described in the text. Thus, increasing the size of the sliding window from 11 to 21 increases the number of weights to be optimized from 550 to 1050 (neglecting the weights for edges between the hidden layer and the two nodes in the output layer as well as the parameters for the activation functions of the nodes in the hidden and output layers). As can be seen from the figure, the cross-validation estimates of the accuracy quickly increase initially in order to reach a plateau and then gradually decrease with the growing size of the sliding window, pointing out problems with overfitting for more complex models. It should be noted that changing the topology of the network we were able to obtain somewhat better results (at the level of MCC=0.74, as reported in Tables 1 and 2 for different sliding windows). We would also like to comment that while very short sliding windows seem to be capable of capturing relatively well the essential information about an amino acid residue and its environment (since not too many residues predicted to be fully buried and in alpha helices are found in the loop domains of TM proteins included in the training set) the overall quality of predictions based on such short windows is low due high level of confusion with globular proteins and low per segment accuracy MCC Sliding Window Size
BIOINFORMATICS. Enhanced Recognition of Protein Transmembrane Domains with Prediction-based Structural Profiles
 BIOINFORMATICS Vol.? no.? 200? Pages 1 1 Enhanced Recognition of Protein Transmembrane Domains with Prediction-based Structural Profiles Baoqiang Cao 2, Aleksey Porollo 1, Rafal Adamczak 1, Mark Jarrell
BIOINFORMATICS Vol.? no.? 200? Pages 1 1 Enhanced Recognition of Protein Transmembrane Domains with Prediction-based Structural Profiles Baoqiang Cao 2, Aleksey Porollo 1, Rafal Adamczak 1, Mark Jarrell
1-D Predictions. Prediction of local features: Secondary structure & surface exposure
 1-D Predictions Prediction of local features: Secondary structure & surface exposure 1 Learning Objectives After today s session you should be able to: Explain the meaning and usage of the following local
1-D Predictions Prediction of local features: Secondary structure & surface exposure 1 Learning Objectives After today s session you should be able to: Explain the meaning and usage of the following local
Neural Networks for Protein Structure Prediction Brown, JMB CS 466 Saurabh Sinha
 Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics
 Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
TMSEG Michael Bernhofer, Jonas Reeb pp1_tmseg
 title: short title: TMSEG Michael Bernhofer, Jonas Reeb pp1_tmseg lecture: Protein Prediction 1 (for Computational Biology) Protein structure TUM summer semester 09.06.2016 1 Last time 2 3 Yet another
title: short title: TMSEG Michael Bernhofer, Jonas Reeb pp1_tmseg lecture: Protein Prediction 1 (for Computational Biology) Protein structure TUM summer semester 09.06.2016 1 Last time 2 3 Yet another
Intro Secondary structure Transmembrane proteins Function End. Last time. Domains Hidden Markov Models
 Last time Domains Hidden Markov Models Today Secondary structure Transmembrane proteins Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL
Last time Domains Hidden Markov Models Today Secondary structure Transmembrane proteins Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL
Today. Last time. Secondary structure Transmembrane proteins. Domains Hidden Markov Models. Structure prediction. Secondary structure
 Last time Today Domains Hidden Markov Models Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL SSLGPVVDAHPEYEEVALLERMVIPERVIE FRVPWEDDNGKVHVNTGYRVQFNGAIGPYK
Last time Today Domains Hidden Markov Models Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL SSLGPVVDAHPEYEEVALLERMVIPERVIE FRVPWEDDNGKVHVNTGYRVQFNGAIGPYK
Protein Secondary Structure Prediction
 Protein Secondary Structure Prediction Doug Brutlag & Scott C. Schmidler Overview Goals and problem definition Existing approaches Classic methods Recent successful approaches Evaluating prediction algorithms
Protein Secondary Structure Prediction Doug Brutlag & Scott C. Schmidler Overview Goals and problem definition Existing approaches Classic methods Recent successful approaches Evaluating prediction algorithms
Improved Protein Secondary Structure Prediction
 Improved Protein Secondary Structure Prediction Secondary Structure Prediction! Given a protein sequence a 1 a 2 a N, secondary structure prediction aims at defining the state of each amino acid ai as
Improved Protein Secondary Structure Prediction Secondary Structure Prediction! Given a protein sequence a 1 a 2 a N, secondary structure prediction aims at defining the state of each amino acid ai as
Presentation Outline. Prediction of Protein Secondary Structure using Neural Networks at Better than 70% Accuracy
 Prediction of Protein Secondary Structure using Neural Networks at Better than 70% Accuracy Burkhard Rost and Chris Sander By Kalyan C. Gopavarapu 1 Presentation Outline Major Terminology Problem Method
Prediction of Protein Secondary Structure using Neural Networks at Better than 70% Accuracy Burkhard Rost and Chris Sander By Kalyan C. Gopavarapu 1 Presentation Outline Major Terminology Problem Method
Protein Structure Prediction Using Neural Networks
 Protein Structure Prediction Using Neural Networks Martha Mercaldi Kasia Wilamowska Literature Review December 16, 2003 The Protein Folding Problem Evolution of Neural Networks Neural networks originally
Protein Structure Prediction Using Neural Networks Martha Mercaldi Kasia Wilamowska Literature Review December 16, 2003 The Protein Folding Problem Evolution of Neural Networks Neural networks originally
Basics of protein structure
 Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Protein structure alignments
 Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
Protein Secondary Structure Prediction
 part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 the goal is the prediction of the secondary structure conformation which is local each amino
part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 the goal is the prediction of the secondary structure conformation which is local each amino
Protein Secondary Structure Assignment and Prediction
 1 Protein Secondary Structure Assignment and Prediction Defining SS features - Dihedral angles, alpha helix, beta stand (Hydrogen bonds) Assigned manually by crystallographers or Automatic DSSP (Kabsch
1 Protein Secondary Structure Assignment and Prediction Defining SS features - Dihedral angles, alpha helix, beta stand (Hydrogen bonds) Assigned manually by crystallographers or Automatic DSSP (Kabsch
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Protein Secondary Structure Prediction using Feed-Forward Neural Network
 COPYRIGHT 2010 JCIT, ISSN 2078-5828 (PRINT), ISSN 2218-5224 (ONLINE), VOLUME 01, ISSUE 01, MANUSCRIPT CODE: 100713 Protein Secondary Structure Prediction using Feed-Forward Neural Network M. A. Mottalib,
COPYRIGHT 2010 JCIT, ISSN 2078-5828 (PRINT), ISSN 2218-5224 (ONLINE), VOLUME 01, ISSUE 01, MANUSCRIPT CODE: 100713 Protein Secondary Structure Prediction using Feed-Forward Neural Network M. A. Mottalib,
SCOP. all-β class. all-α class, 3 different folds. T4 endonuclease V. 4-helical cytokines. Globin-like
 SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
SCOP all-β class 4-helical cytokines T4 endonuclease V all-α class, 3 different folds Globin-like TIM-barrel fold α/β class Profilin-like fold α+β class http://scop.mrc-lmb.cam.ac.uk/scop CATH Class, Architecture,
IT og Sundhed 2010/11
 IT og Sundhed 2010/11 Sequence based predictors. Secondary structure and surface accessibility Bent Petersen 13 January 2011 1 NetSurfP Real Value Solvent Accessibility predictions with amino acid associated
IT og Sundhed 2010/11 Sequence based predictors. Secondary structure and surface accessibility Bent Petersen 13 January 2011 1 NetSurfP Real Value Solvent Accessibility predictions with amino acid associated
An Artificial Neural Network Classifier for the Prediction of Protein Structural Classes
 International Journal of Current Engineering and Technology E-ISSN 2277 4106, P-ISSN 2347 5161 2017 INPRESSCO, All Rights Reserved Available at http://inpressco.com/category/ijcet Research Article An Artificial
International Journal of Current Engineering and Technology E-ISSN 2277 4106, P-ISSN 2347 5161 2017 INPRESSCO, All Rights Reserved Available at http://inpressco.com/category/ijcet Research Article An Artificial
Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction. Sepp Hochreiter
 Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction Institute of Bioinformatics Johannes Kepler University, Linz, Austria Chapter 4 Protein Secondary
Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction Institute of Bioinformatics Johannes Kepler University, Linz, Austria Chapter 4 Protein Secondary
Computational Genomics and Molecular Biology, Fall
 Computational Genomics and Molecular Biology, Fall 2014 1 HMM Lecture Notes Dannie Durand and Rose Hoberman November 6th Introduction In the last few lectures, we have focused on three problems related
Computational Genomics and Molecular Biology, Fall 2014 1 HMM Lecture Notes Dannie Durand and Rose Hoberman November 6th Introduction In the last few lectures, we have focused on three problems related
Bioinformatics: Secondary Structure Prediction
 Bioinformatics: Secondary Structure Prediction Prof. David Jones d.jones@cs.ucl.ac.uk LMLSTQNPALLKRNIIYWNNVALLWEAGSD The greatest unsolved problem in molecular biology:the Protein Folding Problem? Entries
Bioinformatics: Secondary Structure Prediction Prof. David Jones d.jones@cs.ucl.ac.uk LMLSTQNPALLKRNIIYWNNVALLWEAGSD The greatest unsolved problem in molecular biology:the Protein Folding Problem? Entries
Protein Structure Prediction and Display
 Protein Structure Prediction and Display Goal Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each
Protein Structure Prediction and Display Goal Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each
Biochemistry Prof. S. DasGupta Department of Chemistry Indian Institute of Technology Kharagpur. Lecture - 06 Protein Structure IV
 Biochemistry Prof. S. DasGupta Department of Chemistry Indian Institute of Technology Kharagpur Lecture - 06 Protein Structure IV We complete our discussion on Protein Structures today. And just to recap
Biochemistry Prof. S. DasGupta Department of Chemistry Indian Institute of Technology Kharagpur Lecture - 06 Protein Structure IV We complete our discussion on Protein Structures today. And just to recap
addresses: b Department of Mathematics and Statistics, G.N. Khalsa College, University of Mumbai, India. a.
 Reaching Optimized Parameter Set: Protein Secondary Structure Prediction Using Neural Network DongardiveJyotshna* a, Siby Abraham *b a Department of Computer Science, University of Mumbai, Mumbai, India
Reaching Optimized Parameter Set: Protein Secondary Structure Prediction Using Neural Network DongardiveJyotshna* a, Siby Abraham *b a Department of Computer Science, University of Mumbai, Mumbai, India
Protein Structure Prediction using String Kernels. Technical Report
 Protein Structure Prediction using String Kernels Technical Report Department of Computer Science and Engineering University of Minnesota 4-192 EECS Building 200 Union Street SE Minneapolis, MN 55455-0159
Protein Structure Prediction using String Kernels Technical Report Department of Computer Science and Engineering University of Minnesota 4-192 EECS Building 200 Union Street SE Minneapolis, MN 55455-0159
Amino Acid Structures from Klug & Cummings. 10/7/2003 CAP/CGS 5991: Lecture 7 1
 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 1 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 2 Amino Acid Structures from Klug & Cummings
Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 1 Amino Acid Structures from Klug & Cummings 10/7/2003 CAP/CGS 5991: Lecture 7 2 Amino Acid Structures from Klug & Cummings
CHAPTER 29 HW: AMINO ACIDS + PROTEINS
 CAPTER 29 W: AMI ACIDS + PRTEIS For all problems, consult the table of 20 Amino Acids provided in lecture if an amino acid structure is needed; these will be given on exams. Use natural amino acids (L)
CAPTER 29 W: AMI ACIDS + PRTEIS For all problems, consult the table of 20 Amino Acids provided in lecture if an amino acid structure is needed; these will be given on exams. Use natural amino acids (L)
Steps in protein modelling. Structure prediction, fold recognition and homology modelling. Basic principles of protein structure
 Structure prediction, fold recognition and homology modelling Marjolein Thunnissen Lund September 2012 Steps in protein modelling 3-D structure known Comparative Modelling Sequence of interest Similarity
Structure prediction, fold recognition and homology modelling Marjolein Thunnissen Lund September 2012 Steps in protein modelling 3-D structure known Comparative Modelling Sequence of interest Similarity
HMM applications. Applications of HMMs. Gene finding with HMMs. Using the gene finder
 HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Enhanced membrane protein topology prediction using a hierarchical classification method and a new scoring function
 Enhanced membrane protein topology prediction using a hierarchical classification method and a new scoring function Allan Lo 1, 2, Hua-Sheng Chiu 3, Ting-Yi Sung 3, Ping-Chiang Lyu 2, and Wen-Lian Hsu
Enhanced membrane protein topology prediction using a hierarchical classification method and a new scoring function Allan Lo 1, 2, Hua-Sheng Chiu 3, Ting-Yi Sung 3, Ping-Chiang Lyu 2, and Wen-Lian Hsu
SINGLE-SEQUENCE PROTEIN SECONDARY STRUCTURE PREDICTION BY NEAREST-NEIGHBOR CLASSIFICATION OF PROTEIN WORDS
 SINGLE-SEQUENCE PROTEIN SECONDARY STRUCTURE PREDICTION BY NEAREST-NEIGHBOR CLASSIFICATION OF PROTEIN WORDS Item type Authors Publisher Rights text; Electronic Thesis PORFIRIO, DAVID JONATHAN The University
SINGLE-SEQUENCE PROTEIN SECONDARY STRUCTURE PREDICTION BY NEAREST-NEIGHBOR CLASSIFICATION OF PROTEIN WORDS Item type Authors Publisher Rights text; Electronic Thesis PORFIRIO, DAVID JONATHAN The University
Bayesian Models and Algorithms for Protein Beta-Sheet Prediction
 0 Bayesian Models and Algorithms for Protein Beta-Sheet Prediction Zafer Aydin, Student Member, IEEE, Yucel Altunbasak, Senior Member, IEEE, and Hakan Erdogan, Member, IEEE Abstract Prediction of the three-dimensional
0 Bayesian Models and Algorithms for Protein Beta-Sheet Prediction Zafer Aydin, Student Member, IEEE, Yucel Altunbasak, Senior Member, IEEE, and Hakan Erdogan, Member, IEEE Abstract Prediction of the three-dimensional
Review. Membrane proteins. Membrane transport
 Quiz 1 For problem set 11 Q1, you need the equation for the average lateral distance transversed (s) of a molecule in the membrane with respect to the diffusion constant (D) and time (t). s = (4 D t) 1/2
Quiz 1 For problem set 11 Q1, you need the equation for the average lateral distance transversed (s) of a molecule in the membrane with respect to the diffusion constant (D) and time (t). s = (4 D t) 1/2
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Protein 8-class Secondary Structure Prediction Using Conditional Neural Fields
 2010 IEEE International Conference on Bioinformatics and Biomedicine Protein 8-class Secondary Structure Prediction Using Conditional Neural Fields Zhiyong Wang, Feng Zhao, Jian Peng, Jinbo Xu* Toyota
2010 IEEE International Conference on Bioinformatics and Biomedicine Protein 8-class Secondary Structure Prediction Using Conditional Neural Fields Zhiyong Wang, Feng Zhao, Jian Peng, Jinbo Xu* Toyota
Tutorial: Structural Analysis of a Protein-Protein Complex
 Molecular Modeling Section (MMS) Department of Pharmaceutical and Pharmacological Sciences University of Padova Via Marzolo 5-35131 Padova (IT) @contact: stefano.moro@unipd.it Tutorial: Structural Analysis
Molecular Modeling Section (MMS) Department of Pharmaceutical and Pharmacological Sciences University of Padova Via Marzolo 5-35131 Padova (IT) @contact: stefano.moro@unipd.it Tutorial: Structural Analysis
Motif Prediction in Amino Acid Interaction Networks
 Motif Prediction in Amino Acid Interaction Networks Omar GACI and Stefan BALEV Abstract In this paper we represent a protein as a graph where the vertices are amino acids and the edges are interactions
Motif Prediction in Amino Acid Interaction Networks Omar GACI and Stefan BALEV Abstract In this paper we represent a protein as a graph where the vertices are amino acids and the edges are interactions
A New Similarity Measure among Protein Sequences
 A New Similarity Measure among Protein Sequences Kuen-Pin Wu, Hsin-Nan Lin, Ting-Yi Sung and Wen-Lian Hsu * Institute of Information Science Academia Sinica, Taipei 115, Taiwan Abstract Protein sequence
A New Similarity Measure among Protein Sequences Kuen-Pin Wu, Hsin-Nan Lin, Ting-Yi Sung and Wen-Lian Hsu * Institute of Information Science Academia Sinica, Taipei 115, Taiwan Abstract Protein sequence
Secondary Structure. Bioch/BIMS 503 Lecture 2. Structure and Function of Proteins. Further Reading. Φ, Ψ angles alone determine protein structure
 Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Bioinformatics: Secondary Structure Prediction
 Bioinformatics: Secondary Structure Prediction Prof. David Jones d.t.jones@ucl.ac.uk Possibly the greatest unsolved problem in molecular biology: The Protein Folding Problem MWMPPRPEEVARK LRRLGFVERMAKG
Bioinformatics: Secondary Structure Prediction Prof. David Jones d.t.jones@ucl.ac.uk Possibly the greatest unsolved problem in molecular biology: The Protein Folding Problem MWMPPRPEEVARK LRRLGFVERMAKG
A General Method for Combining Predictors Tested on Protein Secondary Structure Prediction
 A General Method for Combining Predictors Tested on Protein Secondary Structure Prediction Jakob V. Hansen Department of Computer Science, University of Aarhus Ny Munkegade, Bldg. 540, DK-8000 Aarhus C,
A General Method for Combining Predictors Tested on Protein Secondary Structure Prediction Jakob V. Hansen Department of Computer Science, University of Aarhus Ny Munkegade, Bldg. 540, DK-8000 Aarhus C,
Molecular Modeling Lecture 7. Homology modeling insertions/deletions manual realignment
 Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Bioinformatics Practical for Biochemists
 Bioinformatics Practical for Biochemists Andrei Lupas, Birte Höcker, Steffen Schmidt WS 2013/14 03. Sequence Features Targeting proteins signal peptide targets proteins to the secretory pathway N-terminal
Bioinformatics Practical for Biochemists Andrei Lupas, Birte Höcker, Steffen Schmidt WS 2013/14 03. Sequence Features Targeting proteins signal peptide targets proteins to the secretory pathway N-terminal
Optimization of the Sliding Window Size for Protein Structure Prediction
 Optimization of the Sliding Window Size for Protein Structure Prediction Ke Chen* 1, Lukasz Kurgan 1 and Jishou Ruan 2 1 University of Alberta, Department of Electrical and Computer Engineering, Edmonton,
Optimization of the Sliding Window Size for Protein Structure Prediction Ke Chen* 1, Lukasz Kurgan 1 and Jishou Ruan 2 1 University of Alberta, Department of Electrical and Computer Engineering, Edmonton,
Model Accuracy Measures
 Model Accuracy Measures Master in Bioinformatics UPF 2017-2018 Eduardo Eyras Computational Genomics Pompeu Fabra University - ICREA Barcelona, Spain Variables What we can measure (attributes) Hypotheses
Model Accuracy Measures Master in Bioinformatics UPF 2017-2018 Eduardo Eyras Computational Genomics Pompeu Fabra University - ICREA Barcelona, Spain Variables What we can measure (attributes) Hypotheses
Improving Protein Secondary-Structure Prediction by Predicting Ends of Secondary-Structure Segments
 Improving Protein Secondary-Structure Prediction by Predicting Ends of Secondary-Structure Segments Uros Midic 1 A. Keith Dunker 2 Zoran Obradovic 1* 1 Center for Information Science and Technology Temple
Improving Protein Secondary-Structure Prediction by Predicting Ends of Secondary-Structure Segments Uros Midic 1 A. Keith Dunker 2 Zoran Obradovic 1* 1 Center for Information Science and Technology Temple
Can protein model accuracy be. identified? NO! CBS, BioCentrum, Morten Nielsen, DTU
 Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Physiochemical Properties of Residues
 Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
PROTEIN SECONDARY STRUCTURE PREDICTION: AN APPLICATION OF CHOU-FASMAN ALGORITHM IN A HYPOTHETICAL PROTEIN OF SARS VIRUS
 Int. J. LifeSc. Bt & Pharm. Res. 2012 Kaladhar, 2012 Research Paper ISSN 2250-3137 www.ijlbpr.com Vol.1, Issue. 1, January 2012 2012 IJLBPR. All Rights Reserved PROTEIN SECONDARY STRUCTURE PREDICTION:
Int. J. LifeSc. Bt & Pharm. Res. 2012 Kaladhar, 2012 Research Paper ISSN 2250-3137 www.ijlbpr.com Vol.1, Issue. 1, January 2012 2012 IJLBPR. All Rights Reserved PROTEIN SECONDARY STRUCTURE PREDICTION:
Analysis and Prediction of Protein Structure (I)
") Analysis and Prediction of Protein Structure (I) Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 2006 Free for academic use. Copyright @ Jianlin Cheng
Analysis and Prediction of Protein Structure (I) Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 2006 Free for academic use. Copyright @ Jianlin Cheng
Conditional Graphical Models
 PhD Thesis Proposal Conditional Graphical Models for Protein Structure Prediction Yan Liu Language Technologies Institute University Thesis Committee Jaime Carbonell (Chair) John Lafferty Eric P. Xing
PhD Thesis Proposal Conditional Graphical Models for Protein Structure Prediction Yan Liu Language Technologies Institute University Thesis Committee Jaime Carbonell (Chair) John Lafferty Eric P. Xing
Protein quality assessment
 Protein quality assessment Speaker: Renzhi Cao Advisor: Dr. Jianlin Cheng Major: Computer Science May 17 th, 2013 1 Outline Introduction Paper1 Paper2 Paper3 Discussion and research plan Acknowledgement
Protein quality assessment Speaker: Renzhi Cao Advisor: Dr. Jianlin Cheng Major: Computer Science May 17 th, 2013 1 Outline Introduction Paper1 Paper2 Paper3 Discussion and research plan Acknowledgement
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
Packing of Secondary Structures
 7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
Towards De Novo Folding of Protein Structures from Cryo-EM 3D Images at Medium Resolutions
 Towards De Novo Folding of Protein Structures from Cryo-EM 3D Images at Medium Resolutions Jing He * and Dong Si Department of Computer Science Old Dominion University Norfolk, VA * jhe@cs.odu.edu Abstract
Towards De Novo Folding of Protein Structures from Cryo-EM 3D Images at Medium Resolutions Jing He * and Dong Si Department of Computer Science Old Dominion University Norfolk, VA * jhe@cs.odu.edu Abstract
TMHMM2.0 User's guide
 TMHMM2.0 User's guide This program is for prediction of transmembrane helices in proteins. July 2001: TMHMM has been rated best in an independent comparison of programs for prediction of TM helices: S.
TMHMM2.0 User's guide This program is for prediction of transmembrane helices in proteins. July 2001: TMHMM has been rated best in an independent comparison of programs for prediction of TM helices: S.
Orientational degeneracy in the presence of one alignment tensor.
 Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Protein Secondary Structure Prediction using Pattern Recognition Neural Network
 Protein Secondary Structure Prediction using Pattern Recognition Neural Network P.V. Nageswara Rao 1 (nagesh@gitam.edu), T. Uma Devi 1, DSVGK Kaladhar 1, G.R. Sridhar 2, Allam Appa Rao 3 1 GITAM University,
Protein Secondary Structure Prediction using Pattern Recognition Neural Network P.V. Nageswara Rao 1 (nagesh@gitam.edu), T. Uma Devi 1, DSVGK Kaladhar 1, G.R. Sridhar 2, Allam Appa Rao 3 1 GITAM University,
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
BCB 444/544 Fall 07 Dobbs 1
 BCB 444/544 Lecture 21 Protein Structure Visualization, Classification & Comparison Secondary Structure #21_Oct10 Required Reading (before lecture) Mon Oct 8 - Lecture 20 Protein Secondary Structure Chp
BCB 444/544 Lecture 21 Protein Structure Visualization, Classification & Comparison Secondary Structure #21_Oct10 Required Reading (before lecture) Mon Oct 8 - Lecture 20 Protein Secondary Structure Chp
2MHR. Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity.
 Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
Protein structure classification is important because it organizes the protein structure universe that is independent of sequence similarity. A global picture of the protein universe will help us to understand
From Amino Acids to Proteins - in 4 Easy Steps
 From Amino Acids to Proteins - in 4 Easy Steps Although protein structure appears to be overwhelmingly complex, you can provide your students with a basic understanding of how proteins fold by focusing
From Amino Acids to Proteins - in 4 Easy Steps Although protein structure appears to be overwhelmingly complex, you can provide your students with a basic understanding of how proteins fold by focusing
Algorithm-Independent Learning Issues
 Algorithm-Independent Learning Issues Selim Aksoy Department of Computer Engineering Bilkent University saksoy@cs.bilkent.edu.tr CS 551, Spring 2007 c 2007, Selim Aksoy Introduction We have seen many learning
Algorithm-Independent Learning Issues Selim Aksoy Department of Computer Engineering Bilkent University saksoy@cs.bilkent.edu.tr CS 551, Spring 2007 c 2007, Selim Aksoy Introduction We have seen many learning
Protein Structures. Sequences of amino acid residues 20 different amino acids. Quaternary. Primary. Tertiary. Secondary. 10/8/2002 Lecture 12 1
 Protein Structures Sequences of amino acid residues 20 different amino acids Primary Secondary Tertiary Quaternary 10/8/2002 Lecture 12 1 Angles φ and ψ in the polypeptide chain 10/8/2002 Lecture 12 2
Protein Structures Sequences of amino acid residues 20 different amino acids Primary Secondary Tertiary Quaternary 10/8/2002 Lecture 12 1 Angles φ and ψ in the polypeptide chain 10/8/2002 Lecture 12 2
Pattern Recognition Prof. P. S. Sastry Department of Electronics and Communication Engineering Indian Institute of Science, Bangalore
 Pattern Recognition Prof. P. S. Sastry Department of Electronics and Communication Engineering Indian Institute of Science, Bangalore Lecture - 27 Multilayer Feedforward Neural networks with Sigmoidal
Pattern Recognition Prof. P. S. Sastry Department of Electronics and Communication Engineering Indian Institute of Science, Bangalore Lecture - 27 Multilayer Feedforward Neural networks with Sigmoidal
Holdout and Cross-Validation Methods Overfitting Avoidance
 Holdout and Cross-Validation Methods Overfitting Avoidance Decision Trees Reduce error pruning Cost-complexity pruning Neural Networks Early stopping Adjusting Regularizers via Cross-Validation Nearest
Holdout and Cross-Validation Methods Overfitting Avoidance Decision Trees Reduce error pruning Cost-complexity pruning Neural Networks Early stopping Adjusting Regularizers via Cross-Validation Nearest
Protein Structure and Function Prediction using Kernel Methods.
 Protein Structure and Function Prediction using Kernel Methods. A THESIS SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA BY Huzefa Rangwala IN PARTIAL FULFILLMENT OF THE
Protein Structure and Function Prediction using Kernel Methods. A THESIS SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA BY Huzefa Rangwala IN PARTIAL FULFILLMENT OF THE
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
Protein Structure Prediction Using Multiple Artificial Neural Network Classifier *
 Protein Structure Prediction Using Multiple Artificial Neural Network Classifier * Hemashree Bordoloi and Kandarpa Kumar Sarma Abstract. Protein secondary structure prediction is the method of extracting
Protein Structure Prediction Using Multiple Artificial Neural Network Classifier * Hemashree Bordoloi and Kandarpa Kumar Sarma Abstract. Protein secondary structure prediction is the method of extracting
Model Mélange. Physical Models of Peptides and Proteins
 Model Mélange Physical Models of Peptides and Proteins In the Model Mélange activity, you will visit four different stations each featuring a variety of different physical models of peptides or proteins.
Model Mélange Physical Models of Peptides and Proteins In the Model Mélange activity, you will visit four different stations each featuring a variety of different physical models of peptides or proteins.
Heteropolymer. Mostly in regular secondary structure
 Heteropolymer - + + - Mostly in regular secondary structure 1 2 3 4 C >N trace how you go around the helix C >N C2 >N6 C1 >N5 What s the pattern? Ci>Ni+? 5 6 move around not quite 120 "#$%&'!()*(+2!3/'!4#5'!1/,#64!#6!,6!
Heteropolymer - + + - Mostly in regular secondary structure 1 2 3 4 C >N trace how you go around the helix C >N C2 >N6 C1 >N5 What s the pattern? Ci>Ni+? 5 6 move around not quite 120 "#$%&'!()*(+2!3/'!4#5'!1/,#64!#6!,6!
Prediction. Emily Wei Xu. A thesis. presented to the University of Waterloo. in fulfillment of the. thesis requirement for the degree of
 The Use of Internal and External Functional Domains to Improve Transmembrane Protein Topology Prediction by Emily Wei Xu A thesis presented to the University of Waterloo in fulfillment of the thesis requirement
The Use of Internal and External Functional Domains to Improve Transmembrane Protein Topology Prediction by Emily Wei Xu A thesis presented to the University of Waterloo in fulfillment of the thesis requirement
) P = 1 if exp # " s. + 0 otherwise
 P = 1 if exp # s. + 0 otherwise") Supplementary Material Monte Carlo algorithm procedures. The Monte Carlo conformational search algorithm has been successfully applied by programs dedicated to finding new folds (Jones 2001; Rohl, Strauss,
Supplementary Material Monte Carlo algorithm procedures. The Monte Carlo conformational search algorithm has been successfully applied by programs dedicated to finding new folds (Jones 2001; Rohl, Strauss,
1. Protein Data Bank (PDB) 1. Protein Data Bank (PDB)
 1. Protein Data Bank (PDB)") Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
10-701/ Machine Learning, Fall
 0-70/5-78 Machine Learning, Fall 2003 Homework 2 Solution If you have questions, please contact Jiayong Zhang .. (Error Function) The sum-of-squares error is the most common training
0-70/5-78 Machine Learning, Fall 2003 Homework 2 Solution If you have questions, please contact Jiayong Zhang .. (Error Function) The sum-of-squares error is the most common training
Protein Structure: Data Bases and Classification Ingo Ruczinski
 Protein Structure: Data Bases and Classification Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Reference Bourne and Weissig Structural Bioinformatics Wiley, 2003 More References
Protein Structure: Data Bases and Classification Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Reference Bourne and Weissig Structural Bioinformatics Wiley, 2003 More References
ARTIFICIAL NEURAL NETWORK PART I HANIEH BORHANAZAD
 ARTIFICIAL NEURAL NETWORK PART I HANIEH BORHANAZAD WHAT IS A NEURAL NETWORK? The simplest definition of a neural network, more properly referred to as an 'artificial' neural network (ANN), is provided
ARTIFICIAL NEURAL NETWORK PART I HANIEH BORHANAZAD WHAT IS A NEURAL NETWORK? The simplest definition of a neural network, more properly referred to as an 'artificial' neural network (ANN), is provided
Identification of Representative Protein Sequence and Secondary Structure Prediction Using SVM Approach
 Identification of Representative Protein Sequence and Secondary Structure Prediction Using SVM Approach Prof. Dr. M. A. Mottalib, Md. Rahat Hossain Department of Computer Science and Information Technology
Identification of Representative Protein Sequence and Secondary Structure Prediction Using SVM Approach Prof. Dr. M. A. Mottalib, Md. Rahat Hossain Department of Computer Science and Information Technology
SUPPLEMENTARY INFORMATION
 Supplementary Figure 1: The HpUreI crystal used for collection of native diffraction data. The crystal belongs to spacegroup P4 2 2 1 2 and has an approximate maximal dimension of 0.25 mm. Supplementary
Supplementary Figure 1: The HpUreI crystal used for collection of native diffraction data. The crystal belongs to spacegroup P4 2 2 1 2 and has an approximate maximal dimension of 0.25 mm. Supplementary
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools. Giri Narasimhan
 CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinff18.html Proteins and Protein Structure
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinff18.html Proteins and Protein Structure
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Supporting Information
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2016 Supporting Information Lipid molecules can induce an opening of membrane-facing
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 2016 Supporting Information Lipid molecules can induce an opening of membrane-facing
Protein Bioinformatics. Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet sandberg.cmb.ki.
 Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Protein Bioinformatics Rickard Sandberg Dept. of Cell and Molecular Biology Karolinska Institutet rickard.sandberg@ki.se sandberg.cmb.ki.se Outline Protein features motifs patterns profiles signals 2 Protein
Advanced Certificate in Principles in Protein Structure. You will be given a start time with your exam instructions
 BIRKBECK COLLEGE (University of London) Advanced Certificate in Principles in Protein Structure MSc Structural Molecular Biology Date: Thursday, 1st September 2011 Time: 3 hours You will be given a start
BIRKBECK COLLEGE (University of London) Advanced Certificate in Principles in Protein Structure MSc Structural Molecular Biology Date: Thursday, 1st September 2011 Time: 3 hours You will be given a start
Predictors (of secondary structure) based on Machine Learning tools
 based on Machine Learning tools") Predictors (of secondary structure) based on Machine Learning tools Predictors of secondary structure 1 Generation methods: propensity of each residue to be in a given conformation Chou-Fasman 2 Generation
Predictors (of secondary structure) based on Machine Learning tools Predictors of secondary structure 1 Generation methods: propensity of each residue to be in a given conformation Chou-Fasman 2 Generation
SUPPLEMENTARY INFORMATION
 SUPPLEMENTARY INFORMATION Structure of human carbamoyl phosphate synthetase: deciphering the on/off switch of human ureagenesis Sergio de Cima, Luis M. Polo, Carmen Díez-Fernández, Ana I. Martínez, Javier
SUPPLEMENTARY INFORMATION Structure of human carbamoyl phosphate synthetase: deciphering the on/off switch of human ureagenesis Sergio de Cima, Luis M. Polo, Carmen Díez-Fernández, Ana I. Martínez, Javier
Protein structure. Protein structure. Amino acid residue. Cell communication channel. Bioinformatics Methods
 Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Section III - Designing Models for 3D Printing
 Section III - Designing Models for 3D Printing In this section of the Jmol Training Guide, you will become familiar with the commands needed to design a model that will be built on a 3D Printer. As you
Section III - Designing Models for 3D Printing In this section of the Jmol Training Guide, you will become familiar with the commands needed to design a model that will be built on a 3D Printer. As you
Getting To Know Your Protein
 Getting To Know Your Protein Comparative Protein Analysis: Part III. Protein Structure Prediction and Comparison Robert Latek, PhD Sr. Bioinformatics Scientist Whitehead Institute for Biomedical Research
Getting To Know Your Protein Comparative Protein Analysis: Part III. Protein Structure Prediction and Comparison Robert Latek, PhD Sr. Bioinformatics Scientist Whitehead Institute for Biomedical Research
AI Programming CS F-20 Neural Networks
 AI Programming CS662-2008F-20 Neural Networks David Galles Department of Computer Science University of San Francisco 20-0: Symbolic AI Most of this class has been focused on Symbolic AI Focus or symbols
AI Programming CS662-2008F-20 Neural Networks David Galles Department of Computer Science University of San Francisco 20-0: Symbolic AI Most of this class has been focused on Symbolic AI Focus or symbols
PROTEIN SUBCELLULAR LOCALIZATION PREDICTION BASED ON COMPARTMENT-SPECIFIC BIOLOGICAL FEATURES
 3251 PROTEIN SUBCELLULAR LOCALIZATION PREDICTION BASED ON COMPARTMENT-SPECIFIC BIOLOGICAL FEATURES Chia-Yu Su 1,2, Allan Lo 1,3, Hua-Sheng Chiu 4, Ting-Yi Sung 4, Wen-Lian Hsu 4,* 1 Bioinformatics Program,
3251 PROTEIN SUBCELLULAR LOCALIZATION PREDICTION BASED ON COMPARTMENT-SPECIFIC BIOLOGICAL FEATURES Chia-Yu Su 1,2, Allan Lo 1,3, Hua-Sheng Chiu 4, Ting-Yi Sung 4, Wen-Lian Hsu 4,* 1 Bioinformatics Program,