Deciphering intrinsic inter-subunit couplings that lead to sequential
|
|
|
- Suzanna Jane Hill
- 6 years ago
- Views:
Transcription
1 Deciphering intrinsic inter-subunit couplings that lead to sequential hydrolysis of F 1 -ATPase ring Liqiang Dai 1, Holger Flechsig 2, and Jin Yu 1 * 1 Beijing Computational Science Research Center, Complex System Research Division, Beijing , China 2 Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima , Japan *Corresponding author: jinyu@csrc.ac.cn(jy) 1
2 Abstract The rotary sequential hydrolysis of metabolic machine F 1 -ATPase is a prominent feature to reveal high coordination among multiple chemical sites on the stator F 1 ring, which also contributes to tight coupling between the chemical reaction and central γ-shaft rotation. High-speed AFM experiments discovered that the sequential hydrolysis was maintained on the F 1 ring even in the absence of the γ rotor. To explore how the intrinsic sequential performance arises, we computationally investigated essential inter-subunit couplings on the hexameric ring of mitochondrial and bacterial F 1. We first reproduced the sequential hydrolysis schemes as experimentally detected, by simulating tri-site ATP hydrolysis cycles on the F 1 ring upon kinetically imposing inter-subunit couplings to substantially promote the hydrolysis products release. We found that it is key for certain ATP binding and hydrolysis events to trigger or accelerate the neighbor-site ADP and Pi release to support the sequential hydrolysis. The kinetically feasible inter-subunit couplings were then scrutinized through atomistic molecular dynamics simulations as well as coarse-grained simulations, in which we enforced targeted conformational changes for the triggering ATP binding or hydrolysis. Notably, we detected the asymmetrical neighbor-site opening that would facilitate the ADP release upon the enforced ATP binding, and computationally captured the complete Pi release through charge hopping upon the enforced neighbor-site ATP hydrolysis. The ATP-hydrolysis triggered Pi release revealed in current TMD simulation confirms a recent prediction made from statistical analyses of single molecule experimental data in regard to the role the ATP hydrolysis plays. Our studies, therefore, elucidate both the concerted chemical kinetics and underlying structural dynamics of the inter-subunit couplings that lead to the rotary sequential hydrolysis of the F 1 stator ring. Keywords: molecular machines, chemical kinetics, stochastic simulation, molecular dynamics simulation, elastic-network relaxation dynamics 2
3 Author Summary Ring-shaped NTPases usually maintain high coordination among multiple subunits on the ring to generate force or torque onto a protein or DNA substrate threading through the center of the ring. Among them, F 1 -ATPase is an exemplary high efficiency nano-machine that tightly couples rotatory sequential hydrolysis on the ring with the central γ-shaft rotation. Experiments found that the sequential hydrolysis maintains on the F 1 stator ring, however, even without the γ rotor in the center. Accordingly, we employed computational methods to investigate how such sequential coordination arises intrinsically, owning to neighbor subunit interactions or couplings on the ring. Kinetically feasible couplings along with structural dynamics basis were elucidated from the combined stochastic modeling, atomistic, and coarse-grained simulations. In particular, we found asymmetrical site openings on the ring to facilitate ADP and Pi releases upon the neighbor-site ATP binding or hydrolysis. The coupling scenarios highlight molecular design principles to synchronize chemical reactions with protein conformational propagations, which may apply to a group of similar types of ring NTPase motors. 3
4 Introduction The coordination on ring-shaped NTPases is fundamental and crucial for diverse physiological functions of these cellular molecule machines [1-5]. In well-studied systems such as F o F 1 -ATP synthase, the binding change mechanism, for example, had been proposed early to illustrate rotary cooperative catalysis around the hexameric F 1 -ATP ring [6,7]. The mechanism indicated that binding and catalytic activities of ATP alternate sequentially on three chemically active sites, formed at the interfaces of the α and β subunits on the ring [8]. According to the mechanism, the central γ-shaft across the F 1 ring dictates the coordinated alternation [9-12]. Nevertheless, experimental studies later on showed that upon significant truncations of the γ-shaft, the F 1 motor still rotates directionally [13-16]. Furthermore, recent experiments conducted in the absence of the central γ-shaft demonstrated that the stator F 1 -ring also maintains the rotary sequential hydrolysis around the three chemical sites [17]. These studies consistently indicated that the sequential coordination arises intrinsically from the F 1 -ATPase ring, though the stator-rotor interactions would likely enhance the rotary cooperativity. In this work, we aimed to investigate how the intrinsic sequential coordination around the F 1 -ATPase ring arises due to inter-subunit or site couplings, in the absence of the central γ-shaft. We assume that the sequential hydrolysis scheme achieved without the γ rotor is similar to that detected originally with the rotor. We focused on two typical reaction schemes, one suggested recently for mitochondrial F 1 (as MF 1 ) [18], and the other determined for bacterial F 1 (as BF 1 ) [19]. For an individual ATPase site, formed by one α-β hetero-dimer, the uni-site reaction rates for the product ADP and Pi releases are particularly low [20,21]. To allow sufficiently fast reactions as that achieved on the tri-site F 1 ring, it is necessary that the inter-subunit couplings significantly accelerate the respective product releases, but how that happen is elusive. Inter-subunit coordination has been widely recognized for ring-like NTPases [1,2], such as viral RNA or DNA packaging motor [22,23], helicases [4,5], and F 1 -ATPase [2,6,24]. For example, in viral phi29 DNA packaging motor, it has been discovered that multiple subunits coordinate ADP releases with ATP bindings one after another or in an alternating fashion during a dwell phase [1,23,25], while Pi releases happen from one subunit to the next in a burst phase. Nevertheless, it is not clear what specific inter-subunit interactions support the coordination, and it remains a common puzzle for various ring-shaped NTPases. Some suggestions had been made, for example, addressing that ATP binding couples with neighbor-site ADP release [1,23-25],arginine-finger insertion from one subunit induces ATP hydrolysis in the next site [22,23,25], ATP hydrolysis may facilitate the Pi release in the neighbor site [26], or the Pi release facilitates the hydrolysis. A basic scenario is that one chemical event triggers certain conformational changes that propagate through the subunit interfaces to the neighbor-site(s), accelerate (or de-accelerate) the site conformational transitions on the chemical reaction path, with both kinetic and structural dynamics consequences. The quantitative studies to reveal these couplings and to determine the kinetics or structural dynamics underneath are nevertheless lack of. 4
5 The major task of our study is, therefore, to present quantitative description and physical understanding of the inter-subunit couplings on the F 1 -ATPase ring that essentially support the sequential hydrolysis. Computational modeling and simulations have contributed significantly to the understanding of molecular mechanisms of F 1 -ATPase [27-36]. In particular, atomistic molecular dynamics (MD) simulations provide detailed structural dynamics to the system, and the simulation time scale moved gradually from several nanoseconds [28] to tens to hundreds of nanoseconds and above [35]. Nevertheless, detecting micro to millisecond long-time conformational changes in such a protein machine is still too hard for conventional all-atom MD. Hence, to probe the inter-subunit conformational couplings, we enforced or accelerated certain conformational transitions to mimic the triggering chemical events in the conventional MD, and combined the atomistic MD with long-time chemical kinetic and coarse-grained structural approaches. While most of previous computational studies focused on quantifying the rotary motion of F 1 -ATPase (α 3 β 3 γ), current work neglected the rotational degree of freedom but studied only a minimal model of the F 1 ring for the sequential hydrolysis (α 3 β 3 ). We started by conducting stochastic kinetics simulations for the three independent ATP hydrolysis cycles, following the uni-site reaction rates obtained for bovine heart mitochondria F 1 [21] and E. coli F 1 [20], respectively. Meanwhile, inter-site couplings that likely happen among the three active sites of the F 1 ring were kinetically incorporated and adjusted in the simulation via tuning the involved kinetic rates. If the sequential hydrolysis emerged as experimentally detected [18,19], we then regard those inter-subunit couplings kinetically feasible. For current practice, we adopted a scenario that Pi releases after the ADP release [19,37]. Through the above quantitative analyses, we were able to determine kinetically feasible inter-subunit couplings that support the sequential hydrolysis. Then, we implemented atomistic MD simulations to probe the physical or structural basis of those couplings. The bovine MF 1 structure was employed in the MD simulation. To accelerate the triggering chemical events in the MD due to the limited simulation time scale (at sub-microseconds), we implemented the targeted MD (TMD) to one chemical site on the ring, mimicking the conformational changes of the ATP binding or hydrolysis event, and then monitored mechanical responses or conformational relaxations in the two neighboring active sites. We consequently found asymmetrical neighbor site openings that may facilitate the product(s) release or bias the ATP binding for the directional coordination. To validate the TMD results obtained under enforcements and accelerations, we further implemented targeted elastic network (TEN) simulations in a coarse-grained model to see whether the slow relaxation dynamics (overall micro to milliseconds e.g.) well captured in the coarse-grained model appear consistently with our hundreds of nanosecond TMD simulations. Results 1. Stochastic simulations identified kinetically feasible inter-subunit couplings that support the sequential hydrolysis on the F 1 ring In order to probe what type of inter-subunit or site couplings that are responsible and 5
6 kinetically feasible for the sequential hydrolysis of the F 1 -ATPase ring, we first conducted the kinetic Monte Carlo (KMC) simulations for three ATP hydrolysis cycles to mimic the chemical kinetics of the three active sites (see Methods). The three chemical cycles proceed independently when there is no inter-subunit coupling connecting the three sites. The coordinated sequential hydrolysis arises kinetically only when certain rates of one hydrolysis cycle are connected with another hydrolysis cycle via some coupling mechanisms or rules. We started by simulating the three independent hydrolysis cycles using uni-site reaction rates obtained experimentally [20,21] (see Supplementary Information SI Table S1 and S2). For each hydrolysis cycle, it goes through ATP binding (from E, the empty, to T, the ATP-bound state), hydrolysis (T or T* to DP, the ADP+Pi bound state), and product release (DP to P, the Pi-bound state, and then to E), under the uni-site rates. Occasionally when two sites among the three are bound with ATP, we refer to T*-site the one binds ATP tighter or earlier, so that it proceeds to an intermediate T* state ready for catalysis [29], i.e., with a higher ATP hydrolysis rate than that of T, an initially ATP bound state. We then imposed coupling rules into the three ATP hydrolysis cycles in the KMC simulations (see SI and Fig S1), e.g., by tuning the product release/unbinding rate of one site upon a certain chemical event in a neighboring site. Only when these rates were tuned sufficiently large at the right moments, can three hydrolysis cycles become sequentially correlated. For that we referred the coupling kinetically feasible. Specifically, the sequential performance was evaluated by a score function (between 0 and 1) based on experimentally detected sequential schemes [18,19]. A higher score shows a better performance (see SI Eq S1 and Fig S2-4). Consistently, in the high performance score case, high correlations between the neighboring sites reveal in their hydrolysis cycles, as one site follows the other closely moving along the chemical reaction path (see SI Eq S2 and Fig S5). Since it is not clear whether the product binding affinities are altered, or only the product release/unbinding rates change upon the chemical triggering event under the inter-subunit couplings, we imposed the coupling rules kinetically in two ways (see schematics in Fig 1A): for the implementation I we altered both the product unbinding and binding rates for the same amount to keep constant binding affinities; for the implementation II we tuned only the product unbinding rates while keeping the binding rates constant, assuming the binding being diffusion limited. Regardless of the way of the implementation, we found that the key couplings that supports the sequential performance are (see Fig 1 and SI Fig S6): 1.1 ATP binding to the E-DP-T ring facilitates the product release from the DP site. Presumably, as the ATP binding (Eà T) induces an open to closed transition in the current site, the next/downstream site (in the counterclockwise direction viewed from the C-terminal side of F 1 ) can be disturbed and the product (ADP and Pi) release rate can be affected [1,23,24]. Accordingly, in the kinetic simulation(s), we allowed the ATP binding 6
7 to the E-DP-T ring to enhance both the ADP and Pi releases in the next site. In MF 1, above ~10 5 fold of the rate enhancement (see SI Fig S3) was applied to both the ADP and Pi release in the next site (from E-DP-T to T-E-T*, see Fig 1B), so that to achieve a sufficiently high sequential performance. In BF 1, (from E-DP-T to T-P-DP, see Fig 1B), the ADP release rate has to be enhanced upon the previous site ATP binding (above ~10 3 fold, see SI Fig S4) to allow the sequential performance; the Pi release rate can be enhanced either along with the ADP release upon the previous ATP binding (as in MF 1 ), or until after the next site ATP hydrolysis (see SI Fig S2B and C), as analyzed below. 1.2 The Pi release can be facilitated right after the ADP release or upon ATP hydrolysis in the T-P-T* ring It has been recently suggested, according to statistical analyses on time series data from high-speed single molecule experiments, that the ATP hydrolysis plays a role to facilitate the neighbor-site Pi release [26]. We tested if this coupling scenario between the ATP hydrolysis and Pi release could support the sequential hydrolysis in the KMC simulation of BF1. We found that by enhancing the Pi release rate ~10 3 fold upon the ATP hydrolysis in the next site (see SI Fig S4), the sequential hydrolysis emerged. Alternatively, the Pi release in BF1 could also be enhanced right after the ADP release as that in MF1 to support the sequential performance. In addition to the most essential couplings identified above, we found it necessary to add a constraint to the MF 1 ring (but not to the BF 1 ring) to prohibit the three-atp bound (3-T) configuration, which was not experimentally detected [17]. In the absence of an explicit constraint, the population of the 3-T was already low in the simulation (e.g. 3~6% in MF 1, below ~1% in BF 1 ), yet the sequential performance score was still low in MF 1 (~ 0.2 to 0.3). Imposing the constraint to MF 1 by reducing the third ATP binding rate for 10 ~ 100 fold (see SI Fig S1 to S3) substantially improved the score (to above ~0.6), and lowered the 3-T population further in MF 1 (below ~1%). On the other hand, there is no need to impose the constraint to BF 1, the 3-T configuration was simply absent with existing tri-site kinetics on the BF 1 ring, and the sequential performance does not require the constraint. As mentioned earlier, in order to ensure that the early ATP bound T*-site hydrolyzes sufficiently fast and faster than the newly bound T-site, we also allowed a rate enhancement (~10 to 100) in T*à DP above that of Tà DP (see SI Fig S1 to S4). To further probe the physical basis of the kinetically feasible couplings identified above, we performed atomistic MD simulations as illustrated below. 2. Atomistic MD simulations demonstrated asymmetrical openings of the neighbor site upon current site closing/tightening induced by ATP binding/hydrolysis We performed four sets of targeted MD (TMD) simulations [38], mimicking the ATP binding or ATP hydrolysis in one chemical site, and monitored the ensued mechanical responses from the two neighboring chemical sites: i) targeting the ATP binding from E-DP-T to T-DP-T* to examine how the downstream DP-site and the upstream T-site 7
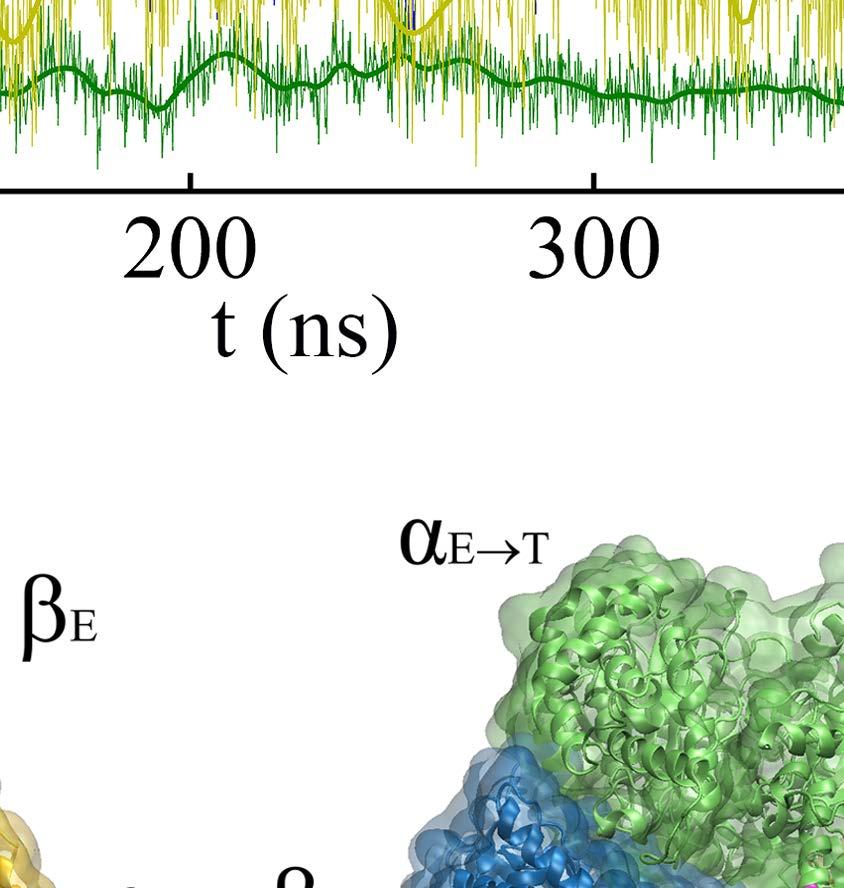
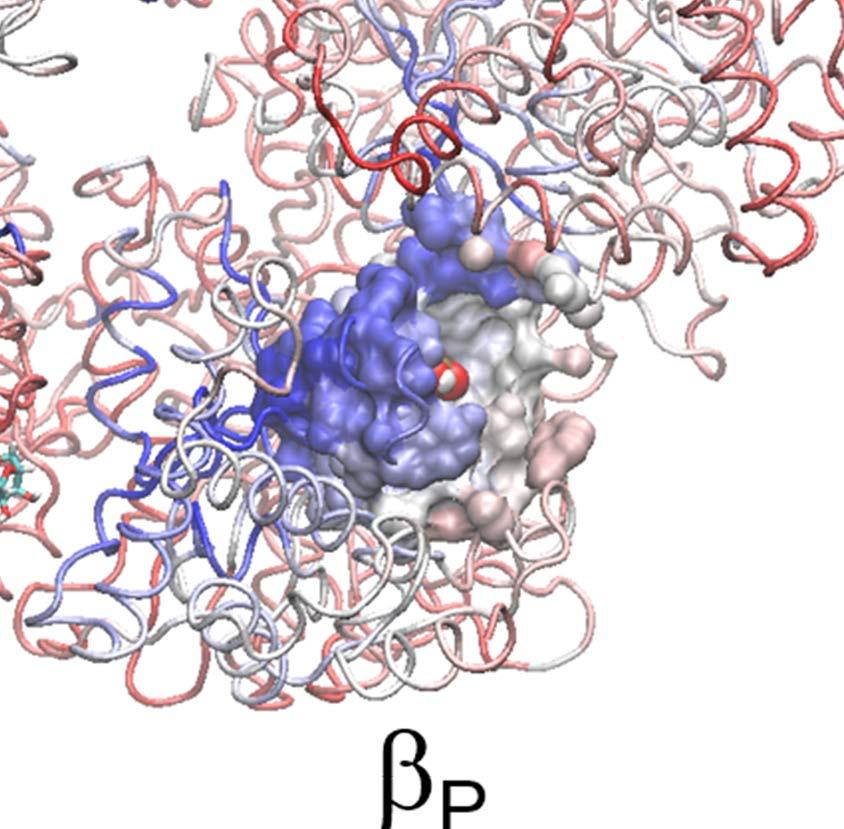
8 would be affected, with a control TMD simulation to include the central γ-shaft; ii) targeting the ATP hydrolysis from T-P-T* to T-P-DP, to examine how the upstream P-site and the downstream T-site would react; iii) targeting the 1 st -ATP binding from E-E-E to E-T-E, to examine the responses without the ligand binding in both neighboring sites; iv) targeting the 3 rd -ATP binding to E-T*-T, to see if the 3-T ring remains stable. Note that in each of the TMD simulations, the protein conformational changes corresponding to the ATP binding or hydrolysis triggering event were enforced to happen to the single chemical site to accelerate the originally slow process. The initial and final conformations of the single targeted site (e.g. T-site for ATP binding and DP-site for hydrolysis) were commonly known. The initial structures of the tri-site ring, however, were often made from existing crystal structures, while the final ring structures were usually unknown but were then generated from our TMD simulations. i. ATP binding to E-DP-T opens the downstream DP-site to facilitate the ADP release We obtained an E-DP-T ring from the P-DP-T structure of the bovine MF 1 (PDB: 1E1R) [39], and performed the TMD simulation to enforce the α E -β E structure to approach to the targeted α T -β T conformation, so that the original E-site was turned into a T-site and a T-DP-T* ring was obtained (see Methods and Fig 2). To determine if the ATP binding site transited from the open to closed form as being enforced to, we calculated the C-terminal protrusion (i.e., on the DELSEED motif) height, the hinge-bending angle on the β subunit, and the size of the ATP binding pocket (see SI and Fig S7). Large values of these measures indicate an open site while small values are for a closed one. In Fig 2A, we see that the β E -protrusion decreased ~ 6 Å during the TMD simulation, indicating clearly a transition from an open site to a closed one (Eà T). In response, the average β DP -protrusion increased slightly (~ 1 Å; also see Fig 2B), the β DP hinge-bending angle increased (~ 5 ), along with the small pocket-size increase (~ 0.2 Å; see SI Fig S8), all indicating a slight opening of the DP-site. In contrast, the average β T -protrusion decreased a little (~ 0.2 Å), the pocket size of the T-site shrunk (~ 0.4 Å), though the hinge-bending angle of β T increased slightly (~ 1 ). Movies were made from the simulation for both the top and side views, highlighting how the β-protrusions shifted (SI Movie S1a and S1b). Since the average magnitudes of the responses were small, we performed an additional TMD simulation for comparison, including the γ-shaft into the center of the E-DP-T ring. The average β DP -protrusion then increased to ~ 3 Å upon the E-site closing (see SI Fig S9), though the changes from other measures remained similarly as in the absence of the γ-shaft. In brief, all three geometric measures consistently suggested that the downstream DP-site opened slightly in response to the ATP binding in the current E-site, while the upstream (clockwise) neighbor T-site did not show such an opening tendency. In particular, one could see that the DELSEED of β Eà T moved toward the center of the ring during the enforced ATP binding transition (see Fig 2C). The trend was kept in the presence of the γ-shaft, though the γ-shaft occupancy prevents the β Eà T protrusion from interacting directly with that of β DP, and the β DP protrusion then increased largely upon the γ-shaft rotation. 8
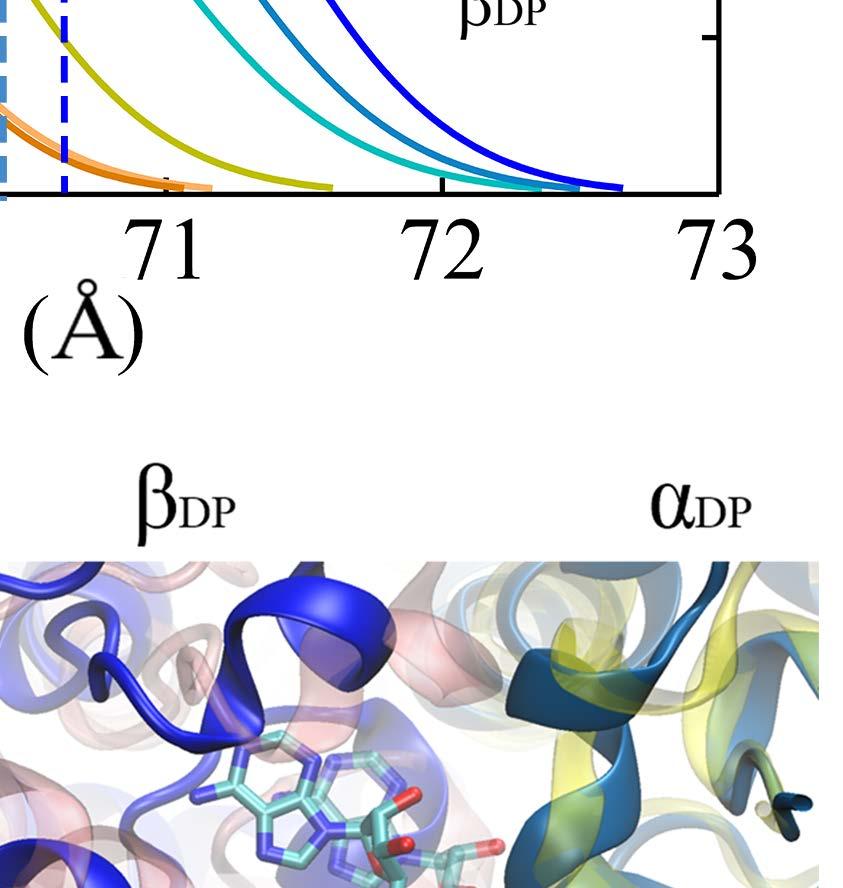
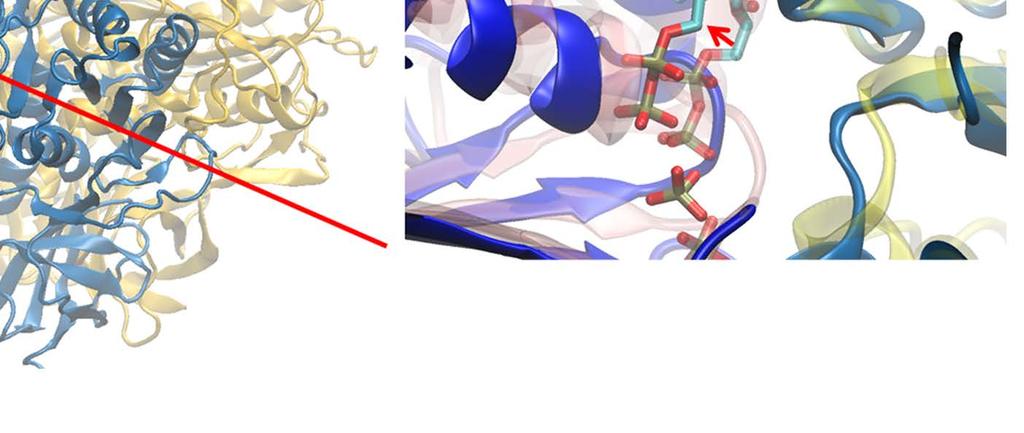
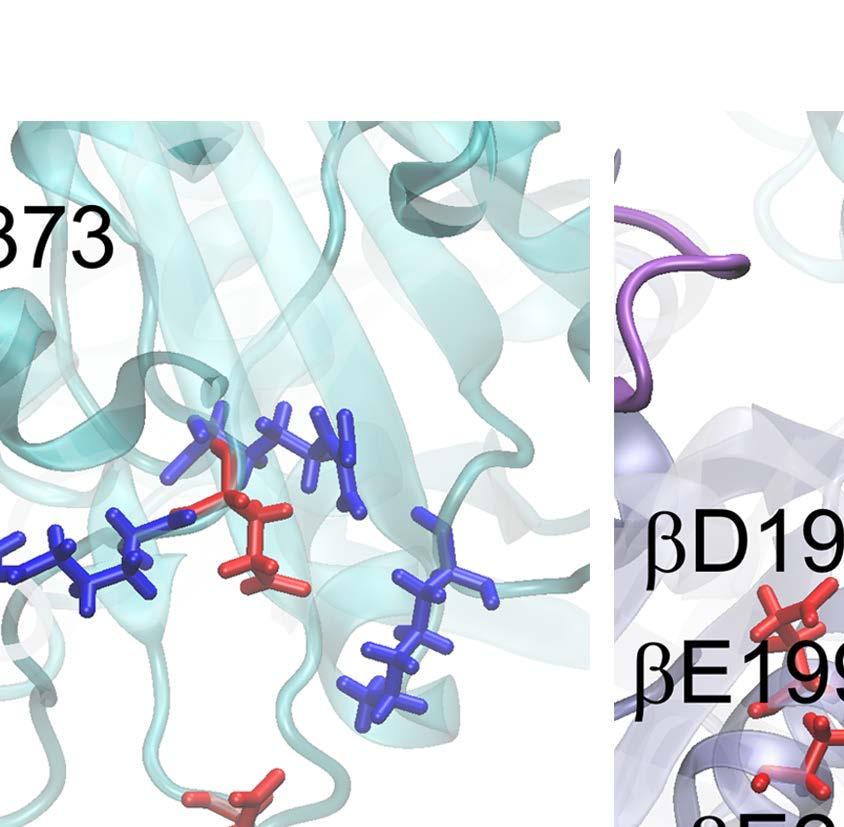
9 Notably, without the γ-shaft, one still sees that the DP-site interface widens between α DP and β DP upon the enforced ATP binding (see Fig 2D). The conformational changes get quite significant in the peripheral region of the DP-site (within 10 Å of the bound ADP), with an RMSD ~ 3 Å upon the enforced ATP binding to the E-site (see SI Fig S10A). The average number of hydrogen bonds in that region also decreased from ~25 (in the equilibrium control) to ~20 in the TMD simulation (SI Fig S10B). In particular, the hydrogen bonds that connect α DP and β DP and ones on the β-strand of α DP frequently broke in the TMD simulation, but not in the equilibrium control. To further probe how the DP-site region responded the enforced E-site ATP binding to facilitate the ADP release, we additionally conducted the steered molecular dynamics (SMD) simulations to pull ADP out of the DP-site from the initial E-DP-T ring (before the TMD) and from the final T-DP-T* structure (after the TMD), respectively. Three directions were chosen for the ADP pulling (see SI Fig S11): upward from the top of the DP bound pocket, outward and inward toward the center of the ring, respectively. Then we conducted three SMD simulations for each direction, to both the E-DP-T and T-DP-T* rings before and after the TMD simulations. From these simulations, we concluded that the enforced E-site closing does impact on the DP-site to make the ADP release comparatively easy, which likely happen along the upward and/or outside pathways (see SI text). Finally we probed how the mechanical responses propagate from the ATP binding E-site to the respective DP- and T-site in the stator F 1 -ring, without the γ-shaft. We calculated individual residue correlations with the E-site ATP binding residues (αi343, αr373, βg161 to V164, βy345, βa421, βf424 and T425) mainly on the β E subunit (see Fig 3A), by evaluating first the pairwise cross-correlation values for all residues (see SI Fig S12) and then for each individual residue away from the ATP binding site, calculating the overall correlation strength between this residue and all those residues within the ATP binding site (see Methods). Close to the E-site, the correlations were maintained similarly high across α E->T and β E->T, respectively. The correlation was still maintained high as being propagated from α E->T to β DP, due largely to a loop region (αq405 to D411; see Fig 3C left) on top of α E that interacts closely with the β DP DELSEED region (βd394 to D400) and a helix underneath (βd383 to M390). On the other hand, the propagation from β E->T to α T and to β T was through a comparatively long distance, and the correlation decayed significantly reaching to β T. Since the ADP/ATP binding site mainly locates on the β subunit, it is reasonable to see that the upstream T-site was affected less than the downstream DP-site by the E-site ATP binding. ii. ATP hydrolysis in T- P-T* facilitates the Pi release from the upstream P-site In order to mimic ATP hydrolysis in the T-P-T* ring (PDB 1W0J) [40], we enforced the T*-site to a targeted DP-site conformation, obtained from the crystal structure (PDB 1E1R) [39]. Interestingly, one notices that in the crystal structure of T-P-T*, the P-site Pi is located around the β P-loop, a marginally stabilized position according to a recent MD study probing free energy of the Pi release [35]. It was indicated that the doubly charged Pi group has a tightly bound state located ~7 Å distance inside, while the P-loop bound state is 9
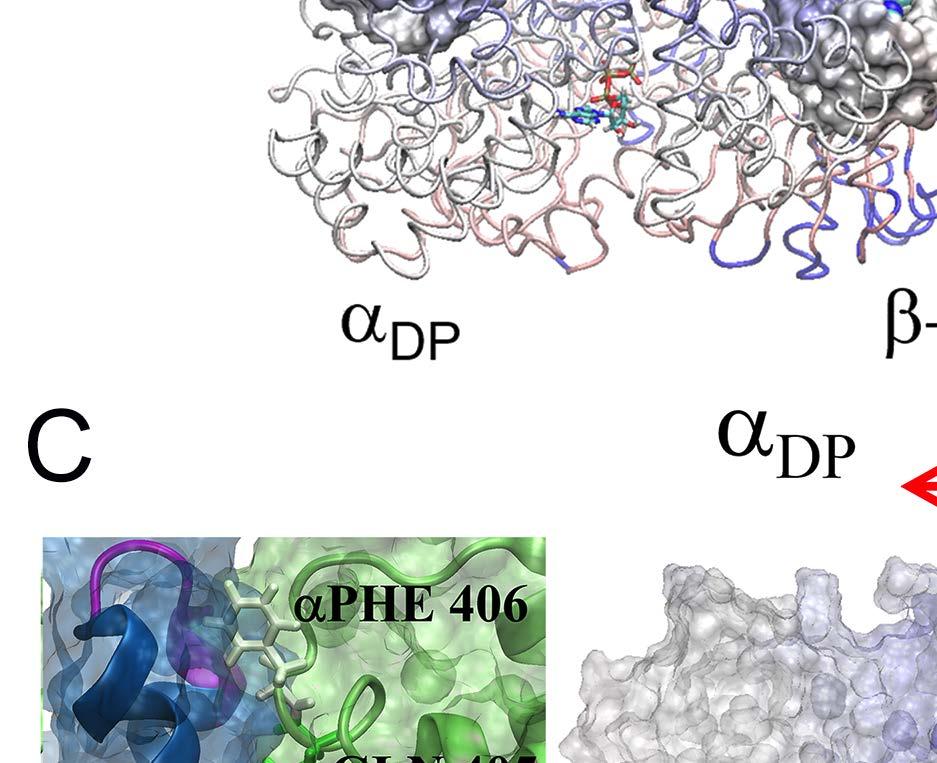
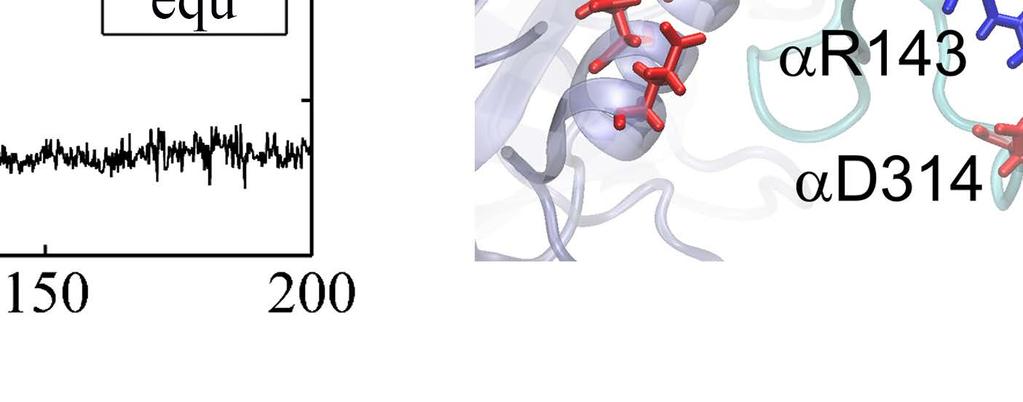
10 a less stabilized intermediate state close to the exit of the Pi release channel. Since the ring structure T-P-T* is obtained from the bovine MF 1, the marginally stabilized positioning of Pi in this structure, by itself, suggests a coupling scenario consistent with our proposal for MF 1 (see SI Fig S1c): the Pi release from the P-site of T-P-T* (to T-E-T*) has been facilitated right along with the ADP release (from T-DP-T* to T-P-T*), upon the ATP binding in the upstream site (E-DP-T to T-DP-T* see Fig 1B top). Moreover, we captured in the TMD simulation of the T-P-T* ring a complete Pi release event from the P-site, upstream of the enforced hydrolysis T*-site. Within 50 ns of the TMD simulation, the protrusion of the T*-site dropped slightly, showing a tightening of the site key to the ATP hydrolysis (T*à DP). Meanwhile, one noticed an up to ~3 Å rise of the β P protrusion for the P-site (see Fig 4A and SI Fig S13), indicating an opening response of the site within 50 ns that consequently led to the Pi release. The finding that the Pi release was facilitated upon the enforced hydrolysis nicely confirmed the prediction from the recent experimental data analyses [26], conducted for BF1 (thermophilic Bacillus PS3). Remarkably, our TMD simulation demonstrated a complete charge-hopping pathway of the Pi release through three arginine residues and one lysine residue (see Fig 4C and SI Movie S2a and S2b), which has not been revealed before. The Pi group was initially positioned through hydrogen bonding with both the P-loop from the β P subunit and the first arginine finger residue Arg373 from the α P subunit. In response to the enforced hydrolysis T*à DP transition, the P-loop from β P retracted slightly from Pi. Meanwhile, Arg143, the second arginine finger from α P which frequently switched its side chain in between Arg373 and Glu144, captured Pi after ~ 50 ns of the simulation, so that Pi dissociated from Arg373. In addition, three negatively charged residues from the β P subunit, Asp195, Glu199 and Glu202, helped to repel Pi away from β P. Later on, Pi was further transferred to Lys196 (~ 140 ns) and Arg161 (~ 180 ns) on α P, and then released fully from the F 1 ring at the end of the 200 ns TMD simulation. In contrast, in an equilibrium control simulation of the T-P-T* ring (for 200 ns), Pi was kept stable in association with the P-loop and Arg373, while the Arg143 side chain was trapped with Asp314 most of time (see SI Movie S2c). To understand why the conformational transition T*à DP impacts on the P-site that is upstream to the enforced T*-site in this case, we probed again the residue-wise correlations on the T-P-T* ring during the enforced hydrolysis transition (see Fig 5). In particular, we see that the correlation decayed quickly right away from the T*-site. Nevertheless, one notices that the local region of the P-site correlates well with the T*-site residues. A close examination shows that the correlation propagates through a restricted region, from the hydrolysis site on β T*, along a beta-sheet structure (αarg164 to Arg171, αile345 to Leu352, and αleu369 to Arg373, see Fig 5C right), and then reaches the P-site on α P -β P. iii. The first ATP binding opens the next/downstream E-site more than the upstream E-site In order to investigate whether asymmetrical responses arise on the early stage of the 10
11 reaction pathway, we conducted the TMD simulation to a fully empty ring E-E-E (see SI and Fig S14 and S15). As we enforced an Eà T transition on one pair of α E -β E in the E-E-E ring, we observed similar trends of mechanical responses as that in the E-DP-T configuration: The downstream E-site opened comparing to the equilibrium control simulation, while the upstream E-site did not (see Fig 6A and SI Fig S15). Further calculations on the cross-correlations also show an asymmetrical pattern (SI Fig S16). Hence, without ligand binding in the neighboring actives sites, the asymmetrical responses toward the ATP binding already exist in the ring. Indeed, the asymmetrical responses triggered by the first ATP binding to the ring can bring a bias in recruiting the second ATP. We found through the KMC simulation that if the second ATP is recruited to the downstream site with a slight bias (e.g. a rate enhancement ~10), then the reaction intermediate quickly converges to E-T*-T, followed by E-DP-T (see Fig 6B), rather than to T-T*-E and then T-DP-E. Once the E-DP-T configuration is reached, the chemical cycles repeat as that demonstrated in Fig 1B, for both MF 1 and BF 1. iv. The third ATP binding to the ring destabilizes both neighboring ATP-bound sites In the KMC simulation, the 3-T configuration in BF 1 was absent due to the coordinated tri-site kinetic, i.e., T-T-T was simply not on the dominant reaction pathway. In contrast, the 3-T configuration in MF 1 needs to be explicitly prohibited to support the sequential performance by reducing the third ATP binding rate (> ~10) (see SI Fig S3A). It indicated that the third ATP binding is unfavorable. In the MD simulation, however, we enforced a third ATP binding to the E-T*-T ring to probe the mechanical (in-)stabilities. We noticed an abnormal increase of the pocket size in the upstream T-site, even though the β T protrusion dropped (see SI Fig S17 and S18). Notably, the bound ATP from the downstream T*-site released in one of the two TMD simulation trials, though the T*-site did not open significantly. Hence, the enforced third ATP binding appears unfavorable as it triggers significant distortions or even destabilizes the bound ATP. Likely, an E-T*-T ring would either resist the third ATP binding, or convert to T-E-T* (rather than T-T*-E) if a third ATP binding succeeds by chance. 3. Targeted elastic network (TEN) simulation reproduced the asymmetrical opening of the neighbor sites upon ATP binding to validate the TMD results Note that the ATP binding and hydrolysis along with induced coupling processes involve substantial conformational transitions with characteristic times of micro- to milliseconds or even longer, which cannot yet be captured by conventional atomistic MD. In order to check whether the accelerated TMD simulations of hundreds of nanoseconds captured essential motions of the original slow conformational transitions, without introducing obvious artifacts, we conducted coarse-grained simulations of the F 1 -ATPase ring. The single bead per residue elastic network (EN) description [41] was employed, which has been widely used to describe the slow or micro- to milliseconds functional domain motions in protein machines [42-44]. To make an easy comparison, we have implemented targeted dynamic schemes as well within the EN model. Conformational changes inside the ring, which 11
12 corresponded to relaxation processes of the protein elastic network (see Methods and SI) [45-47], were followed and the neighbor sites were monitored. In particular, the forced Eà T transition starting from the E-DP-T ring, and that starting from the E-E-E ring, was investigated at the coarse grained level. In the E-DP-T to T-DP-T* case, the C-terminal β protrusion of the downstream DP-site increased, indicating an opening of the site, whereas the upstream T-site did not show such an opening (see SI Fig S19). In the E-E-E to E-T-E situation, the asymmetric responses found in the TMD simulations were also revealed in the TEN simulations, in which the downstream site showed the opening, whereas the upstream site did not (see SI Fig S20). Hence, regarding the conformational responses due to the inter-subunit coupling in the F 1 ring, agreement between the coarse-grained model and the atomistic-level accelerated description is obtained. Discussion To reveal quantitative features and structural dynamics mechanisms supporting high coordination of a prototypical ring-shaped NTPase, we computationally investigated the intrinsic inter-subunit couplings that lead to sequential hydrolysis of the F 1 -ATPase ring, in the absence of the central γ rotor. We first explored which types of inter-subunit couplings are kinetically feasible to allow the sequential hydrolysis of the three active sites on the F 1 -ring through the stochastic simulations. We imposed the couplings to the three ATP hydrolysis cycles by essentially enhancing the ADP and Pi release rates in one cycle/site upon certain neighbor-site ATP binding or hydrolysis event in another cycle. We found that to support the sequential hydrolysis in MF 1, the ADP release needs to be accelerated for above ~10 5 fold upon the upstream-site ATP binding in the E-DP-T ring, and similarly for the followed Pi release from the same site; in BF 1, on the other hand, the ADP release needs to be accelerated for above ~10 3 fold, while the Pi release can be accelerated either along with the ADP release, and/or upon the next-site ATP hydrolysis in the T-P-T* ring. In order to elucidate the physical and structural basis of these kinetically feasible couplings, we next performed atomistic TMD simulations and monitored the ensured mechanical responses on the ring, which followed the enforced conformational transition of the ATP binding or hydrolysis to one chemical site on the ring. We found essentially these features below from the combined stochastic kinetic and structural dynamics simulations. i. The primary inter-subunit coupling that supports the sequential hydrolysis with a directional bias is the ATP binding facilitated downstream-site product release, arising from the asymmetrical site opening inherent to the architecture of the trimer-of-dimer ring Consistently, we found in the atomistic simulation of the E-DP-T ring that the downstream DP-site opens in response to the enforced E-site closing that mimics the ATP binding. The site opening was partially characterized by slight increasing of the β-protrusion, the hinge bending angle, and the pocket size of the binding. Further examinations show notable conformational changes along with weakening of the hydrogen bond network in the peripheral region of the DP-site. All these allosteric impacts induced by the closing E-site 12
13 could possibly facilitate the ADP release from the DP-site. When the central γ rotor was included in the TMD simulation, the β DP -protrusion increase would be easily enhanced. Close examination on the cross-correlation of the E-DP-T ring, in the absence of the γ rotor, shows that the ATP binding induced conformational changes propagate asymmetrically from the E-site to the DP- and T-site, via the α E -β DP and β E -α T -β T paths, respectively. The asymmetrical downstream site opening is also preserved upon the induced site closing that mimics the first ATP binding to the empty E-E-E ring. The asymmetrical responses thus appear to be inherent to the architecture of the trimer-of-heterodimer (α-β) ring, in particular, as α and β subunits differ and alternate around the ring while the chemical active site largely locates on β. The inclusion of the γ rotor into the center of the ring likely enhance the asymmetrical responses, which either promote the ADP release (and initial Pi release) or bias the next ATP binding to downstream of the current ATP binding site. In particular, the bias for the next or second ATP binding downstream allows the sequential hydrolysis to proceed directionally (counterclockwise viewed from the C-terminal side). ii. The sequential hydrolysis is also supported by proper timing and acceleration of Pi release, which either quickly follows the ADP release upon the upstream-site ATP binding, or later upon the downstream-site ATP hydrolysis via minor conformational propagation; the Pi release was then computationally predicted to proceed in charge hopping. On the other hand, the Pi release can be facilitated via two different mechanisms (see Fig 1B), either due to a persistent impact from the upstream-site ATP binding that induced the ADP release (MF 1 [18]), or due to the ATP hydrolysis in the downstream T*-site (BF 1 [19]). The straightforward support of the ATP binding facilitated Pi release comes from the bovine heart MF 1 structure (T-P-T*) per se (PDB 1W0J) [40], in which the Pi group in the P-site (after the ADP release) is located nearby the P-loop, a marginally stabilized state, rather than being located in the innermost tightly bound state [35]. The ATP binding to the ring (from E-DP-T to T-DP-T*) not only destabilized ADP from the DP-site, but also Pi from the same site, so that Pi moved from the tightly bound state to the marginally stabilized state not far from exit of the release channel. The release of Pi thereafter then becomes comparatively easy. Nevertheless, our equilibrium MD simulation of the T-P-T* ring was not yet able to capture the Pi release from the P-site within hundreds of nanoseconds. In the equilibrium simulation, the Pi group was associated with the β P-loop as well as the arginine finger αarg373. The other arginine finger αarg143 was trapped with αasp314 most of time. Nicely, in contrast, a complete Pi release was predicted computationally in the TMD simulation, as the P-site mechanistically responded to the enforced transition mimicking the ATP hydrolysis in the T*-site. The P-loop retracted and Arg143 was able to switch occasionally toward Arg373, while the three negatively charged residues from β P also repelled Pi to assist the release. Consequently, the chance that Pi dissociates from the P-loop to be captured by Arg143 increases significantly, which becomes critical for further charge hopping and the final release of Pi. Mutation of any of residues involved in the process would considerably impact on the Pi release. 13
14 Comparing with the significant open-to-close conformational changes upon the ATP binding, the ATP hydrolysis induced conformational changes are rather subtle. To see how the subtle conformational changes propagate from the hydrolysis T*-site to the upstream P-site, we found that a restricted beta-sheet region bridged the mechanistic pathway from the T*-site to the P-site. By mutating the key residues for the mechanistic propagation, one would expect the hydrolysis induced Pi release to be hindered. In brief, the ATP hydrolysis does not generate the global responses of the ring as that upon the ATP binding; rather, it triggers restricted local conformational propagation. Besides, in the T-P-T* ring, the P-site upstream to the T*-site is more open and likely much more flexible than the downstream T-site, hence, it easier for the mechanical responses to propagate upstream than downstream. iii. The sequential scheme either explicitly requires the third ATP binding inhibited (MF 1 ) or the three-atp bound configuration was simply not on in the corresponding kinetic pathway in the sequential hydrolysis (BF 1 ) Besides, we found additional couplings indispensible for the sequential performances. First, a mechanism aside from the existing tri-site kinetics in MF 1 (but not BF 1 ) ring is needed to prevent three active sites from binding ATP simultaneously. Our chemical kinetic simulations showed that the 3-T configuration in both MF 1 and BF 1 is of low population in the coordinated hydrolysis cycle, even without the mechanical constraint. Nevertheless, the constraint appears to be required further for MF 1 to achieve the sequential hydrolysis. Consistently, the TMD simulation enforcing a third ATP binding to the E-T*-T ring demonstrated significant destabilizations to the ring, as the upstream site abnormally enlarged and the downstream site released the bound ATP. In addition, we adopted a way to enhance the ATP hydrolysis rate in the T*-site, effectively, as a second ATP binds to the ring to form a T-site. Basically, the uni-site hydrolysis rate of F 1 is not sufficiently high to allow for the detected tri-site kinetics. The rate enhancement in the hydrolysis-ready intermediate T* ensures that the site binds ATP early almost always hydrolyzes early. The rotary sequential hydrolysis of F 1 -ring is constantly supported by chemical free energy through the three ATP cycles, which are coordinated with certain phase differences. The rate enhancements under the inter-subunit couplings correspond to lowering the free energy barriers for the facilitated transitions, which are feasible under two ways (or mixed of both): Either a constant binding affinity or free energy of the ligand (ADP or Pi) to the induced site is maintained, or a constant ligand binding rate is kept. To enhance the ADP release rate ~ 10 5 fold (in MF 1 ), for example, while keeping a fairly constant ADP rebinding rate, it requires above ~12 k B T to destabilize the DP-site, with the energy compensation coming from the neighbor-site ATP binding. Since the chemical free energy is mostly dissipated through the rotational degree of freedom in the presence of the γ rotor [48,49], one expects then that the γ rotor plays a significant role to tightly couple the ATP binding to the 14
15 next-site ADP release, both structurally and energetically. Indeed, our studies show that the β DP -protrusion responds to the enforced ATP binding much more significantly in the presence than in the absence of the γ rotor. In comparison, the ATP hydrolysis facilitated Pi release requires less energetic compensation or coupling, if there is any, so we could capture the Pi release event notably in the absence of the γ rotor. As such, we infer that the central γ rotor dictates the inter-subunit coupling more significantly in the ATP binding step than in the ATP hydrolysis step, which leads to stronger inter-subunit couplings or higher coordination in the original F 1 -ATPase than in the stator ring without the rotor. On the other hand, considering the recent MD studies on the γ-shaft rotation and the Pi release [35] together with our simulation results, we suggest that the initial stage of the Pi can also be facilitated by the γ-shaft rotation that accompanies the ATP binding and the facilitated ADP release; further Pi release is then promoted by the neighbor-site ATP hydrolysis in bacterial F 1 ; the complete Pi release couples closely with the further γ-shaft rotation. Based on current work, it would be promising to determine how exactly the central γ rotor contributes to the F 1 inter-subunit coordination in the respective steps of the ATP binding and hydrolysis in further experimental and computational studies. Conclusion Combining stochastic simulations on tri-site chemical kinetic of the F 1 stator ring with targeted atomistic MD and coarse-grained EN simulations, we inferred the most crucial inter-subunit couplings leading to the sequential hydrolysis on the F 1 ring: The ATP binding facilitated downstream ADP release along with the Pi release, as well as the ATP hydrolysis facilitated upstream Pi release. The couplings come from asymmetrical neighbor site opening/loosening responses upon current site closing/tightening during the ATP binding/hydrolysis. The dominant conformational asymmetry, e.g., triggered upon the ATP binding, appears embedded in the trimer-of-heterodimer ring, owning largely to the alternating arrangement of α and β subunits. Alternatively, minor conformational asymmetry reveals upon subtle conformational transitions of the ATP hydrolysis to allow the facilitated Pi release. The mechanistic propagation around the ring, however, is also modulated by the ligand (ATP, ADP or Pi) binding to the neighboring sites. Additionally, preventing a third ATP from binding to the ring, as well as allowing a hydrolysis-ready intermediate to have an elevated hydrolysis rate, also enable the sequential hydrolysis on the F 1 ring. Whether comparable inter-subunit coupling scenarios arise for evolutionarily connected protein enzymes with similar molecular architectures would be of high interest to pursue further. Methods 1. Kinetic Monte Carlo simulation of three chemical sites For each ATP binding or catalytic site at the α-β interface, there are several chemical states: the empty state without any substrate binding (E); the ATP bound state and the hydrolysis ready state (T/T*); the post-hydrolysis state right after the reaction as ATP turns into ADP 15
16 and Pi (DP); and the ADP released but Pi bound state (P). Here we follow the convention that the ADP releases prior to the Pi release [19,37]. As such, the uni-site reaction proceeds in cycles as E T/T* DP P E. We differentiate T* from T by enhancing the ATP hydrolysis rate of the T*-state above that of the T-state. The unite-site reaction rates for forward and backward transitions in the bovine heart mitochondrial and bacterial E. coli F1-ATPase were obtained experimentally [20,21] (see SI Table S1 and S2). We assume that the rate of the ADP release from the DP state is the same as that originally listed from the D state, and the rate of the Pi release from the P state is the same as that originally listed from the DP state. We also assume that the bovine and human mitochondrial F1 behave similarly [18], while E. coli and thermophilic (Bacillus PS3) bacterial F1 perform alike as well [50]. We used the kinetic Monte Carlo (KMC) method [51] to simulate the chemical kinetics of F 1 -ATPase hydrolysis, from the three independent uni-site reactions to the coupled tri-site reactions. In the KMC simulation, one starts with an initial ring configuration (e.g. E-E-E or E-DP-T) of the three chemical sites; each time a transition from one of the three chemical sites was made, either forward or backward along the reaction path, according to rate competitions of all potential transitions in the current ring configuration (i.e., fast transitions get higher chances). When the uni-site reaction rates were adopted for each site, the three sites perform as if they hydrolyze independently. In any coupled tri-site reaction scheme, inter-subunit couplings were imposed at certain ring configurations by tuning certain chemical transition rates upon some neighbor site transitions (e.g. ATP binding Eà T). In particular, we used four parameters to define the strength of the couplings, and we tuned these parameters for optimal sequential performances (see SI). 2. Atomistic targeted molecular dynamics simulations All the MD simulations were performed using the NAMD 3.0 software [52] with CHARMM22 force field for protein and CHARMM27 force field for lipid [53,54], except for the construction of E-DP-T structure using GROMACS [55]. The F 1 -ATPase structures were obtained from the existing crystal structures: The E-DP-T ring was made from the crystal structure of P-DP-T (PDB: 1E1R) [39]; the γ-shaft, when included, was rotated for ~40 accordingly. An E-T-T* structure was obtained directly from the crystal structure (PDB: 2JDI) [56]. An E-E-E structure was made from E-T-T, and a T-P-T* structure was obtained from the crystal structure (PDB: 1W0J) [40] (see detail in SI). The PDB structures were then solvated with TIP3P water in a cubic box and the minimum distance from the protein to the wall was 15 Å. We neutralized the systems with Na + and Cl - to the concentration of 0.15 M. The temperature was set to 310 K and the pressure was 1 bar. The van der Waals (vdw) and short-range electrostatic interactions used a cutoff of 12 Å. The particle-mesh Ewald method was applied to deal with the long-range electrostatic interactions [57]. The solvated system was minimized with the steepest-descent algorithm, followed by 10 ps MD simulation under the canonical ensemble with time-step 1 fs. Then 5 ns equilibrium simulation was performed under the 16
17 NVT ensemble with a time step of 1 fs, and position restraints on the heavy atoms of protein were imposed during the simulation. After the constrained simulation, the unconstrained equilibrium simulations or the targeted MD (TMD) simulations were carried out under the NPT ensemble with a time step of 2 fs. During the TMD simulation, the structure was constantly enforced toward a moving target conformation (according to the heavy-atom RMSD value), geometrically designated on the reaction path toward a final target structure [38]. Further implementations are found in SI. 3. Performing the elastic-network relaxation dynamics The elastic network of the ring was obtained by first replacing each amino acid residue in the atomic structure by a single bead, which was placed at the position of the alpha-carbon atom of the respective residue. These equilibrium positions were denoted by R! (!) for bead i. Then, to determine the pattern of network connections, the distances d (!)!" = R (!)! R!! between equilibrium positions of any two beads i and j were compared with a prescribed interaction radius r!"# = 10Å. If this distance was below r!"#, the two beads were connected by a deformable elastic spring with natural length d (!)!". The network connectivity was stored in matrix A with entries A!" = 1, if beads i and j are connected and A!" = 0, else. The total elastic energy of the network is U = κ!!"!,!;!!! d!" (!) d!!". Here, N is the number of beads in the protein network, d!" = R! R! is the actual length of a spring connecting beads i and j in some deformed network (!) conformation, with R! being the actual position vector of bead i, and d!"!! is the corresponding natural length. The spring stiffness constant κ is assumed to be the same for all network springs. When thermal fluctuations and hydro-dynamical interactions are neglected, the dynamics of the network can be described by a set of Newton s equations in the over-damped limit [45-47]. For bead i the equation of motion is γ d dt R! = U. (1) R! On the left hand side of Eq. (1), γ is the friction coefficient assumed to be equal for all network beads. On the right hand side are the elastic forces exerted by network springs of beads which are connected to bead i. Explicitly, the equations read! d! dt R d!" d!"! = A!" R d! R! + σf!. (2)!!" Here, we have removed the dependencies on γ and κ by an appropriate rescaling of time 17
18 and, after that, added external forces f! which can act on bead i (if σ = 1). In the performed target elastic-network (TEN) simulations external forces were applied only to the beads of one of the catalytic αβ -sites in order to induce the desired nucleotide-induced transition there. Such forces were updated in each integration step and had the form f!,! =!!! RMSD!!! RMSD d!,!!!, for bead i. Here, d!,! is the difference vector between positions of bead i in the target and the actual conformation of the forced αβ-subunit, determined after superposition of the subunits at integration step n (RMSD! is the corresponding root mean square displacement), and RMSD = RMSD!! RMSD!, with S being the total number of integration steps. N!! is the number of beads of the forced αβ-subunit and parameters α = 2,500 and S = 400,000 were chosen in the simulations. The set of equations (2) was numerically integrated to obtain the positions of network beads at all moments in time. In the simulations a first-order scheme with a time-step of 0.1 was used. In particular, we performed TEN to mimic the ATP binding to the E-site of the E-DP-T ring, in comparison with the TMD simulation. Further details are found in SI. 4. Individual residue correlations with the enforced ATP binding site The individual residue correlations with the enforced ATP binding site were calculated to evaluate the overall correlation strength between each residue and the binding pocket. Firstly, we calculated the pairwise correlations for all the residues. The correlation between each pair of residues is given by C!" =!(R i!!r i!) (R j!!r j!)!!(r i!!r i!)!!!(r j!!r j!)!!, where R i and R j are the position vectors of Cα atom of residue i and j, respectively, and the <..> represents the average over the simulation trajectory. Then we obtained a pairwise NxN correlation matrix, where N is the total number of residues. Finally, we calculated the correlation between residue i and binding pocket by C!" =!!!"#$%& C!", where the pocket residues include αile343,αarg373,βgly161, βlys162, βthr163, βval164, βtyr345, βala421, βphe424 and βthr425. Acknowledgements We acknowledge the computational support from the Beijing Computational Science Research Center (CSRC) and Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase). 18
19 References 1. Liu S, Chistol G, Bustamante C (2014) Mechanical Operation and Intersubunit Coordination of Ring-Shaped Molecular Motors: Insights from Single-Molecule Studies. Biophysical Journal 106: Lino R, Noji H (2013) Intersubunit coordination and cooperativity in ring-shaped NTPases. Curr Opin Struct Biol 23: Lyubimov AY, Strycharska M, Berger JM (2011) The nuts and bolts of ring-translocase structure and mechanism. Curr Opin Struct Biol 21: Enemark EJ, Joshua-Tor L (2008) On helicases and other motor proteins. Curr Opin Struct Biol 18: Singleton MR, Dillingham MS, Wigley DB (2007) Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem 76: Boyer PD (1989) A perspective of the binding change mechanism for ATP synthesis. FASEB J 3: Hutton RL, Boyer PD (1979) Subunit Interaction during Catalysis. Alternating Site Cooperativity of Mitochondrial Adenosine Triphosphatase. J Biol Chem 254: Lesile AG, Walker JE (2000) Structural model of F1-ATPase and the implications for rotary catalysis. Philos Trans R Soc Lond B Biol Sci 355: Boyer PD (1993) The binding change mechanism for ATP synthase - Some probabilities and possibilities. Biochimica et Biophysica Arta 1140: Abrahams JP, Leslie AGW, Lutter R, Walker JE (1994) Structure at 2.8 Â resolution of F1-ATPase from bovine heart mitochondria. Nature 370: Noji H, Yasuda R, Yoshida M, Kinosita Jr K (1997) Direct observation of the rotation of F1-ATPase. Nature 386: Adachi K, Oiwa K, Nishizaka T, Furuike S, Noji H, et al. (2007) Coupling of rotation and catalysis in F(1)-ATPase revealed by single-molecule imaging and manipulation. Cell 130: Furuike S, Hossain MD, Maki Y, Adachi K, Suzuki T, et al. (2008) Axle-Less F1-ATPase Rotates in the Correct Direction. Science 319: Hossain MD, Furuike S, Maki Y, Adachi K, Suzuki T, et al. (2008) Neither Helix in the Coiled Coil Region of the Axle of F(1)-ATPase Plays a Significant Role in Torque Production. Biophysical Journal 95: Kohori A, Chiwata R, Hossain Mohammad D, Furuike S, Shiroguchi K, et al. (2011) Torque Generation in F(1)-ATPase Devoid of the Entire Amino-Terminal Helix of the Rotor That Fills Half of the Stator Orifice. Biophysical Journal 101: Chiwata R, Kohori A, Kawakami T, Shiroguchi K, Furuike S, et al. (2014) None of the rotor residues of F1-ATPase are essential for torque generation. Biophys J 106: Uchihashi T, Iino R, Ando T, Noji H (2011) High-speed atomic force microscopy reveals rotary catalysis of rotorless F1-ATPase. Science 333:
20 18. Suzuki T, Tanaka K, Wakabayashi C, Saita E-i, Yoshida M (2014) Chemomechanical coupling of human mitochondrial F1-ATPase motor. Nature chemical biology 10: Watanabe R, Noji H (2014) Timing of inorganic phosphate release modulates the catalytic activity of ATP-driven rotary motor protein. Nat Commun 5: Senior AE (1992) Catalytic sites ofescherichia coli F1-ATPase. Journal of bioenergetics and biomembranes 24: Grubmeyer C, Cross R, Penefsky H (1982) Mechanism of ATP hydrolysis by beef heart mitochondrial ATPase. Rate constants for elementary steps in catalysis at a single site. Journal of Biological Chemistry 257: Lisal J, TUma R (2005) Cooperative mechanismof RNA packaging motor. The Journal of Biological Chemistry 280: Yu J, Moffit J, Hetherington CL, Bustamante C, Oster G (2010) Mechanochemistry of a Viral DNA Packaging Motor. Journal of Molecular Biology 400: Adolfsen R, Moudrianakis EN (1976) Binding of adenine nucleotides to the purified 13S coupling factor of bacterial oxidative phsphorylation. Arch Biochem Biophys 172: Yu J (2014) Coordination and Control Inside Simple Biomolecular Machines. In: Han K, editor. Protein Conformational Dynamics, Advances in Experimental Medicine and Biology: Springer International Publishing Switzerland. pp Li C-B, Ueno H, Watanabe R, Noji H, Komatsuzaki T (2015) ATP hydrolysis assists phosphate release and promotes reaction ordering in F1-ATPase. Nature Communication 6: Wang H, Oster G (1998) Energy transduction in the F1 motor ofatp synthase. Nature 396: Bockmann RA, Grubmuller H (2002) Nanoseconds molecular dynamics simulation of primary mechanical energy transfer steps in F1-ATP synthase. Nature Structural Biology 9: Dittrich M, Hayashi S, Schulten K (2003) On the mechanism of ATP hydrolysis in F1-ATPase. Biophysical Journal 85: Gao YQ, Yang W, Karplus M (2005) The binding change mechanism of F1-ATPase revisited. Cell 123: Koga N, Takada S (2006) Folding-based molecular simulations reveal mechanisms of the rotary motor F1 ATPase. PNAS 103: Gaspard P, Gerritsma E (2007) The stochastic chemomechanics of the F1-ATPase molecular motor. Journal of Theoretical Biology 247: Mukherjee S, Warshel A (2012) Electrostatic origin of the mechanochemical rotary mechanism and the catalytic dwell of F1-ATPase. PNAS 108: Ito Y, Yoshidome T, Matubayasi N, Kinoshita M, Ikeguchi M (2013) Molecular Dynamics Simulations of Yeast F1-ATPase before and after 16 Rotation of the γ Subunit. The Journal of Physical Chemistry B 117: Okazaki K-i, Hummer G (2013) Phosphate release coupled to rotary motion of F1-ATPase. Proceedings of the National Academy of Sciences 110: Okazaki K, Hummer G (2015) Elasticity, friction, and pathway of γ-subunit rotation in 20
21 FoF1-ATP synthase. PNAS 112: Watanabe R, Iino R, Noji H (2010) Phosphate release in F1-ATPase catalytic cycle follows ADP release. Nature chemical biology 6: Schlitter J, Engels M, Krüger P (1994) Targeted molecular dynamics: a new approach for searching pathways of conformational transitions. Journal of molecular graphics 12: Braig K, Menz RI, Montgomery MG, Leslie AGW, Walker JE (2000) Structure of bovine mitochondrial F1-ATPase inhibited by Mg2+ADP and aluminium fluoride. Structure 8: Kagawa R, Montgomery MG, Braig K, Leslie AG, Walker JE (2004) The structure of bovine F1-ATPase inhibited by ADP and beryllium fluoride. The EMBO Journal 23: Bahar I, Atilgan AR, Erman B (1997) Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential Folding and Design 2: Bahar I, Lezon TR, Yang L-W, Eyal E (2010) Global Dynamics of Proteins: Bridging Between Structure and Function. Annual review of biophysics 39: Cui Q, Bahar I, editors (2005) Normal Mode Analysis: Theory and Applications to Biological and Chemical Systems. London/Boca Raton, FL: Chapman and Hall/CRC 44. Zheng W, Doniach S (2003) A comparative study of motor-protein motions by using a simple elastic-network model. PNAS 100: Flechsig H, Mikhailov AS (2010) Tracing entire operation cycles of molecular motor hepatitis C virus helicase in structurally resolved dynamical simulations. Proc Natl Acad Sci USA 107: Togashi Y, Mikhailov AS (2007) Nonlinear relaxation dynamics in elastic networks and design principles of molecular machines. Proc Natl Acad Sci USA 104: Togashi Y, Yanagida T, Mikhailov AS (2010) Nonlinearity of mechanochemical motions in motor proteins. PLos Computational Biology 6: e Toyabe S, Okamoto T, Watanabe-Nakayama T, Taketani H, Kudo S, et al. (2010) Nonequilibrium energetics of a single F1-ATPase molecule. Phys Rev Lett 104: Kawaguchi K, Sasa S, Sagawa T (2014) Nonequilibrium Dissipation-free Transport in F1-ATPase and the Thermodynamic Role of Asymmetric Allosterism. Biophysical Journal 106: Bilyard T, Nakanishi-Matsui M, Steel BC, Pilizota T, Nord AL, et al. (2013) High-resolution single-molecule characterization of the enzymatic states in Escherichia coli F1-ATPase. Philos Trans R Soc Lond B Biol Sci 368: Chatterjee A, Vlachos DG (2007) An overview of spatial microscopic and accelerated kinetic Monte Carlo methods. Journal of Computer-Aided Materials Design 14: Nelson MT, Humphrey W, Gursoy A, Dalke A, Kalé LV, et al. (1996) NAMD: a parallel, object-oriented molecular dynamics program. International Journal of High 21
22 Performance Computing Applications 10: MacKerell AD, Bashford D, Bellott M, Dunbrack R, Evanseck J, et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. The journal of physical chemistry B 102: Foloppe N, MacKerell Jr AD (2000) All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. Journal of computational chemistry 21: Berendsen HJ, van der Spoel D, van Drunen R (1995) GROMACS: A message-passing parallel molecular dynamics implementation. Computer Physics Communications 91: Bowler MW, Montgomery MG, Leslie AGW, Walker JE (2007) Ground State Structure of F1-ATPase from Bovine Heart Mitochondria at 1.9 Å Resolution. Journal of Biological Chemistry 282: Darden T, York D, Pedersen L (1993) Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. The Journal of chemical physics 98:
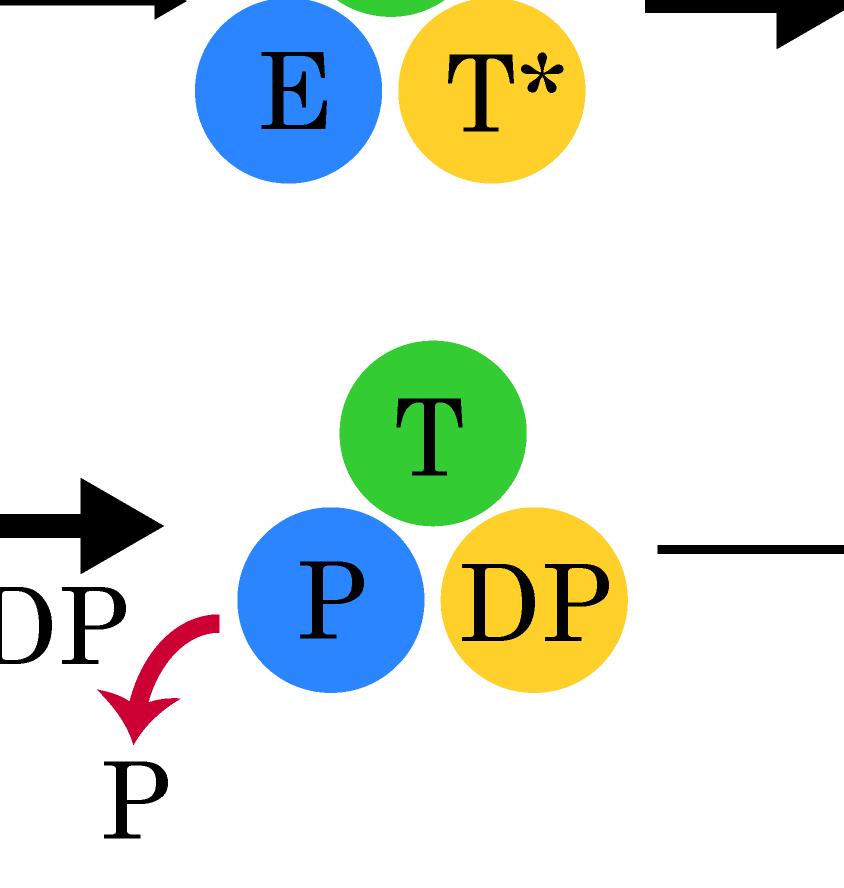
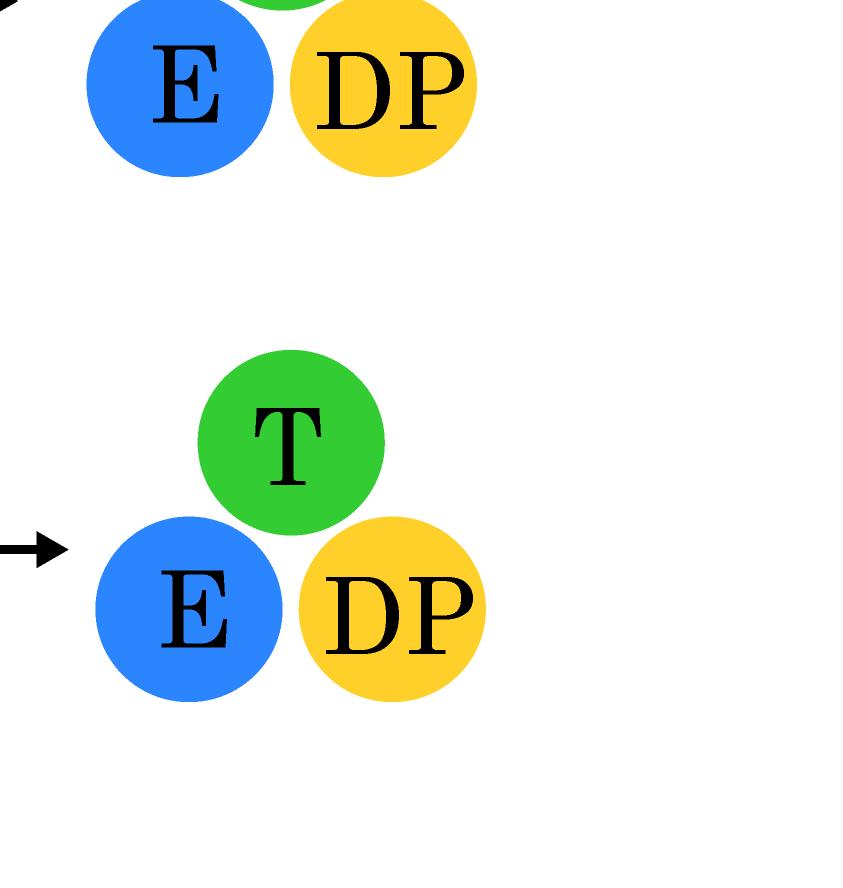
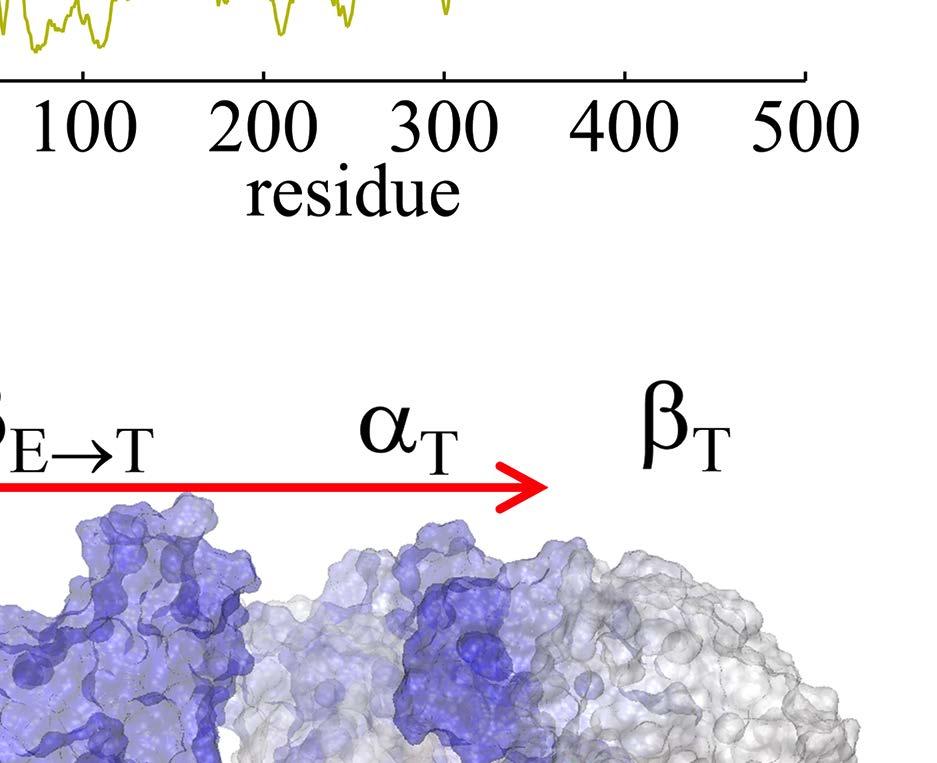
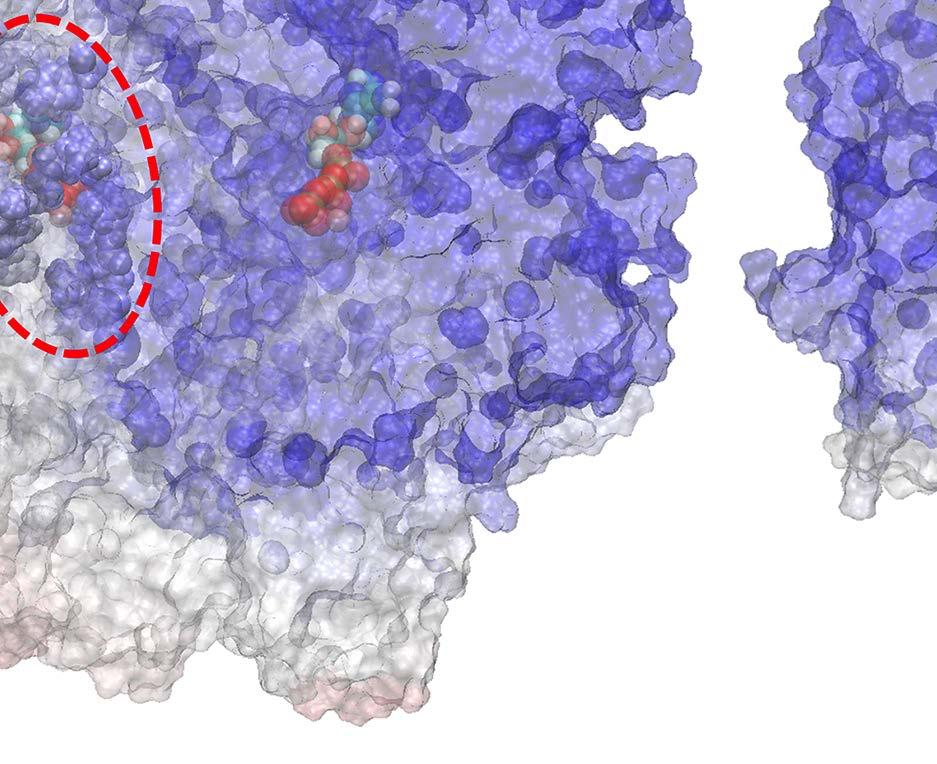
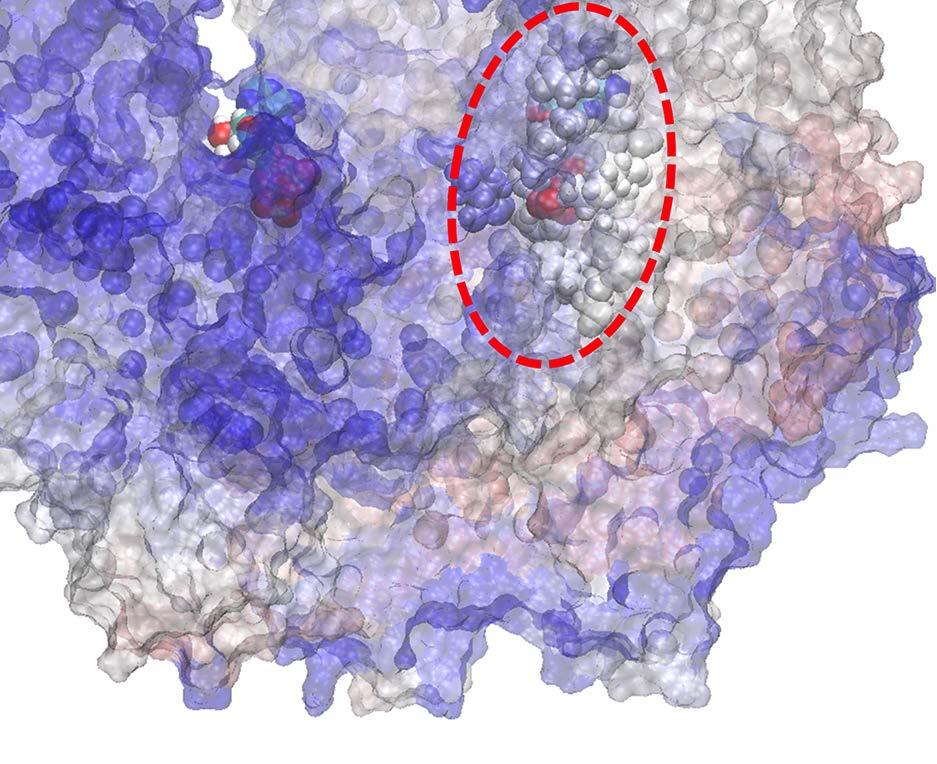
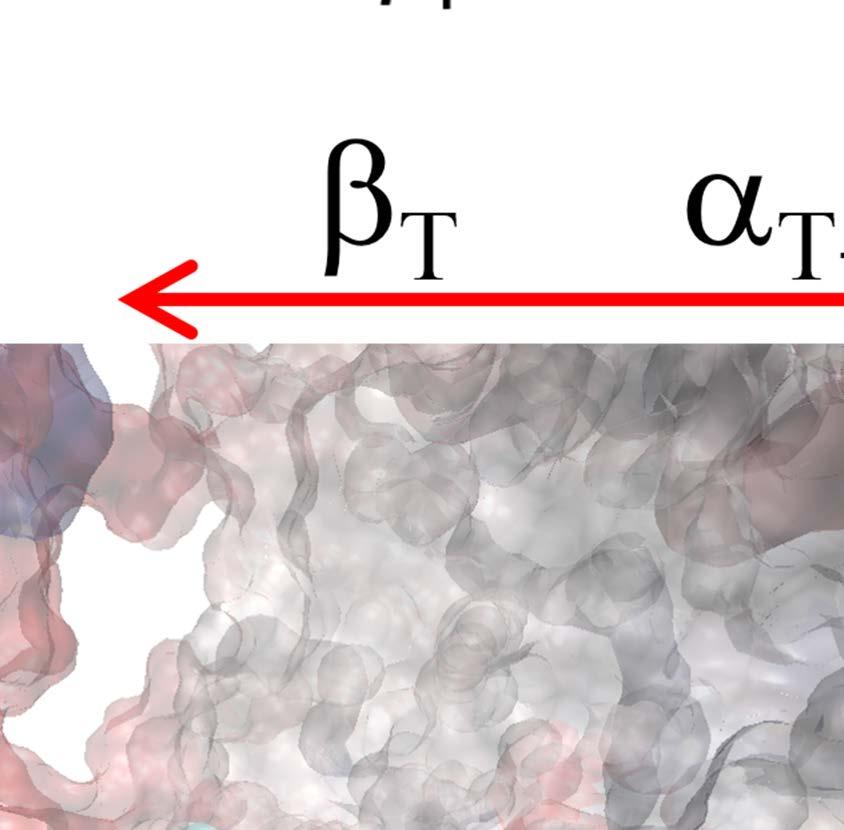
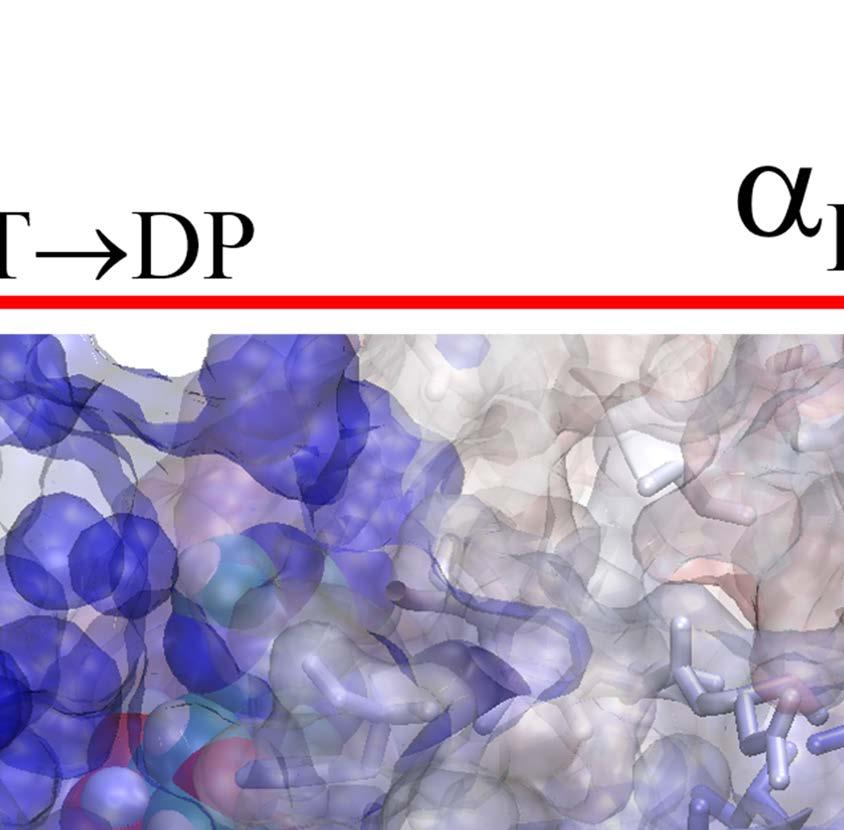
23 Figure Legends Fig 1. Stochastic simulations of tri-site hydrolysis reactions on the F 1 ring. (A) The trajectories show the number of ATP hydrolysis cycles for the three chemical sites in bovine MF1, from the independent (yellow traces, with three different shades shown for the three active sites) to the partially coordinated (blue traces with three shades) and to the highly sequential case (red traces with three shades as well). In the sequential case, each site follows its upstream/counterclockwise site closely along the ATP hydrolysis reaction path as being demonstrated experimentally. The sequential score, which quantify the sequential performance of the three active sites (see SI Eq S1), increased from 0 (independent) to 0.4 (partially coordinated) and to 0.6 (highly sequential), correspondingly. In the independent case, the uni-site reaction rates from the bovine heart MF 1 were used [21]. The ring-shaped structure of a key configuration E-DP-T is shown (E for Empty, T for ATP bound state, DP for ADP*Pi or the hydrolysis state, and P for the Pi bound state), with the most essential inter-subunit coupling illustrated: ATP binding to the E-site facilitates the ADP release from the next DP-site. The coupling is imposed in two ways: implementation I and II (see text), with the implementation I for MF 1 trajectories shown here. The trajectories for MF 1 and BF 1 with both implementations are shown in SI Fig S6. (B) The schemes for the essential inter-subunit couplings to achieve the sequential hydrolysis in MF 1 (top) and BF 1 (bottom): The ATP binding (Eà T) to E-DP-T enhances the ADP release (DPà P), for both MF 1 and BF 1 ; in MF 1, the Pi release (Pà E) is enhanced right after the ADP release upon the ATP binding; in BF 1, the Pi release is enhanced either similarly as that in MF 1 or later upon the ATP hydrolysis (T*à DP). Fig 2. The TMD simulation enforcing an ATP binding to α E -β E in the E-DP-T ring. (A) In the first 150 ns TMD simulation, the Eà T transition was gradually enforced; additional simulations were conducted for relaxation as holding the structure toward the final targeted form. The conformational changes of the chemical sites were monitored by the C-terminal protrusions of the β subunits (green for the E-site, blue for DP, and yellow for T). (Inset) The RMSD of α E -β E with respect to the targeted α T -β T. (B) The histograms of the protrusions from β DP (blue alike colors) and β T (yellow alike), measured from the first 150 ns (light color) TMD, the second and the third 150 ns simulation (darker blue or yellow colors), respectively (with the average changes indicated by arrows). The side view of the conformational changes on the β E->T and β DP C-terminal protrusions were shown, with spheres the DELSEED motif. (C) The top view from the C-terminal side of the F 1 ring, for the initial (E-DP-T, left) and the enforced configuration (T-DP-T*, right). The DELSEED motifs are highlighted in magenta spheres. (D) The widening of the DP-site interface. Left: The side view of the E-DP-T structure. Right: The interface between α DP and β DP around the DP-site before and after the TMD simulation. The initial structure is shown in transparent, with β DP colored pink, α DP colored yellow, and ADP/Pi colored by atoms. The final structure after TMD is colored in blue (β DP ) and light blue (α DP ). Fig 3. The correlation with the ATP binding pocket residues in the TMD simulation of the 23
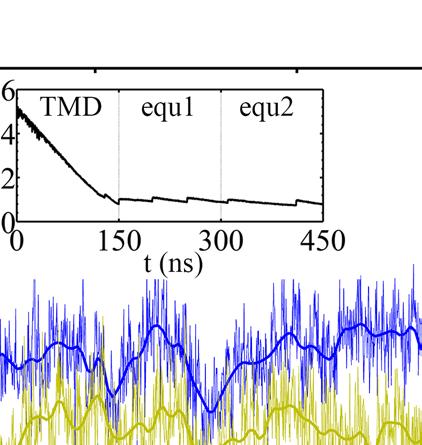
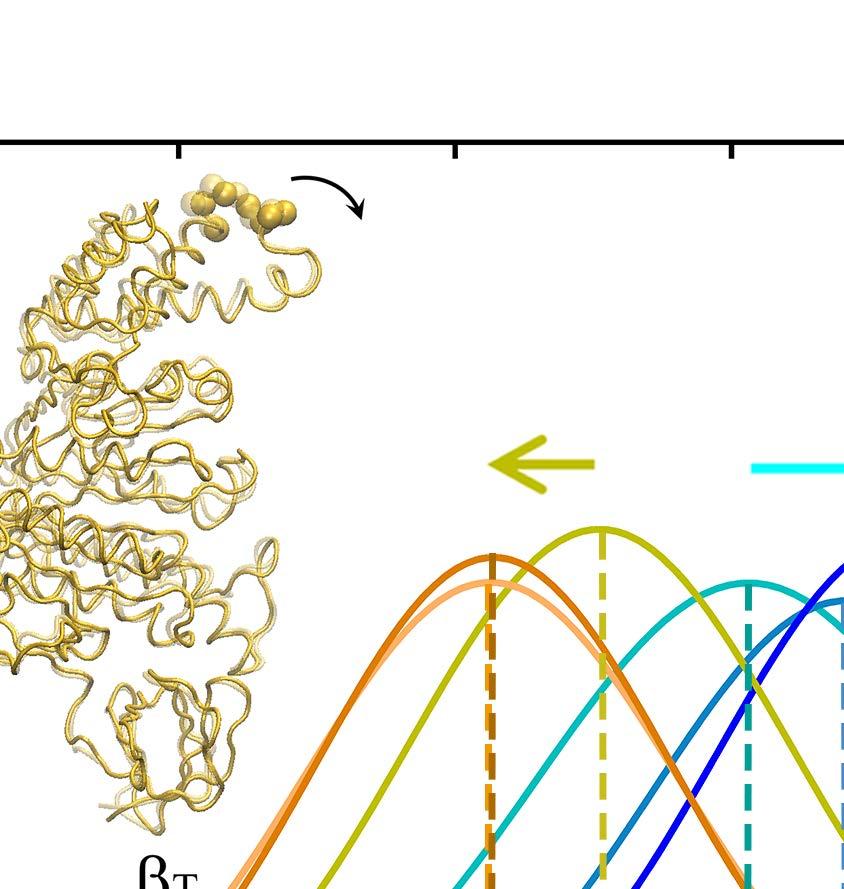
24 E-DP-T ring. (A) The correlation map of the full ring during the enforced Eà T transition starting from E-DP-T, colored according to the correlation values (blue/white/red: high/medium/low) between individual residues and the E-site ATP binding residues (in green spheres). (B) The residue-wise correlation from β DP and β T (blue and gray). (C) An inside view showing the correlation propagation from α E->T to β DP and to α DP (left), and from β E->T to α T and to β T (right). The bound ADP+Pi and ATP at the DP-site and T-site are circled, respectively. The interface region of α E->T to β DP was shown with zoom-in views to the left. Fig 4. The TMD simulation enforcing an ATP hydrolysis transition to the T*-site in the T-P-T* ring. (A) The top views of the ring at an early and a late stage of the simulation (T-site green, P-site blue, and T*-site yellow). (B) The distance between Pi and an inside reference (Cα of αr373) in the TMD and an equilibrium (eq) control simulation of the T-P-T* ring. (C) The full pathway of the Pi release captured in the TMD simulation. The Pi group (spheres) hopped from the arginine finger αr373 to αr143 (~ 80 ns), and then to αk196 (~ 140 ns) and αr161 (~ 180 ns) before the final release. Three negatively charged residues from β P (D195, E199 and E202) also help the Pi release through repulsion. β P is colored in ice blue and its P-loop in magenta, α P in cyan. The key charged residues are colored in blue (positive) and red (negative). Fig 5. The correlation with the ATP hydrolysis pocket in the TMD simulation of the T-P-T* ring. (A) The correlation map of the full ring during the enforced T*à DP transition starting from T-P-T*, colored according to the correlation values (blue/white/red: high/medium/low) between individual residues and the T*-site ATP binding/hydrolysis residues (in green spheres). (B) The residue-wise correlation from β P and β T (blue and gray). (C) An inside side view showing the correlation propagation from α T*à DP to β T and to α T (left), and from β T*à DP to α P and to β P (right). To understand why the conformational transition T*à DP impacts on the P-site that is upstream to the enforced T*-site in this case, we calculated again the residue-wise correlations on the T-P-T* ring during the enforced hydrolysis transition (see Fig 5). In particular, we see that the correlation decayed quickly right away from the T*-site. Nevertheless, one notices that the local region of the P-site correlates well with the T*-site residues. A close examination shows that the correlation propagates through a restricted region, from the hydrolysis site on β T*, along a beta-sheet structure (αarg164 to Arg171, αile345 to Leu352, and αleu369 to Arg373, see Fig 5C right), and then reaches the P-site on α P -β P. Fig 6. The TMD simulation enforcing an Eà T transition to one pair of α E -β E in an empty E-E-E ring. (A) The E-site being forced into a T-site is colored blue, the next/downstream E-site colored yellow, and the previous/upstream E-site green (right). The β-protrusions from both neighboring E-sites, sampled from the TMD simulation (dark) and a control equilibrium simulation (light), was shown, with arrows indicating the opening or closing 24
25 trend (left). (B) An early reaction pathway starting from an empty F 1 -ring (E-E-E) shows a bias after the first ATP binding. The bias likely facilitates the second ATP binding to the downstream site, and further leads to the E-DP-T configuration (instead of T-DP-E). 25





26 Fig 1







27 Fig 2



28 Fig 3


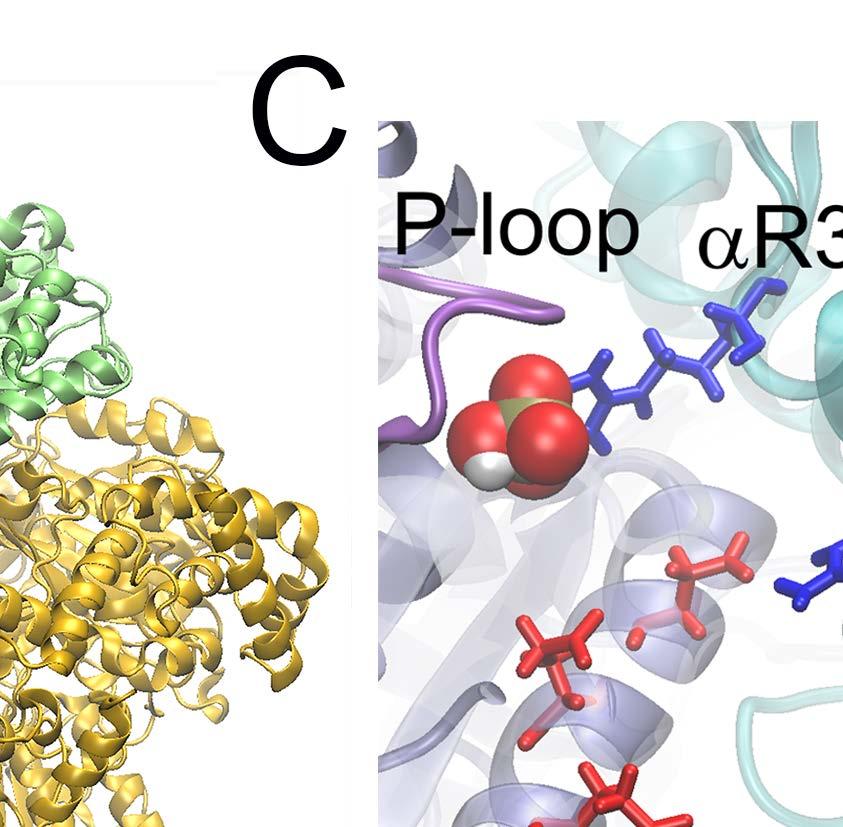





29 Fig 4








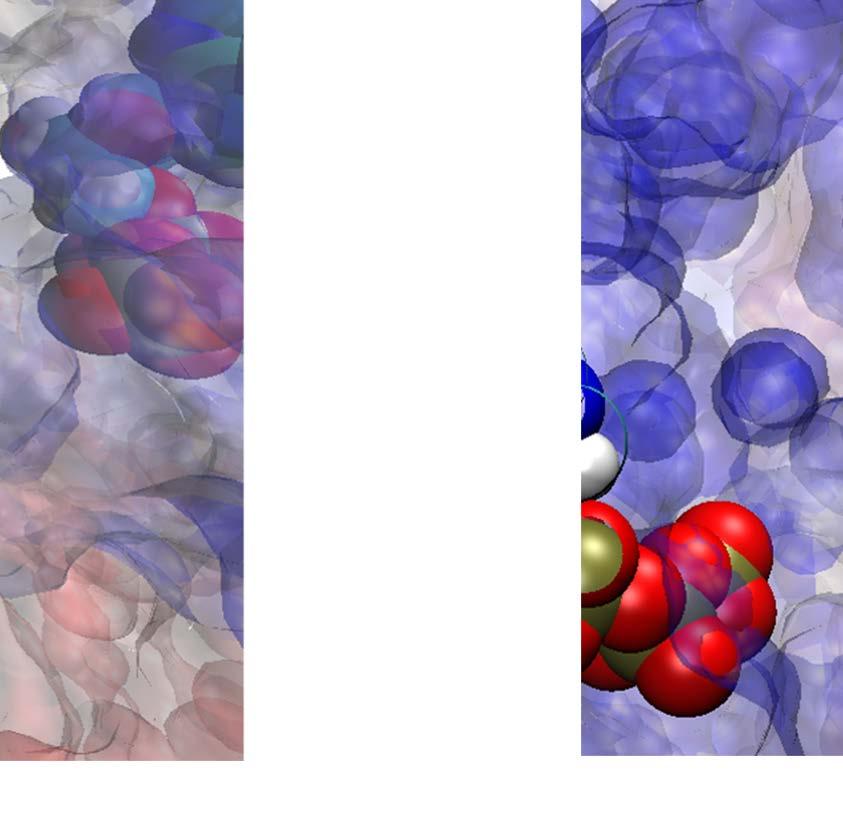


30 Fig 5







31 Fig 6
Molecular dynamics simulation. CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror
 Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
Molecular dynamics simulation CS/CME/BioE/Biophys/BMI 279 Oct. 5 and 10, 2017 Ron Dror 1 Outline Molecular dynamics (MD): The basic idea Equations of motion Key properties of MD simulations Sample applications
ATP hydrolysis 1 1 1
 ATP hydrolysis 1 1 1 ATP hydrolysis 2 2 2 The binding zipper 1 3 3 ATP hydrolysis/synthesis is coupled to a torque Yasuda, R., et al (1998). Cell 93:1117 1124. Abrahams, et al (1994). Nature 370:621-628.
ATP hydrolysis 1 1 1 ATP hydrolysis 2 2 2 The binding zipper 1 3 3 ATP hydrolysis/synthesis is coupled to a torque Yasuda, R., et al (1998). Cell 93:1117 1124. Abrahams, et al (1994). Nature 370:621-628.
Chapter 6- An Introduction to Metabolism*
 Chapter 6- An Introduction to Metabolism* *Lecture notes are to be used as a study guide only and do not represent the comprehensive information you will need to know for the exams. The Energy of Life
Chapter 6- An Introduction to Metabolism* *Lecture notes are to be used as a study guide only and do not represent the comprehensive information you will need to know for the exams. The Energy of Life
ATPase Synthase - A Molecular Double Motor
 ATPase Synthase - A Molecular Double Motor RC bc 1 bc 1 ATPase Photosynthesic Unit of Purple Bacteria Module that converts sun light into chemical energy (ATP) Light in hν H + Q/QH 2 /Q bc 1 ADP ATP out
ATPase Synthase - A Molecular Double Motor RC bc 1 bc 1 ATPase Photosynthesic Unit of Purple Bacteria Module that converts sun light into chemical energy (ATP) Light in hν H + Q/QH 2 /Q bc 1 ADP ATP out
The Molecular Dynamics Method
 The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = d dx U(x) Conformation
Unfolding CspB by means of biased molecular dynamics
 Chapter 4 Unfolding CspB by means of biased molecular dynamics 4.1 Introduction Understanding the mechanism of protein folding has been a major challenge for the last twenty years, as pointed out in the
Chapter 4 Unfolding CspB by means of biased molecular dynamics 4.1 Introduction Understanding the mechanism of protein folding has been a major challenge for the last twenty years, as pointed out in the
A. Reaction Mechanisms and Catalysis (1) proximity effect (2) acid-base catalysts (3) electrostatic (4) functional groups (5) structural flexibility
 proximity effect (2) acid-base catalysts (3) electrostatic (4) functional groups (5) structural flexibility") (P&S Ch 5; Fer Ch 2, 9; Palm Ch 10,11; Zub Ch 9) A. Reaction Mechanisms and Catalysis (1) proximity effect (2) acid-base catalysts (3) electrostatic (4) functional groups (5) structural flexibility B.
(P&S Ch 5; Fer Ch 2, 9; Palm Ch 10,11; Zub Ch 9) A. Reaction Mechanisms and Catalysis (1) proximity effect (2) acid-base catalysts (3) electrostatic (4) functional groups (5) structural flexibility B.
Hands-on Course in Computational Structural Biology and Molecular Simulation BIOP590C/MCB590C. Course Details
 Hands-on Course in Computational Structural Biology and Molecular Simulation BIOP590C/MCB590C Emad Tajkhorshid Center for Computational Biology and Biophysics Email: emad@life.uiuc.edu or tajkhors@uiuc.edu
Hands-on Course in Computational Structural Biology and Molecular Simulation BIOP590C/MCB590C Emad Tajkhorshid Center for Computational Biology and Biophysics Email: emad@life.uiuc.edu or tajkhors@uiuc.edu
SECOND PUBLIC EXAMINATION. Honour School of Physics Part C: 4 Year Course. Honour School of Physics and Philosophy Part C C7: BIOLOGICAL PHYSICS
 2757 SECOND PUBLIC EXAMINATION Honour School of Physics Part C: 4 Year Course Honour School of Physics and Philosophy Part C C7: BIOLOGICAL PHYSICS TRINITY TERM 2013 Monday, 17 June, 2.30 pm 5.45 pm 15
2757 SECOND PUBLIC EXAMINATION Honour School of Physics Part C: 4 Year Course Honour School of Physics and Philosophy Part C C7: BIOLOGICAL PHYSICS TRINITY TERM 2013 Monday, 17 June, 2.30 pm 5.45 pm 15
The Molecular Dynamics Method
 H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
H-bond energy (kcal/mol) - 4.0 The Molecular Dynamics Method Fibronectin III_1, a mechanical protein that glues cells together in wound healing and in preventing tumor metastasis 0 ATPase, a molecular
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar.
 CS490B: Introduction to Bioinformatics Mar.") Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Why Proteins Fold? (Parts of this presentation are based on work of Ashok Kolaskar) CS490B: Introduction to Bioinformatics Mar. 25, 2002 Molecular Dynamics: Introduction At physiological conditions, the
Computational Structural Biology and Molecular Simulation. Introduction to VMD Molecular Visualization and Analysis
 Computational Structural Biology and Molecular Simulation Introduction to VMD Molecular Visualization and Analysis Emad Tajkhorshid Department of Biochemistry, Beckman Institute, Center for Computational
Computational Structural Biology and Molecular Simulation Introduction to VMD Molecular Visualization and Analysis Emad Tajkhorshid Department of Biochemistry, Beckman Institute, Center for Computational
April Barry Isralewitz
 1 April 2002 Barry Isralewitz All enzymes are beautiful, but ATP synthase is one of the most: - Beautiful because of its 3D structure, - Unusual because of its structural complexity and reaction mechanism,
1 April 2002 Barry Isralewitz All enzymes are beautiful, but ATP synthase is one of the most: - Beautiful because of its 3D structure, - Unusual because of its structural complexity and reaction mechanism,
Supplementary Figure S1. Urea-mediated buffering mechanism of H. pylori. Gastric urea is funneled to a cytoplasmic urease that is presumably attached
 Supplementary Figure S1. Urea-mediated buffering mechanism of H. pylori. Gastric urea is funneled to a cytoplasmic urease that is presumably attached to HpUreI. Urea hydrolysis products 2NH 3 and 1CO 2
Supplementary Figure S1. Urea-mediated buffering mechanism of H. pylori. Gastric urea is funneled to a cytoplasmic urease that is presumably attached to HpUreI. Urea hydrolysis products 2NH 3 and 1CO 2
Villa et al. (2005) Structural dynamics of the lac repressor-dna complex revealed by a multiscale simulation. PNAS 102:
 Structural dynamics of the lac repressor-dna complex revealed by a multiscale simulation. PNAS 102:") Villa et al. (2005) Structural dynamics of the lac repressor-dna complex revealed by a multiscale simulation. PNAS 102: 6783-6788. Background: The lac operon is a cluster of genes in the E. coli genome
Villa et al. (2005) Structural dynamics of the lac repressor-dna complex revealed by a multiscale simulation. PNAS 102: 6783-6788. Background: The lac operon is a cluster of genes in the E. coli genome
SINGLE-MOLECULE PHYSIOLOGY
 SINGLE-MOLECULE PHYSIOLOGY Kazuhiko Kinosita, Jr. Center for Integrative Bioscience, Okazaki National Research Institutes Higashiyama 5-1, Myodaiji, Okazaki 444-8585, Japan Single-Molecule Physiology under
SINGLE-MOLECULE PHYSIOLOGY Kazuhiko Kinosita, Jr. Center for Integrative Bioscience, Okazaki National Research Institutes Higashiyama 5-1, Myodaiji, Okazaki 444-8585, Japan Single-Molecule Physiology under
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
What binds to Hb in addition to O 2?
 Reading: Ch5; 158-169, 162-166, 169-174 Problems: Ch5 (text); 3,7,8,10 Ch5 (study guide-facts); 1,2,3,4,5,8 Ch5 (study guide-apply); 2,3 Remember Today at 5:30 in CAS-522 is the second chance for the MB
Reading: Ch5; 158-169, 162-166, 169-174 Problems: Ch5 (text); 3,7,8,10 Ch5 (study guide-facts); 1,2,3,4,5,8 Ch5 (study guide-apply); 2,3 Remember Today at 5:30 in CAS-522 is the second chance for the MB
Chapter 8: An Introduction to Metabolism
 Chapter 8: An Introduction to Metabolism Key Concepts 8.1 An organism s metabolism transforms matter and energy, subject to the laws of thermodynamics 8.2 The free-energy change of a reaction tells us
Chapter 8: An Introduction to Metabolism Key Concepts 8.1 An organism s metabolism transforms matter and energy, subject to the laws of thermodynamics 8.2 The free-energy change of a reaction tells us
Supplementary Information for: Controlling Cellular Uptake of Nanoparticles with ph-sensitive Polymers
 Supplementary Information for: Controlling Cellular Uptake of Nanoparticles with ph-sensitive Polymers Hong-ming Ding 1 & Yu-qiang Ma 1,2, 1 National Laboratory of Solid State Microstructures and Department
Supplementary Information for: Controlling Cellular Uptake of Nanoparticles with ph-sensitive Polymers Hong-ming Ding 1 & Yu-qiang Ma 1,2, 1 National Laboratory of Solid State Microstructures and Department
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Structuring of hydrophobic and hydrophilic polymers at interfaces Stephen Donaldson ChE 210D Final Project Abstract
 Structuring of hydrophobic and hydrophilic polymers at interfaces Stephen Donaldson ChE 210D Final Project Abstract In this work, a simplified Lennard-Jones (LJ) sphere model is used to simulate the aggregation,
Structuring of hydrophobic and hydrophilic polymers at interfaces Stephen Donaldson ChE 210D Final Project Abstract In this work, a simplified Lennard-Jones (LJ) sphere model is used to simulate the aggregation,
Introduction The gramicidin A (ga) channel forms by head-to-head association of two monomers at their amino termini, one from each bilayer leaflet. Th
 channel forms by head-to-head association of two monomers at their amino termini, one from each bilayer leaflet. Th") Abstract When conductive, gramicidin monomers are linked by six hydrogen bonds. To understand the details of dissociation and how the channel transits from a state with 6H bonds to ones with 4H bonds or
Abstract When conductive, gramicidin monomers are linked by six hydrogen bonds. To understand the details of dissociation and how the channel transits from a state with 6H bonds to ones with 4H bonds or
PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS
 TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
TASKQUARTERLYvol.20,No4,2016,pp.353 360 PROTEIN-PROTEIN DOCKING REFINEMENT USING RESTRAINT MOLECULAR DYNAMICS SIMULATIONS MARTIN ZACHARIAS Physics Department T38, Technical University of Munich James-Franck-Str.
Molecular Mechanics, Dynamics & Docking
 Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular Mechanics, Dynamics & Docking Lawrence Hunter, Ph.D. Director, Computational Bioscience Program University of Colorado School of Medicine Larry.Hunter@uchsc.edu http://compbio.uchsc.edu/hunter
Molecular dynamics simulation of Aquaporin-1. 4 nm
 Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Molecular dynamics simulation of Aquaporin-1 4 nm Molecular Dynamics Simulations Schrödinger equation i~@ t (r, R) =H (r, R) Born-Oppenheimer approximation H e e(r; R) =E e (R) e(r; R) Nucleic motion described
Protein Folding & Stability. Lecture 11: Margaret A. Daugherty. Fall How do we go from an unfolded polypeptide chain to a
 Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2004 How do we go from an unfolded polypeptide chain to a compact folded protein? (Folding of thioredoxin, F. Richards) Structure - Function
Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2004 How do we go from an unfolded polypeptide chain to a compact folded protein? (Folding of thioredoxin, F. Richards) Structure - Function
An Introduction to Metabolism
 An Introduction to Metabolism PREFACE The living cell is a chemical factory with thousands of reactions taking place, many of them simultaneously This chapter is about matter and energy flow during life
An Introduction to Metabolism PREFACE The living cell is a chemical factory with thousands of reactions taking place, many of them simultaneously This chapter is about matter and energy flow during life
Problems from Previous Class
 1 Problems from Previous lass 1. What is K m? What are the units of K m? 2. What is V max? What are the units of V max? 3. Write down the Michaelis-Menten equation. 4. What order of reaction is the reaction
1 Problems from Previous lass 1. What is K m? What are the units of K m? 2. What is V max? What are the units of V max? 3. Write down the Michaelis-Menten equation. 4. What order of reaction is the reaction
Structure Investigation of Fam20C, a Golgi Casein Kinase
 Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Introduction to Metabolism (Or Energy Management) Chapter 8
 Chapter 8") Introduction to Metabolism (Or Energy Management) Chapter 8 Metabolism of the chemical reactions in the organism Building up molecules Breaking down molecules Managing energy and materials Route to end-product
Introduction to Metabolism (Or Energy Management) Chapter 8 Metabolism of the chemical reactions in the organism Building up molecules Breaking down molecules Managing energy and materials Route to end-product
MARTINI simulation details
 S1 Appendix MARTINI simulation details MARTINI simulation initialization and equilibration In this section, we describe the initialization of simulations from Main Text section Residue-based coarsegrained
S1 Appendix MARTINI simulation details MARTINI simulation initialization and equilibration In this section, we describe the initialization of simulations from Main Text section Residue-based coarsegrained
An Introduction to Metabolism
 An Introduction to Metabolism I. All of an organism=s chemical reactions taken together is called metabolism. A. Metabolic pathways begin with a specific molecule, which is then altered in a series of
An Introduction to Metabolism I. All of an organism=s chemical reactions taken together is called metabolism. A. Metabolic pathways begin with a specific molecule, which is then altered in a series of
Advanced Certificate in Principles in Protein Structure. You will be given a start time with your exam instructions
 BIRKBECK COLLEGE (University of London) Advanced Certificate in Principles in Protein Structure MSc Structural Molecular Biology Date: Thursday, 1st September 2011 Time: 3 hours You will be given a start
BIRKBECK COLLEGE (University of London) Advanced Certificate in Principles in Protein Structure MSc Structural Molecular Biology Date: Thursday, 1st September 2011 Time: 3 hours You will be given a start
Anatoly B. Kolomeisky. Department of Chemistry CAN WE UNDERSTAND THE COMPLEX DYNAMICS OF MOTOR PROTEINS USING SIMPLE STOCHASTIC MODELS?
 Anatoly B. Kolomeisky Department of Chemistry CAN WE UNDERSTAND THE COMPLEX DYNAMICS OF MOTOR PROTEINS USING SIMPLE STOCHASTIC MODELS? Motor Proteins Enzymes that convert the chemical energy into mechanical
Anatoly B. Kolomeisky Department of Chemistry CAN WE UNDERSTAND THE COMPLEX DYNAMICS OF MOTOR PROTEINS USING SIMPLE STOCHASTIC MODELS? Motor Proteins Enzymes that convert the chemical energy into mechanical
Enhancing Specificity in the Janus Kinases: A Study on the Thienopyridine. JAK2 Selective Mechanism Combined Molecular Dynamics Simulation
 Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is The Royal Society of Chemistry 2015 Supporting Information Enhancing Specificity in the Janus Kinases: A Study on the Thienopyridine
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is The Royal Society of Chemistry 2015 Supporting Information Enhancing Specificity in the Janus Kinases: A Study on the Thienopyridine
Gene regulation II Biochemistry 302. February 27, 2006
 Gene regulation II Biochemistry 302 February 27, 2006 Molecular basis of inhibition of RNAP by Lac repressor 35 promoter site 10 promoter site CRP/DNA complex 60 Lewis, M. et al. (1996) Science 271:1247
Gene regulation II Biochemistry 302 February 27, 2006 Molecular basis of inhibition of RNAP by Lac repressor 35 promoter site 10 promoter site CRP/DNA complex 60 Lewis, M. et al. (1996) Science 271:1247
SUPPLEMENTARY MATERIAL. Supplementary material and methods:
 Electronic Supplementary Material (ESI) for Catalysis Science & Technology. This journal is The Royal Society of Chemistry 2015 SUPPLEMENTARY MATERIAL Supplementary material and methods: - Computational
Electronic Supplementary Material (ESI) for Catalysis Science & Technology. This journal is The Royal Society of Chemistry 2015 SUPPLEMENTARY MATERIAL Supplementary material and methods: - Computational
C a h p a t p e t r e r 6 E z n y z m y e m s
 Chapter 6 Enzymes 4. Examples of enzymatic reactions acid-base catalysis: give and take protons covalent catalysis: a transient covalent bond is formed between the enzyme and the substrate metal ion catalysis:
Chapter 6 Enzymes 4. Examples of enzymatic reactions acid-base catalysis: give and take protons covalent catalysis: a transient covalent bond is formed between the enzyme and the substrate metal ion catalysis:
Destruction of Amyloid Fibrils by Graphene through Penetration and Extraction of Peptides
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2015 Destruction of Amyloid Fibrils by Graphene through Penetration and Extraction of Peptides Zaixing
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2015 Destruction of Amyloid Fibrils by Graphene through Penetration and Extraction of Peptides Zaixing
Structure-Based Model of the Stepping Motor of PcrA Helicase
 Biophysical Journal Volume 91 September 2006 2097 2114 2097 Structure-Based Model of the Stepping Motor of PcrA Helicase Jin Yu,* y Taekjip Ha, yz and Klaus Schulten* y *Beckman Institute, y Department
Biophysical Journal Volume 91 September 2006 2097 2114 2097 Structure-Based Model of the Stepping Motor of PcrA Helicase Jin Yu,* y Taekjip Ha, yz and Klaus Schulten* y *Beckman Institute, y Department
Mechanical Proteins. Stretching imunoglobulin and fibronectin. domains of the muscle protein titin. Adhesion Proteins of the Immune System
 Mechanical Proteins F C D B A domains of the muscle protein titin E Stretching imunoglobulin and fibronectin G NIH Resource for Macromolecular Modeling and Bioinformatics Theoretical Biophysics Group,
Mechanical Proteins F C D B A domains of the muscle protein titin E Stretching imunoglobulin and fibronectin G NIH Resource for Macromolecular Modeling and Bioinformatics Theoretical Biophysics Group,
Mechanochemical Coupling in Myosin: A Theoretical Analysis with Molecular Dynamics and Combined QM/MM Reaction Path Calculations
 3342 J. Phys. Chem. B 2004, 108, 3342-3357 Mechanochemical Coupling in Myosin: A Theoretical Analysis with Molecular Dynamics and Combined QM/MM Reaction Path Calculations Guohui Li and Qiang Cui* Department
3342 J. Phys. Chem. B 2004, 108, 3342-3357 Mechanochemical Coupling in Myosin: A Theoretical Analysis with Molecular Dynamics and Combined QM/MM Reaction Path Calculations Guohui Li and Qiang Cui* Department
An Introduction to Metabolism
 An Introduction to Metabolism PREFACE The living cell is a chemical factory with thousands of reactions taking place, many of them simultaneously This chapter is about matter and energy flow during life
An Introduction to Metabolism PREFACE The living cell is a chemical factory with thousands of reactions taking place, many of them simultaneously This chapter is about matter and energy flow during life
BIOLOGICAL SCIENCE. Lecture Presentation by Cindy S. Malone, PhD, California State University Northridge. FIFTH EDITION Freeman Quillin Allison
 BIOLOGICAL SCIENCE FIFTH EDITION Freeman Quillin Allison 8 Lecture Presentation by Cindy S. Malone, PhD, California State University Northridge Roadmap 8 In this chapter you will learn how Enzymes use
BIOLOGICAL SCIENCE FIFTH EDITION Freeman Quillin Allison 8 Lecture Presentation by Cindy S. Malone, PhD, California State University Northridge Roadmap 8 In this chapter you will learn how Enzymes use
Computer simulation methods (2) Dr. Vania Calandrini
 Dr. Vania Calandrini") Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Computer simulation methods (2) Dr. Vania Calandrini in the previous lecture: time average versus ensemble average MC versus MD simulations equipartition theorem (=> computing T) virial theorem (=> computing
Computational Modeling of Protein Kinase A and Comparison with Nuclear Magnetic Resonance Data
 Computational Modeling of Protein Kinase A and Comparison with Nuclear Magnetic Resonance Data ABSTRACT Keyword Lei Shi 1 Advisor: Gianluigi Veglia 1,2 Department of Chemistry 1, & Biochemistry, Molecular
Computational Modeling of Protein Kinase A and Comparison with Nuclear Magnetic Resonance Data ABSTRACT Keyword Lei Shi 1 Advisor: Gianluigi Veglia 1,2 Department of Chemistry 1, & Biochemistry, Molecular
Department of Physics, University at Buffalo, Buffalo, NY INTRODUCTION
 proteins STRUCTURE O FUNCTION O BIOINFORMATICS Coarse-grained modeling of conformational transitions underlying the processive stepping of myosin V dimer along filamentous actin Wenjun Zheng* Department
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Coarse-grained modeling of conformational transitions underlying the processive stepping of myosin V dimer along filamentous actin Wenjun Zheng* Department
Structural and mechanistic insight into the substrate. binding from the conformational dynamics in apo. and substrate-bound DapE enzyme
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 215 Structural and mechanistic insight into the substrate binding from the conformational
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 215 Structural and mechanistic insight into the substrate binding from the conformational
Biomolecules: lecture 10
 Biomolecules: lecture 10 - understanding in detail how protein 3D structures form - realize that protein molecules are not static wire models but instead dynamic, where in principle every atom moves (yet
Biomolecules: lecture 10 - understanding in detail how protein 3D structures form - realize that protein molecules are not static wire models but instead dynamic, where in principle every atom moves (yet
Hyeyoung Shin a, Tod A. Pascal ab, William A. Goddard III abc*, and Hyungjun Kim a* Korea
 The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
The Scaled Effective Solvent Method for Predicting the Equilibrium Ensemble of Structures with Analysis of Thermodynamic Properties of Amorphous Polyethylene Glycol-Water Mixtures Hyeyoung Shin a, Tod
Potential Energy (hyper)surface
surface") The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
The Molecular Dynamics Method Thermal motion of a lipid bilayer Water permeation through channels Selective sugar transport Potential Energy (hyper)surface What is Force? Energy U(x) F = " d dx U(x) Conformation
Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability
 Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability Dr. Andrew Lee UNC School of Pharmacy (Div. Chemical Biology and Medicinal Chemistry) UNC Med
Proteins are not rigid structures: Protein dynamics, conformational variability, and thermodynamic stability Dr. Andrew Lee UNC School of Pharmacy (Div. Chemical Biology and Medicinal Chemistry) UNC Med
Supporting Information. for An atomistic modeling of F-actin mechanical responses and determination of. mechanical properties
 Supporting Information for An atomistic modeling of F-actin mechanical responses and determination of mechanical properties Authors: Si Li 1, Jin Zhang 2, Chengyuan Wang 1, Perumal Nithiarasu 1 1. Zienkiewicz
Supporting Information for An atomistic modeling of F-actin mechanical responses and determination of mechanical properties Authors: Si Li 1, Jin Zhang 2, Chengyuan Wang 1, Perumal Nithiarasu 1 1. Zienkiewicz
SUPPLEMENTARY INFORMATION
 doi:10.1038/nature10955 Supplementary Figures Supplementary Figure 1. Electron-density maps and crystallographic dimer structures of the motor domain. (a f) Stereo views of the final electron-density maps
doi:10.1038/nature10955 Supplementary Figures Supplementary Figure 1. Electron-density maps and crystallographic dimer structures of the motor domain. (a f) Stereo views of the final electron-density maps
4 Examples of enzymes
 Catalysis 1 4 Examples of enzymes Adding water to a substrate: Serine proteases. Carbonic anhydrase. Restrictions Endonuclease. Transfer of a Phosphoryl group from ATP to a nucleotide. Nucleoside monophosphate
Catalysis 1 4 Examples of enzymes Adding water to a substrate: Serine proteases. Carbonic anhydrase. Restrictions Endonuclease. Transfer of a Phosphoryl group from ATP to a nucleotide. Nucleoside monophosphate
Why study protein dynamics?
 Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
Why study protein dynamics? Protein flexibility is crucial for function. One average structure is not enough. Proteins constantly sample configurational space. Transport - binding and moving molecules
The Unbinding of ATP from F 1 -ATPase
 Biophysical Journal Volume 85 August 2003 695 706 695 The Unbinding of ATP from F 1 -ATPase Iris Antes,* David Chandler,* Hongyun Wang, y and George Oster z *Department of Chemistry, University of California,
Biophysical Journal Volume 85 August 2003 695 706 695 The Unbinding of ATP from F 1 -ATPase Iris Antes,* David Chandler,* Hongyun Wang, y and George Oster z *Department of Chemistry, University of California,
Bioengineering 215. An Introduction to Molecular Dynamics for Biomolecules
 Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
Bioengineering 215 An Introduction to Molecular Dynamics for Biomolecules David Parker May 18, 2007 ntroduction A principal tool to study biological molecules is molecular dynamics simulations (MD). MD
SUPPLEMENTAL MATERIAL
 SUPPLEMENTAL MATERIAL Systematic Coarse-Grained Modeling of Complexation between Small Interfering RNA and Polycations Zonghui Wei 1 and Erik Luijten 1,2,3,4,a) 1 Graduate Program in Applied Physics, Northwestern
SUPPLEMENTAL MATERIAL Systematic Coarse-Grained Modeling of Complexation between Small Interfering RNA and Polycations Zonghui Wei 1 and Erik Luijten 1,2,3,4,a) 1 Graduate Program in Applied Physics, Northwestern
Structural and functional aspects of gastric proton pump, H +,K + -ATPase
 Kazuhiro ABE, Ph. D. E-mail:kabe@cespi.nagoya-u.ac.jp Structural and functional aspects of gastric proton pump, H +,K + -ATPase In response to food intake, ph of our stomach reaches around 1. This highly
Kazuhiro ABE, Ph. D. E-mail:kabe@cespi.nagoya-u.ac.jp Structural and functional aspects of gastric proton pump, H +,K + -ATPase In response to food intake, ph of our stomach reaches around 1. This highly
Supplementary Information
 Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Chapter 15: Enyzmatic Catalysis
 Chapter 15: Enyzmatic Catalysis Voet & Voet: Pages 496-508 Slide 1 Catalytic Mechanisms Catalysis is a process that increases the rate at which a reaction approaches equilibrium Rate enhancement depends
Chapter 15: Enyzmatic Catalysis Voet & Voet: Pages 496-508 Slide 1 Catalytic Mechanisms Catalysis is a process that increases the rate at which a reaction approaches equilibrium Rate enhancement depends
An Introduction to Metabolism
 An Introduction to Metabolism Chapter 8 Objectives Distinguish between the following pairs of terms: catabolic and anabolic pathways; kinetic and potential energy; open and closed systems; exergonic and
An Introduction to Metabolism Chapter 8 Objectives Distinguish between the following pairs of terms: catabolic and anabolic pathways; kinetic and potential energy; open and closed systems; exergonic and
f) Adding an enzyme does not change the Gibbs free energy. It only increases the rate of the reaction by lowering the activation energy.
 Adding an enzyme does not change the Gibbs free energy. It only increases the rate of the reaction by lowering the activation energy.") Problem Set 2-Answer Key BILD1 SP16 1) How does an enzyme catalyze a chemical reaction? Define the terms and substrate and active site. An enzyme lowers the energy of activation so the reaction proceeds
Problem Set 2-Answer Key BILD1 SP16 1) How does an enzyme catalyze a chemical reaction? Define the terms and substrate and active site. An enzyme lowers the energy of activation so the reaction proceeds
Supplemental Data SUPPLEMENTAL FIGURES
 Supplemental Data CRYSTAL STRUCTURE OF THE MG.ADP-INHIBITED STATE OF THE YEAST F 1 C 10 ATP SYNTHASE Alain Dautant*, Jean Velours and Marie-France Giraud* From Université Bordeaux 2, CNRS; Institut de
Supplemental Data CRYSTAL STRUCTURE OF THE MG.ADP-INHIBITED STATE OF THE YEAST F 1 C 10 ATP SYNTHASE Alain Dautant*, Jean Velours and Marie-France Giraud* From Université Bordeaux 2, CNRS; Institut de
Biophysics 490M Project
 Biophysics 490M Project Dan Han Department of Biochemistry Structure Exploration of aa 3 -type Cytochrome c Oxidase from Rhodobacter sphaeroides I. Introduction: All organisms need energy to live. They
Biophysics 490M Project Dan Han Department of Biochemistry Structure Exploration of aa 3 -type Cytochrome c Oxidase from Rhodobacter sphaeroides I. Introduction: All organisms need energy to live. They
Lectures by Kathleen Fitzpatrick
 Chapter 10 Chemotrophic Energy Metabolism: Aerobic Respiration Lectures by Kathleen Fitzpatrick Simon Fraser University Figure 10-1 Figure 10-6 Conversion of pyruvate The conversion of pyruvate to acetyl
Chapter 10 Chemotrophic Energy Metabolism: Aerobic Respiration Lectures by Kathleen Fitzpatrick Simon Fraser University Figure 10-1 Figure 10-6 Conversion of pyruvate The conversion of pyruvate to acetyl
Energy, Enzymes, and Metabolism. Energy, Enzymes, and Metabolism. A. Energy and Energy Conversions. A. Energy and Energy Conversions
 Energy, Enzymes, and Metabolism Lecture Series 6 Energy, Enzymes, and Metabolism B. ATP: Transferring Energy in Cells D. Molecular Structure Determines Enzyme Fxn Energy is the capacity to do work (cause
Energy, Enzymes, and Metabolism Lecture Series 6 Energy, Enzymes, and Metabolism B. ATP: Transferring Energy in Cells D. Molecular Structure Determines Enzyme Fxn Energy is the capacity to do work (cause
T H E J O U R N A L O F G E N E R A L P H Y S I O L O G Y. jgp
 S u p p l e m e n ta l m at e r i a l jgp Lee et al., http://www.jgp.org/cgi/content/full/jgp.201411219/dc1 T H E J O U R N A L O F G E N E R A L P H Y S I O L O G Y S u p p l e m e n ta l D I S C U S
S u p p l e m e n ta l m at e r i a l jgp Lee et al., http://www.jgp.org/cgi/content/full/jgp.201411219/dc1 T H E J O U R N A L O F G E N E R A L P H Y S I O L O G Y S u p p l e m e n ta l D I S C U S
ATP binding controls distinct structural transitions. of Escherichia coli DNA gyrase in complex with DNA
 Supplementary Information ATP binding controls distinct structural transitions of Escherichia coli DNA gyrase in complex with DNA Aakash Basu, Allyn J. Schoeffler, James M. Berger, and Zev Bryant Table
Supplementary Information ATP binding controls distinct structural transitions of Escherichia coli DNA gyrase in complex with DNA Aakash Basu, Allyn J. Schoeffler, James M. Berger, and Zev Bryant Table
It s the amino acids!
 Catalytic Mechanisms HOW do enzymes do their job? Reducing activation energy sure, but HOW does an enzyme catalysis reduce the energy barrier ΔG? Remember: The rate of a chemical reaction of substrate
Catalytic Mechanisms HOW do enzymes do their job? Reducing activation energy sure, but HOW does an enzyme catalysis reduce the energy barrier ΔG? Remember: The rate of a chemical reaction of substrate
BMB Lecture 7. Allostery and Cooperativity
 BMB 178 2017 Lecture 7 October 18, 2017 Allostery and Cooperativity A means for exquisite control Allostery: the basis of enzymatic control From the Greek: allos = other stereos = solid or space Action
BMB 178 2017 Lecture 7 October 18, 2017 Allostery and Cooperativity A means for exquisite control Allostery: the basis of enzymatic control From the Greek: allos = other stereos = solid or space Action
Supplementary Materials for
 advances.sciencemag.org/cgi/content/full/3/5/e1601684/dc1 Supplementary Materials for Directional mechanical stability of Bacteriophage ϕ29 motor s 3WJ-pRNA: Extraordinary robustness along portal axis
advances.sciencemag.org/cgi/content/full/3/5/e1601684/dc1 Supplementary Materials for Directional mechanical stability of Bacteriophage ϕ29 motor s 3WJ-pRNA: Extraordinary robustness along portal axis
Introduction to molecular dynamics
 1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
1 Introduction to molecular dynamics Yves Lansac Université François Rabelais, Tours, France Visiting MSE, GIST for the summer Molecular Simulation 2 Molecular simulation is a computational experiment.
Lipid Regulated Intramolecular Conformational Dynamics of SNARE-Protein Ykt6
 Supplementary Information for: Lipid Regulated Intramolecular Conformational Dynamics of SNARE-Protein Ykt6 Yawei Dai 1, 2, Markus Seeger 3, Jingwei Weng 4, Song Song 1, 2, Wenning Wang 4, Yan-Wen 1, 2,
Supplementary Information for: Lipid Regulated Intramolecular Conformational Dynamics of SNARE-Protein Ykt6 Yawei Dai 1, 2, Markus Seeger 3, Jingwei Weng 4, Song Song 1, 2, Wenning Wang 4, Yan-Wen 1, 2,
A Brief Guide to All Atom/Coarse Grain Simulations 1.1
 A Brief Guide to All Atom/Coarse Grain Simulations 1.1 Contents 1. Introduction 2. AACG simulations 2.1. Capabilities and limitations 2.2. AACG commands 2.3. Visualization and analysis 2.4. AACG simulation
A Brief Guide to All Atom/Coarse Grain Simulations 1.1 Contents 1. Introduction 2. AACG simulations 2.1. Capabilities and limitations 2.2. AACG commands 2.3. Visualization and analysis 2.4. AACG simulation
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
SUPPLEMENTARY INFORMATION
 doi:10.1038/nature17991 Supplementary Discussion Structural comparison with E. coli EmrE The DMT superfamily includes a wide variety of transporters with 4-10 TM segments 1. Since the subfamilies of the
doi:10.1038/nature17991 Supplementary Discussion Structural comparison with E. coli EmrE The DMT superfamily includes a wide variety of transporters with 4-10 TM segments 1. Since the subfamilies of the
Journal of Pharmacology and Experimental Therapy-JPET#172536
 A NEW NON-PEPTIDIC INHIBITOR OF THE 14-3-3 DOCKING SITE INDUCES APOPTOTIC CELL DEATH IN CHRONIC MYELOID LEUKEMIA SENSITIVE OR RESISTANT TO IMATINIB Manuela Mancini, Valentina Corradi, Sara Petta, Enza
A NEW NON-PEPTIDIC INHIBITOR OF THE 14-3-3 DOCKING SITE INDUCES APOPTOTIC CELL DEATH IN CHRONIC MYELOID LEUKEMIA SENSITIVE OR RESISTANT TO IMATINIB Manuela Mancini, Valentina Corradi, Sara Petta, Enza
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Don t forget to bring your MD tutorial. Potential Energy (hyper)surface
surface") Don t forget to bring your MD tutorial Lab session starts at 1pm You will have to finish an MD/SMD exercise on α-conotoxin in oxidized and reduced forms Potential Energy (hyper)surface What is Force? Energy
Don t forget to bring your MD tutorial Lab session starts at 1pm You will have to finish an MD/SMD exercise on α-conotoxin in oxidized and reduced forms Potential Energy (hyper)surface What is Force? Energy
Membrane Protein Channels
 Membrane Protein Channels Potassium ions queuing up in the potassium channel Pumps: 1000 s -1 Channels: 1000000 s -1 Pumps & Channels The lipid bilayer of biological membranes is intrinsically impermeable
Membrane Protein Channels Potassium ions queuing up in the potassium channel Pumps: 1000 s -1 Channels: 1000000 s -1 Pumps & Channels The lipid bilayer of biological membranes is intrinsically impermeable
Signal Transduction Phosphorylation Protein kinases. Misfolding diseases. Protein Engineering Lysozyme variants
 Signal Transduction Phosphorylation Protein kinases Misfolding diseases Protein Engineering Lysozyme variants Cells and Signals Regulation The cell must be able to respond to stimuli Cellular activities
Signal Transduction Phosphorylation Protein kinases Misfolding diseases Protein Engineering Lysozyme variants Cells and Signals Regulation The cell must be able to respond to stimuli Cellular activities
Lots of thanks for many reasons!
 Lots of thanks for many reasons! Structure and function of glutamate transporters from free energy simulations Turgut Baştuğ TOBB ETU Transporter structures Transporters have larger structures, which
Lots of thanks for many reasons! Structure and function of glutamate transporters from free energy simulations Turgut Baştuğ TOBB ETU Transporter structures Transporters have larger structures, which
Multiscale Materials Modeling
 Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Multiscale Materials Modeling Lecture 09 Quantum Mechanics/Molecular Mechanics (QM/MM) Techniques Fundamentals of Sustainable Technology These notes created by David Keffer, University of Tennessee, Knoxville,
Lecture 15: Enzymes & Kinetics. Mechanisms ROLE OF THE TRANSITION STATE. H-O-H + Cl - H-O δ- H Cl δ- HO - + H-Cl. Margaret A. Daugherty.
 Lecture 15: Enzymes & Kinetics Mechanisms Margaret A. Daugherty Fall 2004 ROLE OF THE TRANSITION STATE Consider the reaction: H-O-H + Cl - H-O δ- H Cl δ- HO - + H-Cl Reactants Transition state Products
Lecture 15: Enzymes & Kinetics Mechanisms Margaret A. Daugherty Fall 2004 ROLE OF THE TRANSITION STATE Consider the reaction: H-O-H + Cl - H-O δ- H Cl δ- HO - + H-Cl Reactants Transition state Products
BMB Class 17, November 30, Single Molecule Biophysics (II)
") BMB 178 2018 Class 17, November 30, 2018 15. Single Molecule Biophysics (II) New Advances in Single Molecule Techniques Atomic Force Microscopy Single Molecule Manipulation - optical traps and tweezers
BMB 178 2018 Class 17, November 30, 2018 15. Single Molecule Biophysics (II) New Advances in Single Molecule Techniques Atomic Force Microscopy Single Molecule Manipulation - optical traps and tweezers
Reading for today: Chapter 16 (selections from Sections A, B and C) Friday and Monday: Chapter 17 (Diffusion)
 Friday and Monday: Chapter 17 (Diffusion)") Lecture 29 Enzymes Reading for today: Chapter 6 (selections from Sections, B and C) Friday and Monday: Chapter 7 (Diffusion) 4/3/6 Today s Goals Michaelis-Menten mechanism for simple enzyme reactions:
Lecture 29 Enzymes Reading for today: Chapter 6 (selections from Sections, B and C) Friday and Monday: Chapter 7 (Diffusion) 4/3/6 Today s Goals Michaelis-Menten mechanism for simple enzyme reactions:
Lecture 7: Simple genetic circuits I
 Lecture 7: Simple genetic circuits I Paul C Bressloff (Fall 2018) 7.1 Transcription and translation In Fig. 20 we show the two main stages in the expression of a single gene according to the central dogma.
Lecture 7: Simple genetic circuits I Paul C Bressloff (Fall 2018) 7.1 Transcription and translation In Fig. 20 we show the two main stages in the expression of a single gene according to the central dogma.
Ground Rules of Metabolism CHAPTER 6
 Ground Rules of Metabolism CHAPTER 6 Antioxidants You ve heard the term. What s the big deal? Found naturally in many fruits and vegetables Added to many products What do they actually do? Antioxidants
Ground Rules of Metabolism CHAPTER 6 Antioxidants You ve heard the term. What s the big deal? Found naturally in many fruits and vegetables Added to many products What do they actually do? Antioxidants
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
BMB Lecture 7. Allostery and Cooperativity. A means for exquisite control
 BMB 178 2018 Lecture 7 Allostery and Cooperativity A means for exquisite control Allostery: the basis of enzymatic control From the Greek: allos = other stereos = solid or space Action at a distance Examples
BMB 178 2018 Lecture 7 Allostery and Cooperativity A means for exquisite control Allostery: the basis of enzymatic control From the Greek: allos = other stereos = solid or space Action at a distance Examples
Lecture 11: Protein Folding & Stability
 Structure - Function Protein Folding: What we know Lecture 11: Protein Folding & Stability 1). Amino acid sequence dictates structure. 2). The native structure represents the lowest energy state for a
Structure - Function Protein Folding: What we know Lecture 11: Protein Folding & Stability 1). Amino acid sequence dictates structure. 2). The native structure represents the lowest energy state for a
Protein Folding & Stability. Lecture 11: Margaret A. Daugherty. Fall Protein Folding: What we know. Protein Folding
 Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2003 Structure - Function Protein Folding: What we know 1). Amino acid sequence dictates structure. 2). The native structure represents
Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2003 Structure - Function Protein Folding: What we know 1). Amino acid sequence dictates structure. 2). The native structure represents
Chapter 10: Hemoglobin
 Chapter 10: Hemoglobin Voet & Voet: Pages 320-353 Slide 1 Hemoglobin Function Larger aerobic (oxygen utilizing) organism require an O 2 transport system to deliver sufficient O 2 to tissues Dissolved O
Chapter 10: Hemoglobin Voet & Voet: Pages 320-353 Slide 1 Hemoglobin Function Larger aerobic (oxygen utilizing) organism require an O 2 transport system to deliver sufficient O 2 to tissues Dissolved O
Magnetic tweezers and its application to DNA mechanics
 Biotechnological Center Research group DNA motors (Seidel group) Handout for Practical Course Magnetic tweezers and its application to DNA mechanics When: 9.00 am Where: Biotec, 3 rd Level, Room 317 Tutors:
Biotechnological Center Research group DNA motors (Seidel group) Handout for Practical Course Magnetic tweezers and its application to DNA mechanics When: 9.00 am Where: Biotec, 3 rd Level, Room 317 Tutors:
Chapter 5. Energy Flow in the Life of a Cell
 Chapter 5 Energy Flow in the Life of a Cell Including some materials from lectures by Gregory Ahearn University of North Florida Ammended by John Crocker Copyright 2009 Pearson Education, Inc.. Review
Chapter 5 Energy Flow in the Life of a Cell Including some materials from lectures by Gregory Ahearn University of North Florida Ammended by John Crocker Copyright 2009 Pearson Education, Inc.. Review
Metabolism and enzymes
 Metabolism and enzymes 4-11-16 What is a chemical reaction? A chemical reaction is a process that forms or breaks the chemical bonds that hold atoms together Chemical reactions convert one set of chemical
Metabolism and enzymes 4-11-16 What is a chemical reaction? A chemical reaction is a process that forms or breaks the chemical bonds that hold atoms together Chemical reactions convert one set of chemical
For slowly varying probabilities, the continuum form of these equations is. = (r + d)p T (x) (u + l)p D (x) ar x p T(x, t) + a2 r
p T (x) (u + l)p D (x) ar x p T(x, t) + a2 r") 3.2 Molecular Motors A variety of cellular processes requiring mechanical work, such as movement, transport and packaging material, are performed with the aid of protein motors. These molecules consume
3.2 Molecular Motors A variety of cellular processes requiring mechanical work, such as movement, transport and packaging material, are performed with the aid of protein motors. These molecules consume