Modeling for 3D structure prediction
|
|
|
- Marshall Owen
- 5 years ago
- Views:
Transcription
1 Modeling for 3D structure prediction
2 What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing of preexisting information (data mining/prior knowledge) and newly obtained experimental data is also common in obtaining experimental structures. It is mostly the relative weight of experimental versus computer generated information that determines whether the structure is considered «experimental» or «model»

3 Example: protonation in X-RAY JBC
4 Predicted structures Input needed: amino acid sequence only Which data are going to enter the modelling? What is the expected quality of the structure? How can I identify problems in the structure? How can I improve the structure? What do I need the structure for?
5 100% Predicted structures
6 Data entering the modelling Experimental structures of proteins that differ in sequence from the protein of interest Close sequence homology: homology modelling More distant sequence relationship/shared fold with no obvious sequence relationship : threading. Aim at recognizing a related fold. No fold assigned: Folding from sequence alone.
7 modelling by homology or knowledge-based modelling
8 Why in silico modelling of protein structure? it is the only way to obtain structural information if experimental techniques fail. insoluble proteins like membrane receptors proteins too large for NMR analysis proteins that could not be crystallized for X-ray diffraction slow increase of number of solved 3D-structures, yet ca structures available in the PDB (templates) fast increase of number of protein sequences with no structural informations available
9 Why it is usefull to know 3D structure of a protein, not only its sequence? biochemical function = interaction with other molecules biological function = consequence of these interactions 3D structure is more informative than the sequence because interactions are determined by amino acids that are close in space yet frequently distant in sequence. Function depends more directly on structure than sequence.
10 Examples of 3D structure uses identification of active and binding sites (substrates, co-factors, inhibitors, modulators, proteins, nucleic acids..); designing and improving ligands of a given binding site (e.g., structure-based drug design); understanding functional /physical aspects of multimeric assemblies; testing function hypothesis (e.g. site-directed mutagenesis); rational design of proteins with increased stability or novel functions; characterizing new sequences (e.g., detecting remote relationships between proteins); antigenic behavior (prediction of epitopes); characterization of biological networks
11 Why not all structures experimentally? Many more sequences than structures New structures often «look like» old ones redundancy of structural information in the PDB : less than 3000 unique folds 2 peptidic chains share the same fold whether RMSD < 3.0 Å number of aligned positions 70% total chain lenght

12 The purpose of the Protein Data Bank is to collect and organize 3D structures of proteins, nucleic acids, protein-nucleic acid complexes and complexes with drug molecules and inhibitors. The PDB is the recognized worldwide repository for 3D structures. After validation of deposited structures, the coordinates are made available to all researchers worldwide and free-of-charge.
 & old")
13 PDB : fold number new (blue) & old (red)
14 Homology modelling is based on 2 observations: 1. The structure of a protein is determined by its amino acid sequence. Knowing the sequence should, at least in theory, suffice to obtain the structure. 2. During evolution, the structure is more stable and changes much slower than the associated sequence: similar sequences adopt practically identical structures distantly related sequences may still fold into similar structures

15 SSAP score: measure of structural similarity Protein stucture Alignment, WR Taylor and C Orengo, J. Mol. Biol (1989)
16 When the sequence identity is 30% between 2 sequences, and experimental 3D information is available for one of the sequences, it is possible to build in silico a 3D model for the sequence of unknown structure LIMITS OF IN SILICO MODELS accuracy of a structure predicted from its sequence is much lower than the results achieved experimentally: no homology modelling structures in the PDB! theoretical protein 3D structures represent "low-resolution" models, and hence should be used ACCORDINGLY
17 high sequence similarity results in structure similarity: rmsd of the Cα co-ordinates for protein cores sharing 50% residue identity is expected to be around 1Å two sequences of identical lenght which share more than 30% sequence identity virtually adopt a similar structure. Comparative model building consists of the extrapolation of the structure for a new (target) sequence from the known 3Dstructure of related family members (templates).
18
19 homology modelling workflow 1. Template recognition and initial alignment 2. Alignment correction 3. Conserved regions modelling 4. Loops modelling 5. Side-chain modelling MODEL CONSTRUCT ION 6. Model optimization 7. Model validation
20 Identification of modelling templates query using FastA and BLAST to retrieve from the PDB sequence of target homologous proteins A template must share at least 30% residue identity with the target. whether many template are available best template structure (highest sequence similarity to the target) = reference all structures used to generate structurally correct multiple alignment of the sequences Choose the best structure: the quality of the model could not be better than that of the template
21 Check the template structure The PDBREPORT database (C. Sander, EMBL) indicates problems in structures for every PDB entry. Generally X-ray structures better than NMR structures X-ray structure quality attested by the resolution factor Yet also pay attention to temperature (conformation freezed at low temperature, see local flexibility indicated by B-factor) External conditions: ph, ions, ligand,...
22 Aligning the target sequence with the template sequence Some residues should not be used for model building, for example those located in non-conserved loops Analyzing the alignement Identification of conserved regions no insertion / deletion in conserved regions The quality of alignment determine the quality of the modelled structure!

23 Muliple Steps in constructing sequence a Multiple alignment Alignment

24 BAliBASE reference 3: aldehyde dehydrogenase-like * * * * * * * GSVPTG E C GSTKVG GETRTG E C GSTEVG GSVSAG GSRDVG GSRDVG GSRDVG GSTNVF GSTNVF GSTAVF E E C C Uniprot Annotation NAD binding Active site Active site
25 identity substitution (mutation) PDB template Model target Model insertion deletion PDB PDB Model
26 Lessons from evolution... Upon evolution mutation are observed rather than insertion or deletion (indel) shorter indels are easier to make than longer ones active site residues are conserved Cysteines involved in disulfide bridges are conserved Core residues are better conserved than those exposed at the protein surface
27 Lessons from evolution... Upon evolution residues tend to mutate into similar residues (e.g. V <-> I; S <-> T; see scoring matrix like PAM and BLOSUM ) residues mutate more easily to residues encoded by similar codons Some residues cannot be easily mutated because of their unique structural features (e.g., accessible phi-psi values for Glycine, Proline)
28 Additional informations that can be used to correct sequence alignment... Knowledge about proteolytic cleavage sites metal binding residues ligand binding residues Structure: - structure of homologous protein - predicted secondary structure In any cases, use multiple alignment rather than pairwise alignment
29 building the model " 1. Conserved regions Conserved regions are built using template structural informations, All methods keep the structure of the template in conserved regions with few adjustments (adjustements are necessary when there are deletions in target with respect to template) 2. Non conserved regions 2a. Backbone of inserted sequences (mostly loops), need to be constructed or refined 2b. Side chain of mutated residues need to be reconstructed/refined. This may involve local changes around the mutation in conserved regions (repacking)
30 Example : modelling by spatial restraints Spatial restraints molecular mechanics using distance restraints extracted from the structures of one or several targets PDB template MODEL target


31 Modeller (Andrej Sali)
32 The 3D structure of ECD1 CRF-R2β. First extracellular domain of a type B1 G protein-coupled receptor MODEL (A) A ribbon diagram of the lowest energy conformer highlighting the β- sheets in cyan and the disulfide bonds in yellow. EXPERIMENTAL MODEL (B) Superposition of 20 conformers representing the 3D NMR structure. Only amino acid residues are shown. The bundle is obtained by superimposing the backbone Cα carbons of residues and EXPERIMENTAL Grace C R R et al. PNAS 2004;101:
33 Molecular dynamics as a tools for structure determination Molecular dynamics as a means for finding structures that satisfy restraints. MD explores many structures, and finds stable ones Energy Minimum Minimum Conformations
34 Energy surface: classical MM force field E Total (X N ) = E bonded (X N ) + E nonbonded (X N ) Energy surface with restraints X N : atomic positions! E Total (X N ) = E bonded (X N ) + E nonbonded (X N ) + E RESTRAINTS (X N ) X N : atomic positions! Example of restraint: spring between two atoms that are known to be close in space, from some independant observation (artificial bond) i j i and j are NOT chemically linked, but the artificial bond will pull them close together: advantage of computer simulation: the potential surface can be «manipulated».
35 Example : modelling by spatial restraints «Artificial bonds» will pull atoms close in space, even if there is no real chemical bond between them PDB template MODEL target
36 Incorporate restraints into potential energy function E Total (X N ) = E bonded (X N ) + E nonbonded (X N ) + " i i E RESTRAINTS (X N ) Then return to the formalism developed for unbiased molecular dynamics simulations F i = m i! a i = m i! dv i dt = m! d 2 r i dt 2 F i =!" i E
37 MD with restraints To satisfy experimental observations Most noted example is NMR structure determination: Many experimentally derived restraints: the structure is MOSTLY determined by this data Few experimental restraints: the structure is a «mix» of computational and experimental information Restraints from other source than experiment, expl, from a sequence alignment : HOMOLOGY MODELLING Protein A should «look» like protein B, because the sequence of A and B are similar. If 3D structure of B is known, then A can be modelled «by homology» to B.
38 Restraints obtained from an Nuclear Overhauser effect (NOE) Chemical shifts Hydrogen bonds 3 J scalar couplings... NMR experiment
 A ribbon diagram of the lowest energy conformer highlighting the β- sheets in cyan and the disulfide bonds in yellow.")
39 The 3D structure of ECD1 CRF-R2β. First extracellular domain of a type B1 G protein-coupled receptor MODEL (A) A ribbon diagram of the lowest energy conformer highlighting the β- sheets in cyan and the disulfide bonds in yellow. EXPERIMENTAL MODEL (B) Superposition of 20 conformers representing the 3D NMR structure. Only amino acid residues are shown. The bundle is obtained by superimposing the backbone Cα carbons of residues and EXPERIMENTAL Grace C R R et al. PNAS 2004;101:
40 spatial restraints from sequence alignment template PDKNI SRA target PDKNALTRA restraint = harmonic energy P#A E C"#C" [ ] 2 (t arget) = 1 2 k r P#A C"#C" (t arget) # r P#A C"#C" (template) PDB: template MODEL : target P A
41 Non conserved parts In modelling by spatial restraints, the missing loops and mutated side chain are incorporated automatically in the structure. Yet, since there is no information on these parts in the PDB template, their position is modelled in a less reliable way. Specialized loop/side chain reconstruction algorithms can be applied afterwards, to improve the model.
42 Programmes segment matching / rigid body Software SYBYL (tripos) MOE (CCG) Freeware SwissPDBViewer Serveurs WWW Swiss-model(ExpasY) Spatial constraints Freeware MODELLER (accelrys) WWW tools for structure analysis and validation WHATIF PROCHECK
43 structure evaluation" = validation step geometry analysis: angles length, bonds & torsion values, planarity of aromatic rings, steric clashes pseudo energetical analysis experimental validation : choice of experiments based on structural assumption (e.g., site directed mutagenesis)
44 General Criteria for protein structure validation local Geometry : bond lenght, angle values,chirality, planarity general quality : Ramachandran plot rotamers (χ angle) bumps h-bonds and salt bridges network buried hyrophobic aromatic stacking


45 planarity backbone Torsion Angle Omega Trans-& Cis-conformation side chains Ex: ARG good bad
46 Ramachandran Plot good bad
 along the protein")
47 Reminder Ramanchandran Two degrees of freedom (dihedral angles phi et psi) along the protein backbone)
48 Phi! Backbone conformations

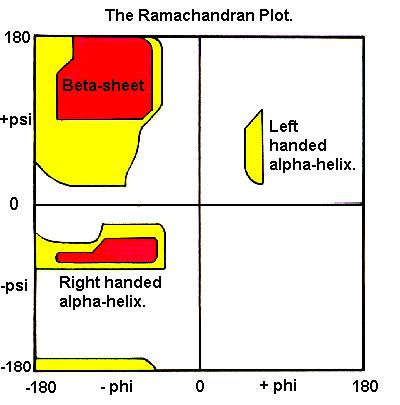
49 Psi! Backbone conformations
50 Ramachandran plot In experimental structures, not all values of phi/psi! are observed.! Some combinations are never seen because they correspond to unstable structures (steric clash - high energy)!

")
51 Ramachandran Plot Each point correspond to ONE AA in the structure Good: most of the AA in Allowed (low energy) regions. Bad: many AA in disallowed (high energy) regions


52 rotamers Bad Good

53 bumps Steric clashes between 2 atoms of the backbone

54 H-bond network Ex: Crambine

55 salt bridge network bad good
56 quality of the packing bad good

57 backbone Conformation normal strange!!
58 pseudo-energy analysis protein folding ΔG < 0 The best model has the lowest free energy ΔG unfolded What is difficult: native native To compute the free energy (NOT THE SAME AS ENERGY) Absolute free energy : very large, yet variation of free energy between 2 folded conformations very small (5-10 kcal / mol )
59 Empirical energy functions e.g. Bowie, Luthy, Eisenberg Science (1991) Definition of 18 classes that describe amino acid properties: Nature (1992) accessibility & secondary structure E: solvant exposed α helix P1, P2: partly exposed * β sheet B1, B2, B3 : buried other For the 20 amino acid types: Score s assigned to the18 classes values from statistical analysis of known 3D structures High score good environment e.g., W: B1. α = 1.00, E α = K: B1. α = -1.82, E α = 0.13
60 Empirical energy functions Application to 3D model evaluation For every amino acid i: Side chain buried surface Percentage of the buried surface facing polar atoms (including water) Local secondary structure score: S(model) = Σ s i N N: number of amino acids S1 > S2 s i Problems! Best model 1 N Sequence (i)
61 Empirical energy functions NOTE that the scale is arbitrary:!!in the e.g. in Bowie, Luthy, Eisenberg energy function, a high score is good!!!in the Modeller DOPE score, which is also an empirical function, although with different parameters, a LOW score is good.!

62 In practice Databases of already built models ModBase, A. Sali

63 In practice Databases of already built models Swiss-Model Repository, T. Schwede
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
HOMOLOGY MODELING. The sequence alignment and template structure are then used to produce a structural model of the target.
 HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling. Roberto Lins EPFL - summer semester 2005
 Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Homology Modeling Roberto Lins EPFL - summer semester 2005 Disclaimer: course material is mainly taken from: P.E. Bourne & H Weissig, Structural Bioinformatics; C.A. Orengo, D.T. Jones & J.M. Thornton,
Procheck output. Bond angles (Procheck) Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.
 Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.") Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Programme Last week s quiz results + Summary Fold recognition Break Exercise: Modelling remote homologues
 Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Introduction to" Protein Structure
 Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Can protein model accuracy be. identified? NO! CBS, BioCentrum, Morten Nielsen, DTU
 Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Can protein model accuracy be identified? Morten Nielsen, CBS, BioCentrum, DTU NO! Identification of Protein-model accuracy Why is it important? What is accuracy RMSD, fraction correct, Protein model correctness/quality
Week 10: Homology Modelling (II) - HHpred
 - HHpred") Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Week 10: Homology Modelling (II) - HHpred Course: Tools for Structural Biology Fabian Glaser BKU - Technion 1 2 Identify and align related structures by sequence methods is not an easy task All comparative
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Homology modeling of Ferredoxin-nitrite reductase from Arabidopsis thaliana
 www.bioinformation.net Hypothesis Volume 6(3) Homology modeling of Ferredoxin-nitrite reductase from Arabidopsis thaliana Karim Kherraz*, Khaled Kherraz, Abdelkrim Kameli Biology department, Ecole Normale
www.bioinformation.net Hypothesis Volume 6(3) Homology modeling of Ferredoxin-nitrite reductase from Arabidopsis thaliana Karim Kherraz*, Khaled Kherraz, Abdelkrim Kameli Biology department, Ecole Normale
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
Bioinformatics. Macromolecular structure
 Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Molecular Modeling lecture 2
 Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
Molecular Modeling 2018 -- lecture 2 Topics 1. Secondary structure 3. Sequence similarity and homology 2. Secondary structure prediction 4. Where do protein structures come from? X-ray crystallography
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Sequence analysis and comparison
 The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
The aim with sequence identification: Sequence analysis and comparison Marjolein Thunnissen Lund September 2012 Is there any known protein sequence that is homologous to mine? Are there any other species
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Biomolecules: lecture 10
 Biomolecules: lecture 10 - understanding in detail how protein 3D structures form - realize that protein molecules are not static wire models but instead dynamic, where in principle every atom moves (yet
Biomolecules: lecture 10 - understanding in detail how protein 3D structures form - realize that protein molecules are not static wire models but instead dynamic, where in principle every atom moves (yet
CS612 - Algorithms in Bioinformatics
 Fall 2017 Protein Structure Detection Methods October 30, 2017 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing
Fall 2017 Protein Structure Detection Methods October 30, 2017 Comparative Modeling Comparative modeling is modeling of the unknown based on comparison to what is known In the context of modeling or computing
Basics of protein structure
 Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Today: 1. Projects a. Requirements: i. Critical review of one paper ii. At least one computational result b. Noon, Dec. 3 rd written report and oral presentation are due; submit via email to bphys101@fas.harvard.edu
Bulk behaviour. Alanine. FIG. 1. Chemical structure of the RKLPDA peptide. Numbers on the left mark alpha carbons.
 Bulk behaviour To characterise the conformational behaviour of the peptide, first we looked at the statistics of alpha carbons and the torsion angles. Then they were correlated with positions of positively
Bulk behaviour To characterise the conformational behaviour of the peptide, first we looked at the statistics of alpha carbons and the torsion angles. Then they were correlated with positions of positively
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
NMR, X-ray Diffraction, Protein Structure, and RasMol
 NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
7.91 Amy Keating. Solving structures using X-ray crystallography & NMR spectroscopy
 7.91 Amy Keating Solving structures using X-ray crystallography & NMR spectroscopy How are X-ray crystal structures determined? 1. Grow crystals - structure determination by X-ray crystallography relies
7.91 Amy Keating Solving structures using X-ray crystallography & NMR spectroscopy How are X-ray crystal structures determined? 1. Grow crystals - structure determination by X-ray crystallography relies
The protein folding problem consists of two parts:
 Energetics and kinetics of protein folding The protein folding problem consists of two parts: 1)Creating a stable, well-defined structure that is significantly more stable than all other possible structures.
Energetics and kinetics of protein folding The protein folding problem consists of two parts: 1)Creating a stable, well-defined structure that is significantly more stable than all other possible structures.
Protein Modeling Methods. Knowledge. Protein Modeling Methods. Fold Recognition. Knowledge-based methods. Introduction to Bioinformatics
 Protein Modeling Methods Introduction to Bioinformatics Iosif Vaisman Ab initio methods Energy-based methods Knowledge-based methods Email: ivaisman@gmu.edu Protein Modeling Methods Ab initio methods:
Protein Modeling Methods Introduction to Bioinformatics Iosif Vaisman Ab initio methods Energy-based methods Knowledge-based methods Email: ivaisman@gmu.edu Protein Modeling Methods Ab initio methods:
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein Structure Basics
 Protein Structure Basics Presented by Alison Fraser, Christine Lee, Pradhuman Jhala, Corban Rivera Importance of Proteins Muscle structure depends on protein-protein interactions Transport across membranes
Protein Structure Basics Presented by Alison Fraser, Christine Lee, Pradhuman Jhala, Corban Rivera Importance of Proteins Muscle structure depends on protein-protein interactions Transport across membranes
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
Building 3D models of proteins
 Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Building 3D models of proteins Why make a structural model for your protein? The structure can provide clues to the function through structural similarity with other proteins With a structure it is easier
Useful background reading
 Overview of lecture * General comment on peptide bond * Discussion of backbone dihedral angles * Discussion of Ramachandran plots * Description of helix types. * Description of structures * NMR patterns
Overview of lecture * General comment on peptide bond * Discussion of backbone dihedral angles * Discussion of Ramachandran plots * Description of helix types. * Description of structures * NMR patterns
Protein Structures: Experiments and Modeling. Patrice Koehl
 Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Protein Structures: Experiments and Modeling Patrice Koehl Structural Bioinformatics: Proteins Proteins: Sources of Structure Information Proteins: Homology Modeling Proteins: Ab initio prediction Proteins:
Protein structure analysis. Risto Laakso 10th January 2005
 Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Secondary Structure. Bioch/BIMS 503 Lecture 2. Structure and Function of Proteins. Further Reading. Φ, Ψ angles alone determine protein structure
 Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Bioch/BIMS 503 Lecture 2 Structure and Function of Proteins August 28, 2008 Robert Nakamoto rkn3c@virginia.edu 2-0279 Secondary Structure Φ Ψ angles determine protein structure Φ Ψ angles are restricted
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University
 Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
09/06/25. Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Non-uniform distribution of folds. Scheme of protein structure predicition
 Non-uniform distribution of folds. Scheme of protein structure predicition") Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Sequence identity Structural similarity Computergestützte Strukturbiologie (Strukturelle Bioinformatik) Fold recognition Sommersemester 2009 Peter Güntert Structural similarity X Sequence identity Non-uniform
Francisco Melo, Damien Devos, Eric Depiereux and Ernest Feytmans
 From: ISMB-97 Proceedings. Copyright 1997, AAAI (www.aaai.org). All rights reserved. ANOLEA: A www Server to Assess Protein Structures Francisco Melo, Damien Devos, Eric Depiereux and Ernest Feytmans Facultés
From: ISMB-97 Proceedings. Copyright 1997, AAAI (www.aaai.org). All rights reserved. ANOLEA: A www Server to Assess Protein Structures Francisco Melo, Damien Devos, Eric Depiereux and Ernest Feytmans Facultés
Orientational degeneracy in the presence of one alignment tensor.
 Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Orientational degeneracy in the presence of one alignment tensor. Rotation about the x, y and z axes can be performed in the aligned mode of the program to examine the four degenerate orientations of two
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche
 Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Protein Structure Prediction II Lecturer: Serafim Batzoglou Scribe: Samy Hamdouche The molecular structure of a protein can be broken down hierarchically. The primary structure of a protein is simply its
Visualization of Macromolecular Structures
 Visualization of Macromolecular Structures Present by: Qihang Li orig. author: O Donoghue, et al. Structural biology is rapidly accumulating a wealth of detailed information. Over 60,000 high-resolution
Visualization of Macromolecular Structures Present by: Qihang Li orig. author: O Donoghue, et al. Structural biology is rapidly accumulating a wealth of detailed information. Over 60,000 high-resolution
Secondary and sidechain structures
 Lecture 2 Secondary and sidechain structures James Chou BCMP201 Spring 2008 Images from Petsko & Ringe, Protein Structure and Function. Branden & Tooze, Introduction to Protein Structure. Richardson, J.
Lecture 2 Secondary and sidechain structures James Chou BCMP201 Spring 2008 Images from Petsko & Ringe, Protein Structure and Function. Branden & Tooze, Introduction to Protein Structure. Richardson, J.
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program)
") Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Syllabus of BIOINF 528 (2017 Fall, Bioinformatics Program) Course Name: Structural Bioinformatics Course Description: Instructor: This course introduces fundamental concepts and methods for structural
Protein Structure Determination
 Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Protein Structure Determination Given a protein sequence, determine its 3D structure 1 MIKLGIVMDP IANINIKKDS SFAMLLEAQR RGYELHYMEM GDLYLINGEA 51 RAHTRTLNVK QNYEEWFSFV GEQDLPLADL DVILMRKDPP FDTEFIYATY 101
Bi 8 Midterm Review. TAs: Sarah Cohen, Doo Young Lee, Erin Isaza, and Courtney Chen
 Bi 8 Midterm Review TAs: Sarah Cohen, Doo Young Lee, Erin Isaza, and Courtney Chen The Central Dogma Biology Fundamental! Prokaryotes and Eukaryotes Nucleic Acid Components Nucleic Acid Structure DNA Base
Bi 8 Midterm Review TAs: Sarah Cohen, Doo Young Lee, Erin Isaza, and Courtney Chen The Central Dogma Biology Fundamental! Prokaryotes and Eukaryotes Nucleic Acid Components Nucleic Acid Structure DNA Base
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence
 Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
Number sequence representation of protein structures based on the second derivative of a folded tetrahedron sequence Naoto Morikawa (nmorika@genocript.com) October 7, 2006. Abstract A protein is a sequence
Conformational Geometry of Peptides and Proteins:
 Conformational Geometry of Peptides and Proteins: Before discussing secondary structure, it is important to appreciate the conformational plasticity of proteins. Each residue in a polypeptide has three
Conformational Geometry of Peptides and Proteins: Before discussing secondary structure, it is important to appreciate the conformational plasticity of proteins. Each residue in a polypeptide has three
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Protein Modeling. Generating, Evaluating and Refining Protein Homology Models
 Protein Modeling Generating, Evaluating and Refining Protein Homology Models Troy Wymore and Kristen Messinger Biomedical Initiatives Group Pittsburgh Supercomputing Center Homology Modeling of Proteins
Protein Modeling Generating, Evaluating and Refining Protein Homology Models Troy Wymore and Kristen Messinger Biomedical Initiatives Group Pittsburgh Supercomputing Center Homology Modeling of Proteins
Homology Modeling I. Growth of the Protein Data Bank PDB. Basel, September 30, EMBnet course: Introduction to Protein Structure Bioinformatics
 Swiss Institute of Bioinformatics EMBnet course: Introduction to Protein Structure Bioinformatics Homology Modeling I Basel, September 30, 2004 Torsten Schwede Biozentrum - Universität Basel Swiss Institute
Swiss Institute of Bioinformatics EMBnet course: Introduction to Protein Structure Bioinformatics Homology Modeling I Basel, September 30, 2004 Torsten Schwede Biozentrum - Universität Basel Swiss Institute
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Molecular Modeling Lecture 7. Homology modeling insertions/deletions manual realignment
 Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Molecular Modeling 2018-- Lecture 7 Homology modeling insertions/deletions manual realignment Homology modeling also called comparative modeling Sequences that have similar sequence have similar structure.
Physiochemical Properties of Residues
 Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
SUPPLEMENTARY INFORMATION
 www.nature.com/nature 1 Figure S1 Sequence alignment. a Structure based alignment of the plgic of E. chrysanthemi (ELIC), the acetylcholine binding protein from the snail Lymnea stagnalis (AchBP, PDB code
www.nature.com/nature 1 Figure S1 Sequence alignment. a Structure based alignment of the plgic of E. chrysanthemi (ELIC), the acetylcholine binding protein from the snail Lymnea stagnalis (AchBP, PDB code
Giri Narasimhan. CAP 5510: Introduction to Bioinformatics. ECS 254; Phone: x3748
 CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
CAP 5510: Introduction to Bioinformatics Giri Narasimhan ECS 254; Phone: x3748 giri@cis.fiu.edu www.cis.fiu.edu/~giri/teach/bioinfs07.html 2/15/07 CAP5510 1 EM Algorithm Goal: Find θ, Z that maximize Pr
Building a Homology Model of the Transmembrane Domain of the Human Glycine α-1 Receptor
 Building a Homology Model of the Transmembrane Domain of the Human Glycine α-1 Receptor Presented by Stephanie Lee Research Mentor: Dr. Rob Coalson Glycine Alpha 1 Receptor (GlyRa1) Member of the superfamily
Building a Homology Model of the Transmembrane Domain of the Human Glycine α-1 Receptor Presented by Stephanie Lee Research Mentor: Dr. Rob Coalson Glycine Alpha 1 Receptor (GlyRa1) Member of the superfamily
Protein Structure. W. M. Grogan, Ph.D. OBJECTIVES
 Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Protein Structures. 11/19/2002 Lecture 24 1
 Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Protein Structures 11/19/2002 Lecture 24 1 All 3 figures are cartoons of an amino acid residue. 11/19/2002 Lecture 24 2 Peptide bonds in chains of residues 11/19/2002 Lecture 24 3 Angles φ and ψ in the
Supplementary Figure 1. Aligned sequences of yeast IDH1 (top) and IDH2 (bottom) with isocitrate
 and IDH2 (bottom) with isocitrate") SUPPLEMENTARY FIGURE LEGENDS Supplementary Figure 1. Aligned sequences of yeast IDH1 (top) and IDH2 (bottom) with isocitrate dehydrogenase from Escherichia coli [ICD, pdb 1PB1, Mesecar, A. D., and Koshland,
SUPPLEMENTARY FIGURE LEGENDS Supplementary Figure 1. Aligned sequences of yeast IDH1 (top) and IDH2 (bottom) with isocitrate dehydrogenase from Escherichia coli [ICD, pdb 1PB1, Mesecar, A. D., and Koshland,
Preparing a PDB File
 Figure 1: Schematic view of the ligand-binding domain from the vitamin D receptor (PDB file 1IE9). The crystallographic waters are shown as small spheres and the bound ligand is shown as a CPK model. HO
Figure 1: Schematic view of the ligand-binding domain from the vitamin D receptor (PDB file 1IE9). The crystallographic waters are shown as small spheres and the bound ligand is shown as a CPK model. HO
Table S1. Primers used for the constructions of recombinant GAL1 and λ5 mutants. GAL1-E74A ccgagcagcgggcggctgtctttcc ggaaagacagccgcccgctgctcgg
 SUPPLEMENTAL DATA Table S1. Primers used for the constructions of recombinant GAL1 and λ5 mutants Sense primer (5 to 3 ) Anti-sense primer (5 to 3 ) GAL1 mutants GAL1-E74A ccgagcagcgggcggctgtctttcc ggaaagacagccgcccgctgctcgg
SUPPLEMENTAL DATA Table S1. Primers used for the constructions of recombinant GAL1 and λ5 mutants Sense primer (5 to 3 ) Anti-sense primer (5 to 3 ) GAL1 mutants GAL1-E74A ccgagcagcgggcggctgtctttcc ggaaagacagccgcccgctgctcgg
Better Bond Angles in the Protein Data Bank
 Better Bond Angles in the Protein Data Bank C.J. Robinson and D.B. Skillicorn School of Computing Queen s University {robinson,skill}@cs.queensu.ca Abstract The Protein Data Bank (PDB) contains, at least
Better Bond Angles in the Protein Data Bank C.J. Robinson and D.B. Skillicorn School of Computing Queen s University {robinson,skill}@cs.queensu.ca Abstract The Protein Data Bank (PDB) contains, at least
Copyright Mark Brandt, Ph.D A third method, cryogenic electron microscopy has seen increasing use over the past few years.
 Structure Determination and Sequence Analysis The vast majority of the experimentally determined three-dimensional protein structures have been solved by one of two methods: X-ray diffraction and Nuclear
Structure Determination and Sequence Analysis The vast majority of the experimentally determined three-dimensional protein structures have been solved by one of two methods: X-ray diffraction and Nuclear
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Structure Comparison
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
CMPS 6630: Introduction to Computational Biology and Bioinformatics Structure Comparison Protein Structure Comparison Motivation Understand sequence and structure variability Understand Domain architecture
Gerd Krause, Structural bioinformatics and protein design Leibniz-Institute of molecular Pharmacology
 Basics of Molecular modelling _2 History, Interdisciplinary classification, Goals, Model definition, Molecular mechanics forcefields, Energy minimization algorithms Molecular dynamics simulation Comparative
Basics of Molecular modelling _2 History, Interdisciplinary classification, Goals, Model definition, Molecular mechanics forcefields, Energy minimization algorithms Molecular dynamics simulation Comparative
Central Dogma. modifications genome transcriptome proteome
 entral Dogma DA ma protein post-translational modifications genome transcriptome proteome 83 ierarchy of Protein Structure 20 Amino Acids There are 20 n possible sequences for a protein of n residues!
entral Dogma DA ma protein post-translational modifications genome transcriptome proteome 83 ierarchy of Protein Structure 20 Amino Acids There are 20 n possible sequences for a protein of n residues!
Prediction and refinement of NMR structures from sparse experimental data
 Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
Prediction and refinement of NMR structures from sparse experimental data Jeff Skolnick Director Center for the Study of Systems Biology School of Biology Georgia Institute of Technology Overview of talk
Figure 1. Molecules geometries of 5021 and Each neutral group in CHARMM topology was grouped in dash circle.
 Project I Chemistry 8021, Spring 2005/2/23 This document was turned in by a student as a homework paper. 1. Methods First, the cartesian coordinates of 5021 and 8021 molecules (Fig. 1) are generated, in
Project I Chemistry 8021, Spring 2005/2/23 This document was turned in by a student as a homework paper. 1. Methods First, the cartesian coordinates of 5021 and 8021 molecules (Fig. 1) are generated, in
RNA and Protein Structure Prediction
 RNA and Protein Structure Prediction Bioinformatics: Issues and Algorithms CSE 308-408 Spring 2007 Lecture 18-1- Outline Multi-Dimensional Nature of Life RNA Secondary Structure Prediction Protein Structure
RNA and Protein Structure Prediction Bioinformatics: Issues and Algorithms CSE 308-408 Spring 2007 Lecture 18-1- Outline Multi-Dimensional Nature of Life RNA Secondary Structure Prediction Protein Structure
Summary of Experimental Protein Structure Determination. Key Elements
 Programme 8.00-8.20 Summary of last week s lecture and quiz 8.20-9.00 Structure validation 9.00-9.15 Break 9.15-11.00 Exercise: Structure validation tutorial 11.00-11.10 Break 11.10-11.40 Summary & discussion
Programme 8.00-8.20 Summary of last week s lecture and quiz 8.20-9.00 Structure validation 9.00-9.15 Break 9.15-11.00 Exercise: Structure validation tutorial 11.00-11.10 Break 11.10-11.40 Summary & discussion
Sequence Alignment: A General Overview. COMP Fall 2010 Luay Nakhleh, Rice University
 Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
Sequence Alignment: A General Overview COMP 571 - Fall 2010 Luay Nakhleh, Rice University Life through Evolution All living organisms are related to each other through evolution This means: any pair of
NMR Assay of Purity and Folding
 NMR Assay of Purity and Folding Don t Need Resonance Assignments or Labeling 1D requires only 10-50 µm protein concentration 2D Provides A More Detailed Assay 15 N- 1 H HSQC 1 H COSY 13 C HSQC also! Analyze
NMR Assay of Purity and Folding Don t Need Resonance Assignments or Labeling 1D requires only 10-50 µm protein concentration 2D Provides A More Detailed Assay 15 N- 1 H HSQC 1 H COSY 13 C HSQC also! Analyze
We used the PSI-BLAST program (http://www.ncbi.nlm.nih.gov/blast/) to search the
 to search the") SUPPLEMENTARY METHODS - in silico protein analysis We used the PSI-BLAST program (http://www.ncbi.nlm.nih.gov/blast/) to search the Protein Data Bank (PDB, http://www.rcsb.org/pdb/) and the NCBI non-redundant
SUPPLEMENTARY METHODS - in silico protein analysis We used the PSI-BLAST program (http://www.ncbi.nlm.nih.gov/blast/) to search the Protein Data Bank (PDB, http://www.rcsb.org/pdb/) and the NCBI non-redundant
Details of Protein Structure
 Details of Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Anne Mølgaard, Kemisk Institut, Københavns Universitet Learning Objectives
Details of Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Anne Mølgaard, Kemisk Institut, Københavns Universitet Learning Objectives
Protein Structure Prediction and Protein-Ligand Docking
 Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
Protein Structure Prediction and Protein-Ligand Docking Björn Wallner bjornw@ifm.liu.se Jan. 24, 2014 Todays topics Protein Folding Intro Protein structure prediction How can we predict the structure of
1) NMR is a method of chemical analysis. (Who uses NMR in this way?) 2) NMR is used as a method for medical imaging. (called MRI )
 NMR is a method of chemical analysis. (Who uses NMR in this way?) 2) NMR is used as a method for medical imaging. (called MRI )") Uses of NMR: 1) NMR is a method of chemical analysis. (Who uses NMR in this way?) 2) NMR is used as a method for medical imaging. (called MRI ) 3) NMR is used as a method for determining of protein, DNA,
Uses of NMR: 1) NMR is a method of chemical analysis. (Who uses NMR in this way?) 2) NMR is used as a method for medical imaging. (called MRI ) 3) NMR is used as a method for determining of protein, DNA,
Homology models of the tetramerization domain of six eukaryotic voltage-gated potassium channels Kv1.1-Kv1.6
 Homology models of the tetramerization domain of six eukaryotic voltage-gated potassium channels Kv1.1-Kv1.6 Hsuan-Liang Liu* and Chin-Wen Chen Department of Chemical Engineering and Graduate Institute
Homology models of the tetramerization domain of six eukaryotic voltage-gated potassium channels Kv1.1-Kv1.6 Hsuan-Liang Liu* and Chin-Wen Chen Department of Chemical Engineering and Graduate Institute
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Protein Structure & Motifs
 & Motifs Biochemistry 201 Molecular Biology January 12, 2000 Doug Brutlag Introduction Proteins are more flexible than nucleic acids in structure because of both the larger number of types of residues
& Motifs Biochemistry 201 Molecular Biology January 12, 2000 Doug Brutlag Introduction Proteins are more flexible than nucleic acids in structure because of both the larger number of types of residues
Tools for Cryo-EM Map Fitting. Paul Emsley MRC Laboratory of Molecular Biology
 Tools for Cryo-EM Map Fitting Paul Emsley MRC Laboratory of Molecular Biology April 2017 Cryo-EM model-building typically need to move more atoms that one does for crystallography the maps are lower resolution
Tools for Cryo-EM Map Fitting Paul Emsley MRC Laboratory of Molecular Biology April 2017 Cryo-EM model-building typically need to move more atoms that one does for crystallography the maps are lower resolution
Supplementary Information
 1 Supplementary Information Figure S1 The V=0.5 Harker section of an anomalous difference Patterson map calculated using diffraction data from the NNQQNY crystal at 1.3 Å resolution. The position of the
1 Supplementary Information Figure S1 The V=0.5 Harker section of an anomalous difference Patterson map calculated using diffraction data from the NNQQNY crystal at 1.3 Å resolution. The position of the
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Nature Structural and Molecular Biology: doi: /nsmb.2938
 Supplementary Figure 1 Characterization of designed leucine-rich-repeat proteins. (a) Water-mediate hydrogen-bond network is frequently visible in the convex region of LRR crystal structures. Examples
Supplementary Figure 1 Characterization of designed leucine-rich-repeat proteins. (a) Water-mediate hydrogen-bond network is frequently visible in the convex region of LRR crystal structures. Examples
Protein structure (and biomolecular structure more generally) CS/CME/BioE/Biophys/BMI 279 Sept. 28 and Oct. 3, 2017 Ron Dror
 CS/CME/BioE/Biophys/BMI 279 Sept. 28 and Oct. 3, 2017 Ron Dror") Protein structure (and biomolecular structure more generally) CS/CME/BioE/Biophys/BMI 279 Sept. 28 and Oct. 3, 2017 Ron Dror Please interrupt if you have questions, and especially if you re confused! Assignment
Protein structure (and biomolecular structure more generally) CS/CME/BioE/Biophys/BMI 279 Sept. 28 and Oct. 3, 2017 Ron Dror Please interrupt if you have questions, and especially if you re confused! Assignment
Bioengineering & Bioinformatics Summer Institute, Dept. Computational Biology, University of Pittsburgh, PGH, PA
 Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
Pharmacophore Model Development for the Identification of Novel Acetylcholinesterase Inhibitors Edwin Kamau Dept Chem & Biochem Kennesa State Uni ersit Kennesa GA 30144 Dept. Chem. & Biochem. Kennesaw
BCMP 201 Protein biochemistry
 BCMP 201 Protein biochemistry BCMP 201 Protein biochemistry with emphasis on the interrelated roles of protein structure, catalytic activity, and macromolecular interactions in biological processes. The
BCMP 201 Protein biochemistry BCMP 201 Protein biochemistry with emphasis on the interrelated roles of protein structure, catalytic activity, and macromolecular interactions in biological processes. The
COMP 598 Advanced Computational Biology Methods & Research. Introduction. Jérôme Waldispühl School of Computer Science McGill University
 COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
COMP 598 Advanced Computational Biology Methods & Research Introduction Jérôme Waldispühl School of Computer Science McGill University General informations (1) Office hours: by appointment Office: TR3018
1. Protein Data Bank (PDB) 1. Protein Data Bank (PDB)
 1. Protein Data Bank (PDB)") Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
Protein structure databases; visualization; and classifications 1. Introduction to Protein Data Bank (PDB) 2. Free graphic software for 3D structure visualization 3. Hierarchical classification of protein
Template-Based Modeling of Protein Structure
 Template-Based Modeling of Protein Structure David Constant Biochemistry 218 December 11, 2011 Introduction. Much can be learned about the biology of a protein from its structure. Simply put, structure
Template-Based Modeling of Protein Structure David Constant Biochemistry 218 December 11, 2011 Introduction. Much can be learned about the biology of a protein from its structure. Simply put, structure
Student Questions and Answers October 8, 2002
 Student Questions and Answers October 8, 2002 Q l. Is the Cα of Proline also chiral? Answer: FK: Yes, there are 4 different residues bound to this C. Only in a strictly planar molecule this would not hold,
Student Questions and Answers October 8, 2002 Q l. Is the Cα of Proline also chiral? Answer: FK: Yes, there are 4 different residues bound to this C. Only in a strictly planar molecule this would not hold,
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009
 114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
114 Grundlagen der Bioinformatik, SS 09, D. Huson, July 6, 2009 9 Protein tertiary structure Sources for this chapter, which are all recommended reading: D.W. Mount. Bioinformatics: Sequences and Genome
Theory and Applications of Residual Dipolar Couplings in Biomolecular NMR
 Theory and Applications of Residual Dipolar Couplings in Biomolecular NMR Residual Dipolar Couplings (RDC s) Relatively new technique ~ 1996 Nico Tjandra, Ad Bax- NIH, Jim Prestegard, UGA Combination of
Theory and Applications of Residual Dipolar Couplings in Biomolecular NMR Residual Dipolar Couplings (RDC s) Relatively new technique ~ 1996 Nico Tjandra, Ad Bax- NIH, Jim Prestegard, UGA Combination of