Bioinformatics: Secondary Structure Prediction
|
|
|
- Laurence Hood
- 5 years ago
- Views:
Transcription
1 Bioinformatics: Secondary Structure Prediction Prof. David Jones

2 Possibly the greatest unsolved problem in molecular biology: The Protein Folding Problem MWMPPRPEEVARK LRRLGFVERMAKG GHRLYTHPDGRIV VVPFHSGELPKGT FKRILRDAGLTEE EFHNL?








3 biological multimeric state evolutionary relationships INTERACTIONS MULTIMERS FOLD MUTANTS & SNPs 3D STRUCTURE SURFACE SHAPE LIGANDS ELECTROSTATICS CLUSTERS ligand & functional sites catalytic clusters, mechanisms & motifs enzyme active sites Original image by J.M. Thornton
4 Why predict structure? Growth of sequence and structure data banks Structures Sequences Structures Sequences Number of Proteins Number of Proteins Year Year
5 Growth of sequence and structure data banks Structures Sequences Structures Sequences Number of Proteins Number of Proteins Year Year



6 Protein Secondary Structure ALPHA HELIX BETA SHEET (STRAND) COIL
7 Secondary Structure Prediction L M L S T Q N P A L L K R N I I Y W N N V A L L W E A G S D INPUT: 20-letter Amino Acid Alphabet? LMLSTQNPALL HHHEEEECCCC OUTPUT: 3-letter Secondary Structure Alphabet H = Helix E = Extended (strand) C = Coil
8 Goal Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure (helix/strand/coil) that is most likely to be adopted by each residue
9 1st Generation Methods Based on statistical analysis of single amino acid residues Examples: Chou & Fasman (1974) Lim (1974) Garnier, Osguthorpe & Robson (GOR) (1978) Although these methods were important early contributions to the field and are a good starting point for teaching purposes, in practice these methods are now considered ENTIRELY OBSOLETE DON T USE THEM!
10 Chou-Fasman method Uses table of conformational parameters (propensities) determined primarily from measurements of secondary structure by CD spectroscopy Table consists of one likelihood for each structure for each amino acid For amino acid type A (e.g. leucine/serine/proline etc.) and structure type S (e.g. α helix), a propensity score can be calculated as follows: p( A S) na, S ns PS = = p( A) n n A
11 Chou-Fasman propensities (partial table) A m i n o A c i d P α P β P t G l u M e t A l a V a l I l e T y r P r o G l y
12 Chou-Fasman method Calculation rules are somewhat ad hoc Example: Method for helix Search for nucleating region where 4 out of 6 a.a. have P α > 1.03 Extend until 4 consecutive a.a. have an average P α < 1.00 If region is at least 6 a.a. long, has an average P α > 1.03, and average P α > average P β, consider region to be helix Unclear in what order the rules should be applied
13 2nd Generation Methods Secondary structure effects strongly depend on neighbouring residues 2 nd Gen methods therefore based on residue pairs or peptide segments Example: GOR III (1987) The BIG NEWS, however, was the appearance of the first examples of MACHINE LEARNING in secondary structure prediction Neural Networks: Qian & Sejnowski (1988), Bohr et al. (1988), Holley & Karplus (1989)
14 Neural Networks Originally, neural network methods were developed to model brain function i.e. they were intended to allow simulations of real networks of neurons. Hence the term Artificial Neural Network. Today these very simple models are obsolete in neuroscience research but instead have become very useful tools for finding patterns in data (data mining).
15 Artificial Neural Networks Input Layer Hidden Layer Output Layer Inputs Output Usually, each layer is fully connected to the next layer An artificial neural network (ANN) is a set of linked mathematical functions, which can be thought of as a network of switching units (artificial neurons) connected together according to a specific architecture. The objective of an artificial neural network is to learn how to transform inputs into meaningful outputs.
16 This is the activation function Trivial Example The Perceptron Input variables Inputs % Alpha helix 34 % Beta sheet 24 Protein length Σ Connection Weights Output = e -(Σ + C) Neuron Output 0.8 Probability of being a TIM barrel enzyme Prediction
17 Training and Using ANNs The procedure for training a neural network is called BACKPROPAGATION We start with complete random connection weights Then we present an input pattern from our training set to the network and compare the calculated output to what it should be We then repeat this for all the examples we have Pseudocode for ANN training: initialize network weights (usually small random values) do foreach training example x prediction = neural-net-output(network, x) // forward pass actual = expected-output(x) // Calculate what output you expect to see compute error (prediction - actual) at the output units compute w h for all weights from hidden layer to output layer // backward pass compute w i for all weights from input layer to hidden layer // backward pass continued update network weights // input layer is not modified by the error estimate! until all training examples classified correctly or another stopping criterion satisfied return the final set of network weights
18 Training and Testing Sets The data we use to adjust the network weights is called the TRAINING SET To make sure we have not over-fitted our network to our training set, we should test the network on a completely separate TESTING SET This splitting of training and testing data is called CROSS- VALIDATION and is an important concept in statistics and machine learning as it allows us to predict how well a method is likely to work on entirely new data
19 Trivial example of Neural Network Training Data input output The OUTPUT is 1 when both INPUTS are different. This simple function CANNOT be learned by a perceptron because perceptrons can only learn linearly separable functions.
20 XOR Function input output w 5 w 6 θ = 0.5 for all units w 1 w 4 w 3 w 2 f(w 1, w 2, w 3, w 4, w 5, w 6 ) By using hidden units we can extend perceptrons to cover more complex functions e.g. the XOR function. a possible set of values for w s (w 1, w 2, w 3, w 4, w 5, w 6 ) (0.6,-0.6,-0.7,0.8,1,1) θ f(a) = 1, for a > θ 0, for a θ
21 Some real data... We randomly select some of the data for use as our TESTING SET and keep the rest for our TRAINING SET. Inputs A R N D C Q E G H I L K M F P S T W Y V Coil Strand Coil Strand Strand Helix Helix Coil Strand Helix Helix Helix Strand Helix Helix Strand Strand Strand Coil Helix Helix Coil Coil Coil... And so on... Desired Outputs
22 Predicting Secondary Structure by Machine Learning Rather than just one residue at a time, we look at neighbouring residues as well! Window of 15 residues Classifier (neural network) Helix Strand Coil MLSPQAMSDFHEELKWLLCNIPGQKLASLANREYT The goal is to predict the secondary structure for this central residue
23 Representation Usual representation is to use 21 inputs per amino acid: Ala : Val : :
24 3rd Generation Neural Network Methods PHD First method to exceed average accuracy of 70% - pioneered the field, but now lags slightly behind best modern methods. Still available as a Web server. Rost B, Sander, C. (1993) Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 232, PSIPRED ( Currently the best method for predicting secondary structure (>82% accuracy on average for latest version). Uses PSI-BLAST and a more sophisticated neural network model than PHD. Available both as a Web server and as a downloadable program. If you are only going to use one method, use this one! Jones, D.T. (1999) Protein secondary structure prediction based on positionspecific scoring matrices. J. Mol. Biol. 292:
25 3rd Generation Methods Exploit evolutionary information Based on conservation analysis of multiple sequence alignments (profiles or HMMs) Can extract some long-range information via accessibility patterns Conserved Hydrophobic Residues -> BURIED Variable Polar Residues -> EXPOSED


26 Residue Variability and Solvent Accessibility Number of a.a. types Relative Solvent Exposure In a family of proteins, positions that tolerate the widest choice of amino acid types across evolution are likely to have high solvent exposure.
27 PROs Pros & Cons of 3rd Generation High residue accuracy Methods Less underprediction of strands Good quality segment end predictions CONs Provides prediction for average FAMILY structure NOT THE STRUCTURE OF THE SPECIFIC TARGET SEQUENCE
28 PSIPRED Works directly on PSI-BLAST profiles (PSSMs) Uses 2 separate stages of neural networks First network predicts secondary structure Second network cleans outputs from 1st net Trained on profile from >3000 different proteins of known structure (taken from PDB) Observed secondary structures are defined by DSSP
29 DSSP DSSP (Define Secondary Structure of Proteins ) is the de facto standard method for assigning secondary structure to the amino acids of a protein, given its atomic coordinates. It begins by identifying the intra-backbone hydrogen bonds of the protein using a simple electrostatic definition. Eight types of secondary structure are assigned. The 3 10 helix (G), α-helix (H) and π- helix (I) are recognised by having a particular sequence of hydrogen bonds three, four, or five residues apart respectively. Two types of beta sheet structure are recognised: an isolated beta bridge is denoted with a symbol B, and long runs of bridges are given the symbol E (for extended structure). Finally, T is used for turns, and S is used for regions of high curvature i.e. sharp bends in the polypeptide chain. A blank space indicates coil. In secondary structure prediction, we typically consider G & H both as Helix states, B and E as Strand states and all other symbols as coil. Kabsch W, Sander C (1983). "Dictionary of protein secondary structure: pattern recognition of hydrogenbonded and geometrical features". Biopolymers 22 (12):
30 PSIPRED Neural Networks 1st level network 15x21 input units 75 hidden units 3 output units (H/E/C) 2nd level network 15x4 input units 55 hidden units 3 output units (H/E/C) These are BIG neural networks!
")


31 The PSIPRED Method (slightly simplified) This is the raw Position Specific Score Matrix (profile) generated by PSI-BLAST The raw values are normalised so that they range between 0 and 1 1 st Network 315 inputs 75 hidden units 3 outputs 2 nd Network 60 inputs 55 hidden units 3 outputs Note: 60 inputs rather than 45 why?
32 2 nd Network: Filtering Raw Predictions from 1 st Network Stage 1 outputs Stage 2 outputs Actual Oops! 49 T E L E V E R E V E P C G C S C W E E H I H P H V H A H C H E 49 T E L E V E R E V E P C G C S H W H E H I H P H V H A H C H E 49 T E 50 L E 51 V E 52 R E 53 V E 54 P C 55 G C 56 S H 57 W H 58 E H 59 I H 60 P H 61 V H 62 A H The second network is able to clean up many of the little blips produced by the first network! Essentially this is an example of DATA SMOOTHING.
33 Common Measures of Secondary Structure Prediction Accuracy Q 3 scores give the percentage of correctly predicted residues across 3 states (H,E,C) This is the most commonly used measure Other scores such as Matthew s Correlation Coefficient try to identify accuracy for individual states (Coil, Strand, Helix) and are more sensitive to over-prediction e.g. if you predict all residues to be random coil you will get a Q 3 score of around 50% just because around 50% of residues in proteins are in random coil regions. However, the MCC scores will be close to zero!
34 Matthews Correlation Coefficient Probably the more robust metric is the Matthews Correlation Coefficient (MCC). Here we calculate the MCC for predicted helix states: Methods are now so accurate (>80%) that it makes little difference whether you compare methods by Q 3 or MCC. Q 3 tends to win out because it s so easy to interpret!
35 Original (1999) PSIPRED Benchmark Results Mean Q 3 score: 77.8% (>82% today) Number Q3 Score (%)

")
36 Comparison of Generations by Average Q3 Scores GEN 1 GEN 2 GEN First use of neural networks Current best methods Chou & Fasman GOR I Qian & Sejnowski PHD (1994) PSIPRED 0

37 PSIPRED Example
38
39 Bioinformatics: Threading and Protein Fold Recognition Prof. David Jones
40 Sequence Comparison > 30% Identity between two protein sequences usually implies common structure and possibly common function However, there are many exceptions to this rule of thumb 1PLC 1NIN IDVLLGADDGSLAFVPSEFSISPGEKIVFKNNAGFPHNIVFDEDSIPS-GVDASKISM : X:.: : :.: :...:.::.. : :: :::.::: :...:.: :. ETYTVKLGSDKGLLVFEPAKLTIKPGDTVEFLNNKVPPHNVVFDAALNPAKSADLAK-SL PLC 1NIN SEEDLLNAKGETFEVAL---SNKGEYSFYCSPHQGAGMVGKVTVN- :...:X. : :::.::: ::.:::::::.:: SHKQLLMSPGQSTSTTFPADAPAGEYTFYCEPHRGAGMVGKITVAG

 Ribonuclease")
41 Similar Sequence Similar 3-D Structure (RMSD = 2.1 A, Seq. ID = 30%) Ribonuclease MC1 Ribonuclease Rh
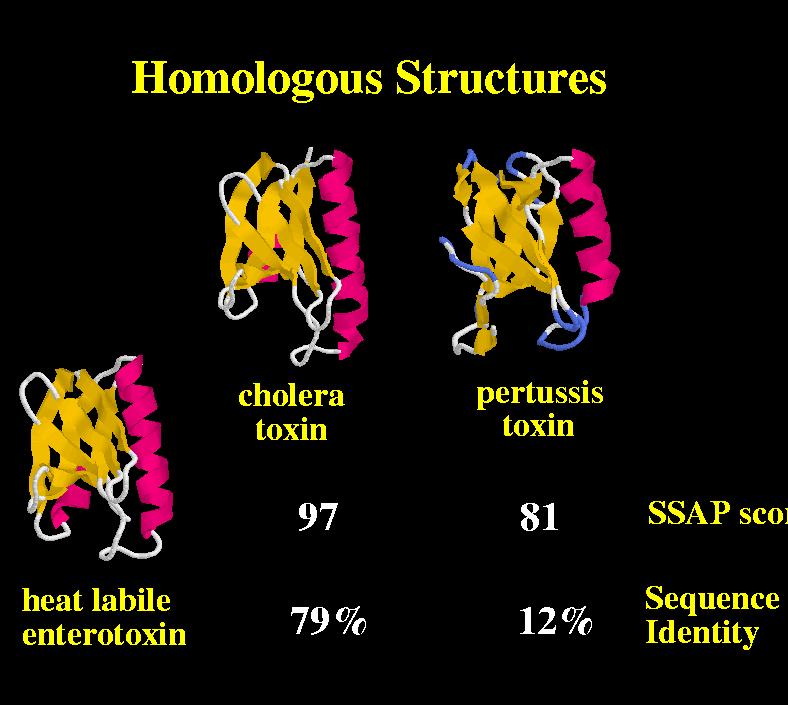
42 Structure similarity
43 Protein Structure Prediction Methods Increasing accuracy/reliability Comparative modelling Requires: Known fold + clear homology Fold recognition Requires: Known fold Ab initio / new fold methods Requires: only target sequence Increasing Difficulty
44 Tertiary Structure Prediction BASIS: native fold is expected to be the conformation of lowest (free) energy True for small molecules Almost certainly true for small protein domains May well be true for larger protein domains but larger proteins may end up kinetically trapped in a low but not lowest free energy state IMPLICATION: native fold can be found by defining a potential energy function and searching all conformations for the one with lowest energy
45 The Levinthal Paradox For a protein sequence of length l, the total number of possible chain conformations N is given by: N 10 l >> Even if a protein was able to rearrange itself at the speed of light it would take ~10 75 years to locate the global energy minimum for a 100 residue protein. >> Proteins evolve to have predefined folding pathways which they follow to the native structure
46 Fold recognition a short cut to predicting protein tertiary structure Although there are vast number of possible protein structures, only a few have been observed in Nature The chance of a newly solved structure having a previously unknown fold is only ~10% >> We might be able to predict protein structure by selecting from already observed folds (a Multiple Choice Question version of the protein folding problem!)

47 Protein Structure Prediction by Threading
48 A Scoring Function for Threading We want a way of assessing the energy of a model i.e. a model based on an alignment of a sequence with a structure Energy functions based on physics do not work Let s use a KNOWLEDGE-BASED STATISTICAL APPROACH Look at known structures and see how often particular features (e.g. atomic contacts between amino acids) occur. In other words, we start by estimating probabilities.
49 Native template structure (fragment) T G P A S K Native structure oppositely charged side chains help stabilise the fold. D I Q
50 First Attempt Threading alignment - W R T M E D Y Ouch! Two negatively charged side chains can t fit here because they would tend to destabilize this structure! S
51 Threading alignment 2 - W - T M E R D Ah, that s better - opposite charges again! Y
52 Converting Probabilities to Energy Estimates Probabilities are inconvenient for computer algorithms as we need to multiply them to combine them and we end up with very small numbers Additive quantities (e.g. energies) are much easier to handle >> For many applications it is common to transform probabilities into additive energy-like quantities
53 The Inverse Boltzmann Principle The basis of generating statistical potentials from probabilities comes from the Inverse Boltzmann principle. According to the normal Boltzmann principle, the probability of occurrence of a given conformational state of energy E scales with the Boltzmann factor e -E /RT, where R is the gas constant (1.987 x 10-3 kcal.mol -1 K -1 ) and T is the absolute temperature (e.g. room temperature).
54 Potentials of Mean Force Count interactions of given type (e.g. alanine->serine beta-carbon to alpha-carbon) in real protein structures Count interactions of same type in randomly generated protein structures or randomly selected sites (the reference state) Ratio of probabilities provides an estimate of the free energy change according to the inverse Boltzmann equation: G = RT log p( interaction in real proteins) p( interaction in decoy structures) RT log Z These energies are sometimes referred to as POTENTIALS OF MEAN FORCE because we are not considering all of the different component forces (e.g. electrostatic/van der Waals etc.), but instead just an average overall force. This bit is usually left out as it doesn t involve information from the structure! G High Energy State Low Energy State
55 Calculating Potentials of Mean Force Separation in Sequence = 4 Spatial Distance = 10 Å A e.g. Cα atoms (but could be any pair of atom types) S How often do we see this configuration in folded proteins? What about in unfolded protein chains?

56 EXAMPLE: Short-range Cβ Pair Potential of Mean Force Sequence Separation 4) Potential What creates this apparently favourable interaction? Distance (A) Here we have done the same calculation shown on the previous slide, but for a range of different spatial distances (4-10 Angstroms)
 terms usually included Can scale terms according to protein size This method can also be used to spot mistakes in X-ray or NMR")
57 We can estimate the stability of a given protein model by summing potentials of mean force for all residue pairs Loops are sometimes ignored Single residue (solvation) terms usually included Can scale terms according to protein size This method can also be used to spot mistakes in X-ray or NMR structures!

58 Partially correct Immunoglobulin Heavy Chain Fab Fragment Homology Model
59 Partially correct Immunoglobulin Heavy Chain Fab Fragment Model Coloured by a Potential of Mean Force + HIGH ENERGY SCALE - LOW
60 Search Problem: Threading Searching for the optimum mapping of sequence to structure while optimizing the sum of pair interactions is NP-complete (proven by R. Lathrop in 1994) i.e. an exhaustive search is needed to guarantee an optimal solution This search process is called THREADING i.e. what is the best way of threading this sequence through this structure In practice due to short range of pair potentials, heuristic solutions work fairly well: Exhaustive search (not practical) Dynamic programming. Double dynamic programming. Branch and bound. Monte Carlo methods: simulated annealing, Gibbs sampling, genetic algorithms.
61 Threading Methods in Practice Compared to comparative modelling, threading methods can produce models where there is no detectable homologue to be found in PDB. The simplifying assumption that the backbone of the structure does not change when the sequence changes also results in poor recognition of folds (~30% reliability) and inaccurate models (i.e. inaccurate alignments). For distant homologues, however, we can combine information from sequence profiles and secondary structure to improve alignments and make much better models than from threading potentials alone. This is what most modern fold recognition or threading methods do.
62 Examples of Fold Recognition Methods THREADER The original (1992) threading method Uses threading potentials and predicted secondary structure GenTHREADER Update to the original threading methods Combines threading potentials and predicted secondary structure of THREADER with profile-profile matching 3D-PSSM Combines sequence profiles and predicted secondary structure HHpred Combines HMMs with predicted secondary structure MUSTER Combines multiple sources of information, including secondary structure and sequence-profile information
 Although slower, fold recognition can find many more homologues than sequence comparison methods even")
63 Comparing Sequence Alignment to Fold Recognition in Homology Searching Run times to search a set of bacterial sequences for matches to Adenylate Kinase structure Method Hits Time Taken BLAST 61 8 sec PSIBLAST min GenTHREADER min Adenylate Kinase All hits at same error rate (1%) Although slower, fold recognition can find many more homologues than sequence comparison methods even PSI-BLAST.
64 An Early Example of Success in applying Fold Recognition to Genome Annotation
65 Our Example ORF HI0073 from H. influenzae 114 a.a. long Function was UNKNOWN BLAST against UNIPROT just turned up the following: E64000 hypothetical protein HI Haemophilus influenzae (str e-46 A71149 hypothetical protein PH Pyrococcus horikoshii 101 7e-22 H70345 conserved hypothetical protein aq_507 - Aquifex aeolicus 99 2e-21 F72600 hypothetical protein APE Aeropyrum pernix (strain K1) 50 1e-06 C75046 hypothetical protein PAB Pyrococcus abyssi (strain e-04 C64354 hypothetical protein MJ Methanococcus jannaschii D64375 hypothetical protein MJ Methanococcus jannaschii H90346 conserved hypothetical protein [imported] - Sulfolobus so S62544 hypothetical protein SPAC12G12.13c - fission yeast (Schiz C90279 conserved hypothetical protein [imported] - Sulfolobus so H64462 hypothetical protein MJ Methanococcus jannaschii C69282 conserved hypothetical protein AF Archaeoglobus ful
66
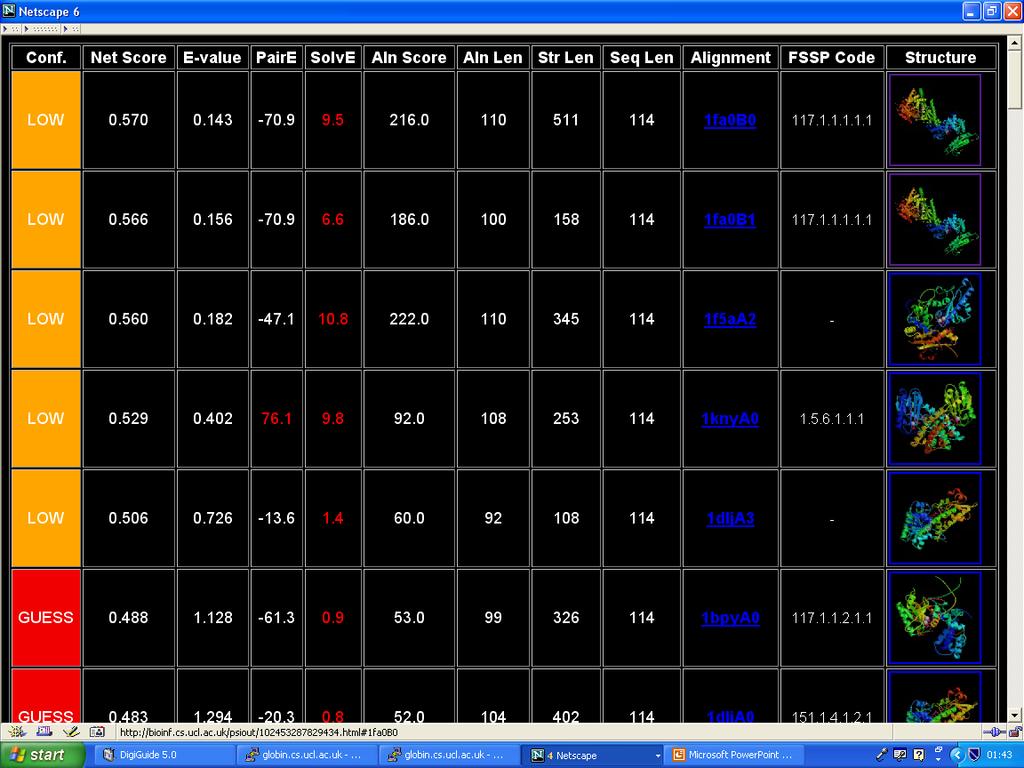
67
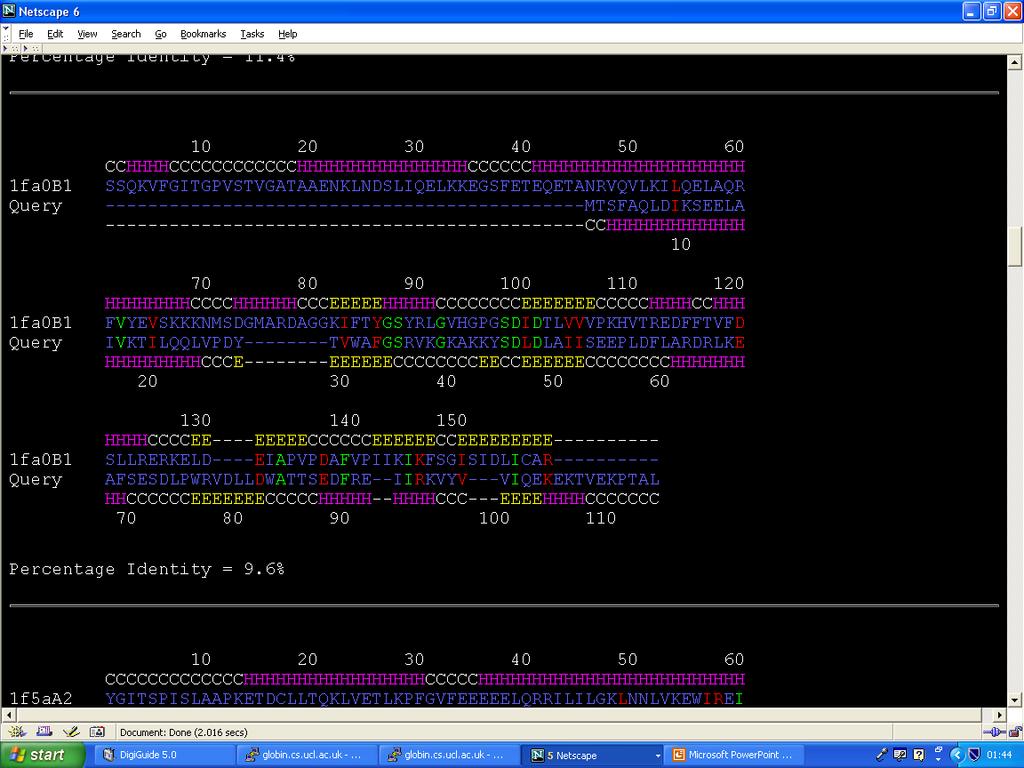
68

69
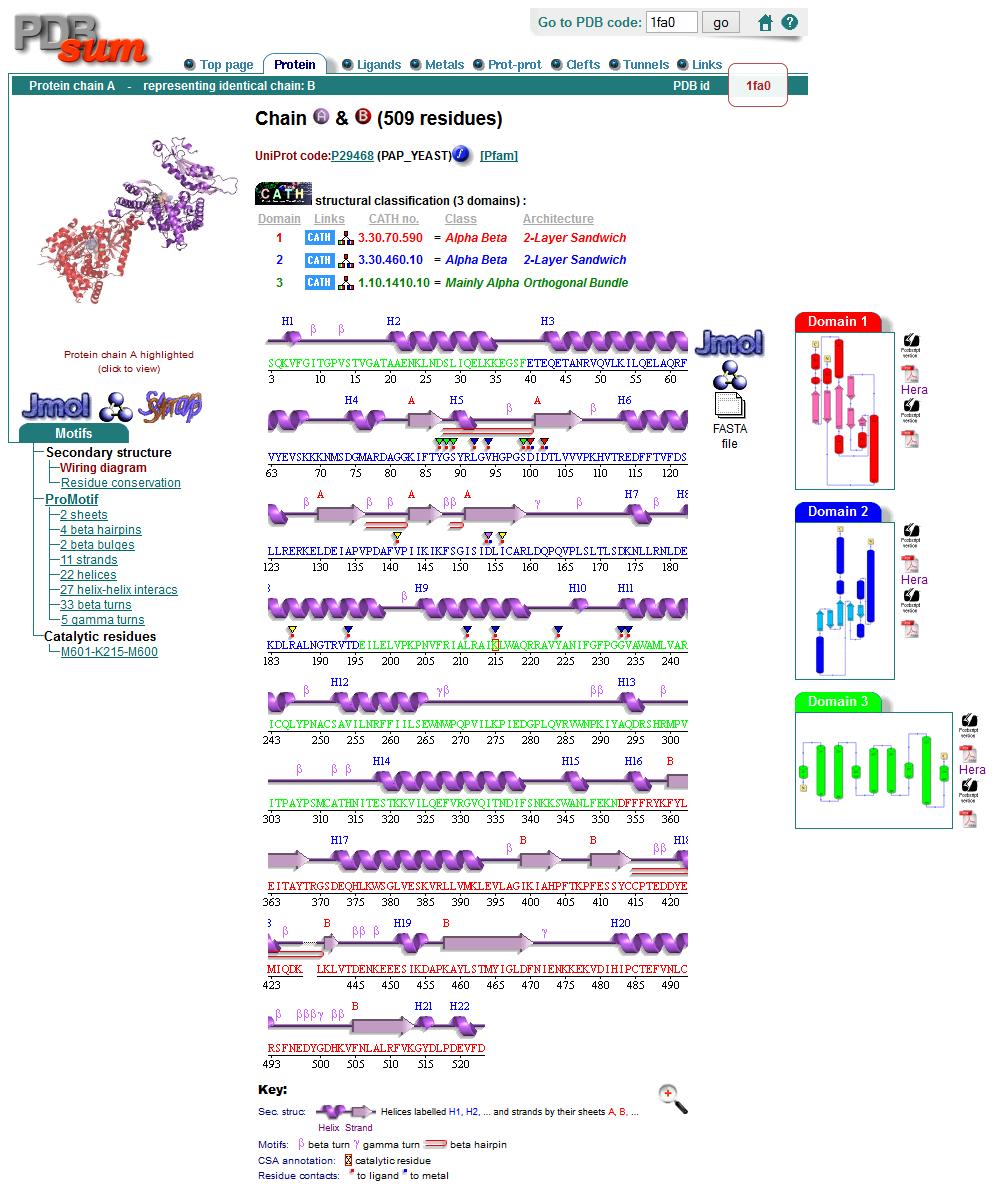
70 Conclusions We predicted that HI0073 was a probable nucleotidyl transferase (now confirmed) Consistent fold recognition results Match to 1FA0 Chain B Secondary structure in reasonable agreement Functionally Important Residues were CONSERVED
Bioinformatics: Secondary Structure Prediction
 Bioinformatics: Secondary Structure Prediction Prof. David Jones d.jones@cs.ucl.ac.uk LMLSTQNPALLKRNIIYWNNVALLWEAGSD The greatest unsolved problem in molecular biology:the Protein Folding Problem? Entries
Bioinformatics: Secondary Structure Prediction Prof. David Jones d.jones@cs.ucl.ac.uk LMLSTQNPALLKRNIIYWNNVALLWEAGSD The greatest unsolved problem in molecular biology:the Protein Folding Problem? Entries
Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction. Sepp Hochreiter
 Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction Institute of Bioinformatics Johannes Kepler University, Linz, Austria Chapter 4 Protein Secondary
Bioinformatics III Structural Bioinformatics and Genome Analysis Part Protein Secondary Structure Prediction Institute of Bioinformatics Johannes Kepler University, Linz, Austria Chapter 4 Protein Secondary
Protein Structure Prediction and Display
 Protein Structure Prediction and Display Goal Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each
Protein Structure Prediction and Display Goal Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
Physiochemical Properties of Residues
 Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
Protein Secondary Structure Prediction
 part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 the goal is the prediction of the secondary structure conformation which is local each amino
part of Bioinformatik von RNA- und Proteinstrukturen Computational EvoDevo University Leipzig Leipzig, SS 2011 the goal is the prediction of the secondary structure conformation which is local each amino
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics
 Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Statistical Machine Learning Methods for Bioinformatics IV. Neural Network & Deep Learning Applications in Bioinformatics Jianlin Cheng, PhD Department of Computer Science University of Missouri, Columbia
Basics of protein structure
 Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Protein Structures: Experiments and Modeling. Patrice Koehl
 Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
IT og Sundhed 2010/11
 IT og Sundhed 2010/11 Sequence based predictors. Secondary structure and surface accessibility Bent Petersen 13 January 2011 1 NetSurfP Real Value Solvent Accessibility predictions with amino acid associated
IT og Sundhed 2010/11 Sequence based predictors. Secondary structure and surface accessibility Bent Petersen 13 January 2011 1 NetSurfP Real Value Solvent Accessibility predictions with amino acid associated
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
Neural Networks for Protein Structure Prediction Brown, JMB CS 466 Saurabh Sinha
 Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
Neural Networks for Protein Structure Prediction Brown, JMB 1999 CS 466 Saurabh Sinha Outline Goal is to predict secondary structure of a protein from its sequence Artificial Neural Network used for this
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Programme Last week s quiz results + Summary Fold recognition Break Exercise: Modelling remote homologues
 Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
3D Structure. Prediction & Assessment Pt. 2. David Wishart 3-41 Athabasca Hall
 3D Structure Prediction & Assessment Pt. 2 David Wishart 3-41 Athabasca Hall david.wishart@ualberta.ca Objectives Become familiar with methods and algorithms for secondary Structure Prediction Become familiar
3D Structure Prediction & Assessment Pt. 2 David Wishart 3-41 Athabasca Hall david.wishart@ualberta.ca Objectives Become familiar with methods and algorithms for secondary Structure Prediction Become familiar
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools. Giri Narasimhan
 CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinff18.html Proteins and Protein Structure
CAP 5510: Introduction to Bioinformatics CGS 5166: Bioinformatics Tools Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinff18.html Proteins and Protein Structure
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Protein Secondary Structure Prediction using Pattern Recognition Neural Network
 Protein Secondary Structure Prediction using Pattern Recognition Neural Network P.V. Nageswara Rao 1 (nagesh@gitam.edu), T. Uma Devi 1, DSVGK Kaladhar 1, G.R. Sridhar 2, Allam Appa Rao 3 1 GITAM University,
Protein Secondary Structure Prediction using Pattern Recognition Neural Network P.V. Nageswara Rao 1 (nagesh@gitam.edu), T. Uma Devi 1, DSVGK Kaladhar 1, G.R. Sridhar 2, Allam Appa Rao 3 1 GITAM University,
1-D Predictions. Prediction of local features: Secondary structure & surface exposure
 1-D Predictions Prediction of local features: Secondary structure & surface exposure 1 Learning Objectives After today s session you should be able to: Explain the meaning and usage of the following local
1-D Predictions Prediction of local features: Secondary structure & surface exposure 1 Learning Objectives After today s session you should be able to: Explain the meaning and usage of the following local
Protein Structure Prediction
 Protein Structure Prediction Michael Feig MMTSB/CTBP 2006 Summer Workshop From Sequence to Structure SEALGDTIVKNA Ab initio Structure Prediction Protocol Amino Acid Sequence Conformational Sampling to
Protein Structure Prediction Michael Feig MMTSB/CTBP 2006 Summer Workshop From Sequence to Structure SEALGDTIVKNA Ab initio Structure Prediction Protocol Amino Acid Sequence Conformational Sampling to
Analysis and Prediction of Protein Structure (I)
") Analysis and Prediction of Protein Structure (I) Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 2006 Free for academic use. Copyright @ Jianlin Cheng
Analysis and Prediction of Protein Structure (I) Jianlin Cheng, PhD School of Electrical Engineering and Computer Science University of Central Florida 2006 Free for academic use. Copyright @ Jianlin Cheng
BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer Protein Structure Prediction I
 BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 9. Protein Structure Prediction I Structure Prediction Overview Overview of problem variants Secondary structure prediction
BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 9. Protein Structure Prediction I Structure Prediction Overview Overview of problem variants Secondary structure prediction
SUPPLEMENTARY MATERIALS
 SUPPLEMENTARY MATERIALS Enhanced Recognition of Transmembrane Protein Domains with Prediction-based Structural Profiles Baoqiang Cao, Aleksey Porollo, Rafal Adamczak, Mark Jarrell and Jaroslaw Meller Contact:
SUPPLEMENTARY MATERIALS Enhanced Recognition of Transmembrane Protein Domains with Prediction-based Structural Profiles Baoqiang Cao, Aleksey Porollo, Rafal Adamczak, Mark Jarrell and Jaroslaw Meller Contact:
Protein Secondary Structure Prediction
 Protein Secondary Structure Prediction Doug Brutlag & Scott C. Schmidler Overview Goals and problem definition Existing approaches Classic methods Recent successful approaches Evaluating prediction algorithms
Protein Secondary Structure Prediction Doug Brutlag & Scott C. Schmidler Overview Goals and problem definition Existing approaches Classic methods Recent successful approaches Evaluating prediction algorithms
Protein Secondary Structure Prediction using Feed-Forward Neural Network
 COPYRIGHT 2010 JCIT, ISSN 2078-5828 (PRINT), ISSN 2218-5224 (ONLINE), VOLUME 01, ISSUE 01, MANUSCRIPT CODE: 100713 Protein Secondary Structure Prediction using Feed-Forward Neural Network M. A. Mottalib,
COPYRIGHT 2010 JCIT, ISSN 2078-5828 (PRINT), ISSN 2218-5224 (ONLINE), VOLUME 01, ISSUE 01, MANUSCRIPT CODE: 100713 Protein Secondary Structure Prediction using Feed-Forward Neural Network M. A. Mottalib,
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Structure Prediction Using Multiple Artificial Neural Network Classifier *
 Protein Structure Prediction Using Multiple Artificial Neural Network Classifier * Hemashree Bordoloi and Kandarpa Kumar Sarma Abstract. Protein secondary structure prediction is the method of extracting
Protein Structure Prediction Using Multiple Artificial Neural Network Classifier * Hemashree Bordoloi and Kandarpa Kumar Sarma Abstract. Protein secondary structure prediction is the method of extracting
Presentation Outline. Prediction of Protein Secondary Structure using Neural Networks at Better than 70% Accuracy
 Prediction of Protein Secondary Structure using Neural Networks at Better than 70% Accuracy Burkhard Rost and Chris Sander By Kalyan C. Gopavarapu 1 Presentation Outline Major Terminology Problem Method
Prediction of Protein Secondary Structure using Neural Networks at Better than 70% Accuracy Burkhard Rost and Chris Sander By Kalyan C. Gopavarapu 1 Presentation Outline Major Terminology Problem Method
Protein Structure. W. M. Grogan, Ph.D. OBJECTIVES
 Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
PROTEIN SECONDARY STRUCTURE PREDICTION USING NEURAL NETWORKS AND SUPPORT VECTOR MACHINES
 PROTEIN SECONDARY STRUCTURE PREDICTION USING NEURAL NETWORKS AND SUPPORT VECTOR MACHINES by Lipontseng Cecilia Tsilo A thesis submitted to Rhodes University in partial fulfillment of the requirements for
PROTEIN SECONDARY STRUCTURE PREDICTION USING NEURAL NETWORKS AND SUPPORT VECTOR MACHINES by Lipontseng Cecilia Tsilo A thesis submitted to Rhodes University in partial fulfillment of the requirements for
Improved Protein Secondary Structure Prediction
 Improved Protein Secondary Structure Prediction Secondary Structure Prediction! Given a protein sequence a 1 a 2 a N, secondary structure prediction aims at defining the state of each amino acid ai as
Improved Protein Secondary Structure Prediction Secondary Structure Prediction! Given a protein sequence a 1 a 2 a N, secondary structure prediction aims at defining the state of each amino acid ai as
PROTEIN SECONDARY STRUCTURE PREDICTION: AN APPLICATION OF CHOU-FASMAN ALGORITHM IN A HYPOTHETICAL PROTEIN OF SARS VIRUS
 Int. J. LifeSc. Bt & Pharm. Res. 2012 Kaladhar, 2012 Research Paper ISSN 2250-3137 www.ijlbpr.com Vol.1, Issue. 1, January 2012 2012 IJLBPR. All Rights Reserved PROTEIN SECONDARY STRUCTURE PREDICTION:
Int. J. LifeSc. Bt & Pharm. Res. 2012 Kaladhar, 2012 Research Paper ISSN 2250-3137 www.ijlbpr.com Vol.1, Issue. 1, January 2012 2012 IJLBPR. All Rights Reserved PROTEIN SECONDARY STRUCTURE PREDICTION:
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence
 Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
HIV protease inhibitor. Certain level of function can be found without structure. But a structure is a key to understand the detailed mechanism.
 Proteins are linear polypeptide chains (one or more) Building blocks: 20 types of amino acids. Range from a few 10s-1000s They fold into varying three-dimensional shapes structure medicine Certain level
Proteins are linear polypeptide chains (one or more) Building blocks: 20 types of amino acids. Range from a few 10s-1000s They fold into varying three-dimensional shapes structure medicine Certain level
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Steps in protein modelling. Structure prediction, fold recognition and homology modelling. Basic principles of protein structure
 Structure prediction, fold recognition and homology modelling Marjolein Thunnissen Lund September 2012 Steps in protein modelling 3-D structure known Comparative Modelling Sequence of interest Similarity
Structure prediction, fold recognition and homology modelling Marjolein Thunnissen Lund September 2012 Steps in protein modelling 3-D structure known Comparative Modelling Sequence of interest Similarity
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein structure. Protein structure. Amino acid residue. Cell communication channel. Bioinformatics Methods
 Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Cell communication channel Bioinformatics Methods Iosif Vaisman Email: ivaisman@gmu.edu SEQUENCE STRUCTURE DNA Sequence Protein Sequence Protein Structure Protein structure ATGAAATTTGGAAACTTCCTTCTCACTTATCAGCCACCT...
Lecture 7. Protein Secondary Structure Prediction. Secondary Structure DSSP. Master Course DNA/Protein Structurefunction.
 C N T R F O R N T G R A T V B O N F O R M A T C S V U Master Course DNA/Protein Structurefunction Analysis and Prediction Lecture 7 Protein Secondary Structure Prediction Protein primary structure 20 amino
C N T R F O R N T G R A T V B O N F O R M A T C S V U Master Course DNA/Protein Structurefunction Analysis and Prediction Lecture 7 Protein Secondary Structure Prediction Protein primary structure 20 amino
Intro Secondary structure Transmembrane proteins Function End. Last time. Domains Hidden Markov Models
 Last time Domains Hidden Markov Models Today Secondary structure Transmembrane proteins Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL
Last time Domains Hidden Markov Models Today Secondary structure Transmembrane proteins Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL
Today. Last time. Secondary structure Transmembrane proteins. Domains Hidden Markov Models. Structure prediction. Secondary structure
 Last time Today Domains Hidden Markov Models Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL SSLGPVVDAHPEYEEVALLERMVIPERVIE FRVPWEDDNGKVHVNTGYRVQFNGAIGPYK
Last time Today Domains Hidden Markov Models Structure prediction NAD-specific glutamate dehydrogenase Hard Easy >P24295 DHE2_CLOSY MSKYVDRVIAEVEKKYADEPEFVQTVEEVL SSLGPVVDAHPEYEEVALLERMVIPERVIE FRVPWEDDNGKVHVNTGYRVQFNGAIGPYK
09/06/25. Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Non-uniform distribution of folds. Scheme of protein structure predicition
 Non-uniform distribution of folds. Scheme of protein structure predicition") Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Protein Secondary Structure Assignment and Prediction
 1 Protein Secondary Structure Assignment and Prediction Defining SS features - Dihedral angles, alpha helix, beta stand (Hydrogen bonds) Assigned manually by crystallographers or Automatic DSSP (Kabsch
1 Protein Secondary Structure Assignment and Prediction Defining SS features - Dihedral angles, alpha helix, beta stand (Hydrogen bonds) Assigned manually by crystallographers or Automatic DSSP (Kabsch
Protein 8-class Secondary Structure Prediction Using Conditional Neural Fields
 2010 IEEE International Conference on Bioinformatics and Biomedicine Protein 8-class Secondary Structure Prediction Using Conditional Neural Fields Zhiyong Wang, Feng Zhao, Jian Peng, Jinbo Xu* Toyota
2010 IEEE International Conference on Bioinformatics and Biomedicine Protein 8-class Secondary Structure Prediction Using Conditional Neural Fields Zhiyong Wang, Feng Zhao, Jian Peng, Jinbo Xu* Toyota
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Secondary Structure. Bioch/BIMS 503 Lecture 2. Structure and Function of Proteins. Further Reading. Φ, Ψ angles alone determine protein structure
 Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Protein folding. Today s Outline
 Protein folding Today s Outline Review of previous sessions Thermodynamics of folding and unfolding Determinants of folding Techniques for measuring folding The folding process The folding problem: Prediction
Protein folding Today s Outline Review of previous sessions Thermodynamics of folding and unfolding Determinants of folding Techniques for measuring folding The folding process The folding problem: Prediction
Presenter: She Zhang
 Presenter: She Zhang Introduction Dr. David Baker Introduction Why design proteins de novo? It is not clear how non-covalent interactions favor one specific native structure over many other non-native
Presenter: She Zhang Introduction Dr. David Baker Introduction Why design proteins de novo? It is not clear how non-covalent interactions favor one specific native structure over many other non-native
Getting To Know Your Protein
 Getting To Know Your Protein Comparative Protein Analysis: Part III. Protein Structure Prediction and Comparison Robert Latek, PhD Sr. Bioinformatics Scientist Whitehead Institute for Biomedical Research
Getting To Know Your Protein Comparative Protein Analysis: Part III. Protein Structure Prediction and Comparison Robert Latek, PhD Sr. Bioinformatics Scientist Whitehead Institute for Biomedical Research
Protein Structure Prediction
 Protein Structure Prediction Michael Feig MMTSB/CTBP 2009 Summer Workshop From Sequence to Structure SEALGDTIVKNA Folding with All-Atom Models AAQAAAAQAAAAQAA All-atom MD in general not succesful for real
Protein Structure Prediction Michael Feig MMTSB/CTBP 2009 Summer Workshop From Sequence to Structure SEALGDTIVKNA Folding with All-Atom Models AAQAAAAQAAAAQAA All-atom MD in general not succesful for real
BCH 4053 Spring 2003 Chapter 6 Lecture Notes
 BCH 4053 Spring 2003 Chapter 6 Lecture Notes 1 CHAPTER 6 Proteins: Secondary, Tertiary, and Quaternary Structure 2 Levels of Protein Structure Primary (sequence) Secondary (ordered structure along peptide
BCH 4053 Spring 2003 Chapter 6 Lecture Notes 1 CHAPTER 6 Proteins: Secondary, Tertiary, and Quaternary Structure 2 Levels of Protein Structure Primary (sequence) Secondary (ordered structure along peptide
From Amino Acids to Proteins - in 4 Easy Steps
 From Amino Acids to Proteins - in 4 Easy Steps Although protein structure appears to be overwhelmingly complex, you can provide your students with a basic understanding of how proteins fold by focusing
From Amino Acids to Proteins - in 4 Easy Steps Although protein structure appears to be overwhelmingly complex, you can provide your students with a basic understanding of how proteins fold by focusing
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
DATE A DAtabase of TIM Barrel Enzymes
 DATE A DAtabase of TIM Barrel Enzymes 2 2.1 Introduction.. 2.2 Objective and salient features of the database 2.2.1 Choice of the dataset.. 2.3 Statistical information on the database.. 2.4 Features....
DATE A DAtabase of TIM Barrel Enzymes 2 2.1 Introduction.. 2.2 Objective and salient features of the database 2.2.1 Choice of the dataset.. 2.3 Statistical information on the database.. 2.4 Features....
Optimization of the Sliding Window Size for Protein Structure Prediction
 Optimization of the Sliding Window Size for Protein Structure Prediction Ke Chen* 1, Lukasz Kurgan 1 and Jishou Ruan 2 1 University of Alberta, Department of Electrical and Computer Engineering, Edmonton,
Optimization of the Sliding Window Size for Protein Structure Prediction Ke Chen* 1, Lukasz Kurgan 1 and Jishou Ruan 2 1 University of Alberta, Department of Electrical and Computer Engineering, Edmonton,
Procheck output. Bond angles (Procheck) Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.
 Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.") Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Can protein model accuracy be. identified? NO! CBS, BioCentrum, Morten Nielsen, DTU
 Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Motif Prediction in Amino Acid Interaction Networks
 Motif Prediction in Amino Acid Interaction Networks Omar GACI and Stefan BALEV Abstract In this paper we represent a protein as a graph where the vertices are amino acids and the edges are interactions
Motif Prediction in Amino Acid Interaction Networks Omar GACI and Stefan BALEV Abstract In this paper we represent a protein as a graph where the vertices are amino acids and the edges are interactions
HMM applications. Applications of HMMs. Gene finding with HMMs. Using the gene finder
 HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
HMM applications Applications of HMMs Gene finding Pairwise alignment (pair HMMs) Characterizing protein families (profile HMMs) Predicting membrane proteins, and membrane protein topology Gene finding
Packing of Secondary Structures
 7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
7.88 Lecture Notes - 4 7.24/7.88J/5.48J The Protein Folding and Human Disease Professor Gossard Retrieving, Viewing Protein Structures from the Protein Data Base Helix helix packing Packing of Secondary
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
Structure to Function. Molecular Bioinformatics, X3, 2006
 Structure to Function Molecular Bioinformatics, X3, 2006 Structural GeNOMICS Structural Genomics project aims at determination of 3D structures of all proteins: - organize known proteins into families
Structure to Function Molecular Bioinformatics, X3, 2006 Structural GeNOMICS Structural Genomics project aims at determination of 3D structures of all proteins: - organize known proteins into families
Protein Structure Analysis and Verification. Course S Basics for Biosystems of the Cell exercise work. Maija Nevala, BIO, 67485U 16.1.
 Protein Structure Analysis and Verification Course S-114.2500 Basics for Biosystems of the Cell exercise work Maija Nevala, BIO, 67485U 16.1.2008 1. Preface When faced with an unknown protein, scientists
Protein Structure Analysis and Verification Course S-114.2500 Basics for Biosystems of the Cell exercise work Maija Nevala, BIO, 67485U 16.1.2008 1. Preface When faced with an unknown protein, scientists
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Computational Genomics and Molecular Biology, Fall
 Computational Genomics and Molecular Biology, Fall 2014 1 HMM Lecture Notes Dannie Durand and Rose Hoberman November 6th Introduction In the last few lectures, we have focused on three problems related
Computational Genomics and Molecular Biology, Fall 2014 1 HMM Lecture Notes Dannie Durand and Rose Hoberman November 6th Introduction In the last few lectures, we have focused on three problems related
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Prediction of protein secondary structure by mining structural fragment database
 Polymer 46 (2005) 4314 4321 www.elsevier.com/locate/polymer Prediction of protein secondary structure by mining structural fragment database Haitao Cheng a, Taner Z. Sen a, Andrzej Kloczkowski a, Dimitris
Polymer 46 (2005) 4314 4321 www.elsevier.com/locate/polymer Prediction of protein secondary structure by mining structural fragment database Haitao Cheng a, Taner Z. Sen a, Andrzej Kloczkowski a, Dimitris
Protein Structure Prediction using String Kernels. Technical Report
 Protein Structure Prediction using String Kernels Technical Report Department of Computer Science and Engineering University of Minnesota 4-192 EECS Building 200 Union Street SE Minneapolis, MN 55455-0159
Protein Structure Prediction using String Kernels Technical Report Department of Computer Science and Engineering University of Minnesota 4-192 EECS Building 200 Union Street SE Minneapolis, MN 55455-0159
Bioinformatics. Macromolecular structure
 Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009
 114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
Protein Secondary Structure Prediction
 C E N T R F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U E Master Course DNA/Protein Structurefunction Analysis and Prediction Lecture 7 Protein Secondary Structure Prediction Protein primary
C E N T R F O R I N T E G R A T I V E B I O I N F O R M A T I C S V U E Master Course DNA/Protein Structurefunction Analysis and Prediction Lecture 7 Protein Secondary Structure Prediction Protein primary
Building 3D models of proteins
 Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation
 Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Predicting Protein Structural Features With Artificial Neural Networks
 CHAPTER 4 Predicting Protein Structural Features With Artificial Neural Networks Stephen R. Holbrook, Steven M. Muskal and Sung-Hou Kim 1. Introduction The prediction of protein structure from amino acid
CHAPTER 4 Predicting Protein Structural Features With Artificial Neural Networks Stephen R. Holbrook, Steven M. Muskal and Sung-Hou Kim 1. Introduction The prediction of protein structure from amino acid
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015,
 Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Biochemistry,530:,, Introduc5on,to,Structural,Biology, Autumn,Quarter,2015, Course,Informa5on, BIOC%530% GraduateAlevel,discussion,of,the,structure,,func5on,,and,chemistry,of,proteins,and, nucleic,acids,,control,of,enzyma5c,reac5ons.,please,see,the,course,syllabus,and,
Conditional Graphical Models
 PhD Thesis Proposal Conditional Graphical Models for Protein Structure Prediction Yan Liu Language Technologies Institute University Thesis Committee Jaime Carbonell (Chair) John Lafferty Eric P. Xing
PhD Thesis Proposal Conditional Graphical Models for Protein Structure Prediction Yan Liu Language Technologies Institute University Thesis Committee Jaime Carbonell (Chair) John Lafferty Eric P. Xing
We used the PSI-BLAST program (http://www.ncbi.nlm.nih.gov/blast/) to search the
 to search the") SUPPLEMENTARY METHODS - in silico protein analysis We used the PSI-BLAST program (http://www.ncbi.nlm.nih.gov/blast/) to search the Protein Data Bank (PDB, http://www.rcsb.org/pdb/) and the NCBI non-redundant
SUPPLEMENTARY METHODS - in silico protein analysis We used the PSI-BLAST program (http://www.ncbi.nlm.nih.gov/blast/) to search the Protein Data Bank (PDB, http://www.rcsb.org/pdb/) and the NCBI non-redundant
An Artificial Neural Network Classifier for the Prediction of Protein Structural Classes
 International Journal of Current Engineering and Technology E-ISSN 2277 4106, P-ISSN 2347 5161 2017 INPRESSCO, All Rights Reserved Available at http://inpressco.com/category/ijcet Research Article An Artificial
International Journal of Current Engineering and Technology E-ISSN 2277 4106, P-ISSN 2347 5161 2017 INPRESSCO, All Rights Reserved Available at http://inpressco.com/category/ijcet Research Article An Artificial
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Outline. The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation. Unfolded Folded. What is protein folding?
 The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation By Jun Shimada and Eugine Shaknovich Bill Hawse Dr. Bahar Elisa Sandvik and Mehrdad Safavian Outline Background on protein
The ensemble folding kinetics of protein G from an all-atom Monte Carlo simulation By Jun Shimada and Eugine Shaknovich Bill Hawse Dr. Bahar Elisa Sandvik and Mehrdad Safavian Outline Background on protein
Dihedral Angles. Homayoun Valafar. Department of Computer Science and Engineering, USC 02/03/10 CSCE 769
 Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Dihedral Angles Homayoun Valafar Department of Computer Science and Engineering, USC The precise definition of a dihedral or torsion angle can be found in spatial geometry Angle between to planes Dihedral
Improving Protein Secondary-Structure Prediction by Predicting Ends of Secondary-Structure Segments
 Improving Protein Secondary-Structure Prediction by Predicting Ends of Secondary-Structure Segments Uros Midic 1 A. Keith Dunker 2 Zoran Obradovic 1* 1 Center for Information Science and Technology Temple
Improving Protein Secondary-Structure Prediction by Predicting Ends of Secondary-Structure Segments Uros Midic 1 A. Keith Dunker 2 Zoran Obradovic 1* 1 Center for Information Science and Technology Temple
Master s Thesis June 2018 Supervisor: Christian Nørgaard Storm Pedersen Aarhus University
 P R O T E I N S E C O N D A RY S T R U C T U R E P R E D I C T I O N U S I N G A RT I F I C I A L N E U R A L N E T W O R K S judit kisistók, 201602119 bakhtawar noor, 201602561 Master s Thesis June 2018
P R O T E I N S E C O N D A RY S T R U C T U R E P R E D I C T I O N U S I N G A RT I F I C I A L N E U R A L N E T W O R K S judit kisistók, 201602119 bakhtawar noor, 201602561 Master s Thesis June 2018
Student Questions and Answers October 8, 2002
 Student Questions and Answers October 8, 2002 Q l. Is the Cα of Proline also chiral? Answer: FK: Yes, there are 4 different residues bound to this C. Only in a strictly planar molecule this would not hold,
Student Questions and Answers October 8, 2002 Q l. Is the Cα of Proline also chiral? Answer: FK: Yes, there are 4 different residues bound to this C. Only in a strictly planar molecule this would not hold,
Protein structure alignments
 Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
Protein structure alignments Proteins that fold in the same way, i.e. have the same fold are often homologs. Structure evolves slower than sequence Sequence is less conserved than structure If BLAST gives
Protein Structure Determination
 Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Protein Folding Prof. Eugene Shakhnovich
 Protein Folding Eugene Shakhnovich Department of Chemistry and Chemical Biology Harvard University 1 Proteins are folded on various scales As of now we know hundreds of thousands of sequences (Swissprot)
Protein Folding Eugene Shakhnovich Department of Chemistry and Chemical Biology Harvard University 1 Proteins are folded on various scales As of now we know hundreds of thousands of sequences (Swissprot)
Protein Structure Basics
 Protein Structure Basics Presented by Alison Fraser, Christine Lee, Pradhuman Jhala, Corban Rivera Importance of Proteins Muscle structure depends on protein-protein interactions Transport across membranes
Protein Structure Basics Presented by Alison Fraser, Christine Lee, Pradhuman Jhala, Corban Rivera Importance of Proteins Muscle structure depends on protein-protein interactions Transport across membranes
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions Van der Waals Interactions
Lecture 2-3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions Van der Waals Interactions