Gary Kleiger, Anjanabha Saha, Steven Lewis, Brian Kuhlman, and Raymond J. Deshaies
|
|
|
- Virgil Hood
- 5 years ago
- Views:
Transcription
1 Cell, Volume 139 Supplemental Data Rapid E2-E3 Assembly and Disassembly Enable Processive Ubiquitylation of Cullin-RING Ubiquitin Ligase Substrates Gary Kleiger, Anjanabha Saha, Steven Lewis, Brian Kuhlman, and Raymond J. Deshaies Docking of the acidic tail to the Cul1 and Cul5 CTD Generation of starting models No crystal structure exists of the Cul1-Rbx1-Cdc34 complex, with or without Nedd8 attached. Additionally, no structure exists of the flexible Cdc34 tail we were interested in modeling. To create the requisite starting models, we used a combination of structural alignments to homologous structures and computational docking. Cul1-Rbx1-Cd34 was pieced together from PDB files 2OB4 (human Cdc34) and 1LDJ (Cul1-Rbx1) (Zheng et al., 2002). Cul1 was trimmed to residues To approximate the E2 RING interaction, these two PDB files were aligned against the E2 RING complex CBL-UBCH7, 1FBV (Zheng et al., 2000). This docking alignment was then refined using Rosetta's docking tools (Gray et al., 2003). A similar model complex was generated in the same fashion from PDB files 2OB4 (Cdc34) and 3DQV (chains A, C, and R) (Cul5-Rbx1- Nedd8) (Duda et al., 2008). The final step of generating starting models was attaching the tail sequence to Cdc34 (not seen in the crystal structure). We used the simple expedient of extending the tail straight out into space from the C-terminal residue in the Cdc34 crystal structure. Modeling the flexible tail To explore how the flexible tail might interact with the E3 in this system, we built a new protocol within the Rosetta framework. Figure (S8) demonstrates 1
2 the flow of this protocol. The protocol has a reduced-representation centroid phase and a second refinement phase with all atoms present. The centroid phase is designed to collapse the long tail, which initially points straight into space, into some reasonable conformation. The reduced representation has only single large centroid atoms replacing the side-chains (Rohl et al., 2004), obviating the need to consider packing details. The advantage of using this representation in the early part of the trajectory is that we no longer need to repack side-chains to smooth out minor side-chain/side-chain clashes, thus speeding up the computation. This phase consisted of 5,000 Metropolis Monte Carlo cycles. Most cycles were used to perturb the tail structure with one of three Rosetta tools (Rohl et al., 2004). The first tool, marked as Small_180 on Figure S8, randomly perturbed a phi and psi angle of a tail residue, weighted to choose good Ramachandran values. The second, Shear_180 on Figure S8, modifies a phi angle at position n and psi at position n-1 in opposite directions. This has the effect of tweaking local structure without longer-distance effect. Again, this was a random perturbation subject to Ramachandran constraints. Shear movements are designed to minimize perturbation of the backbone far from the move, so they were disabled for the first third of the protocol to give the tail a chance to develop interactions before their use. The third type of perturbation was a 3-residue fragment insertion. Fragment insertion consists of replacing the backbone torsional parameters with parameters from a small protein fragment derived from the PDB. Fragments were generated via the Robetta server (Kim et al., 2004). This allowed for somewhat faster sampling of local conformational and secondary structure preferences. After 19 cycles, the protocol performed a gradient-based energy minimization of the structure. The available degrees of freedom were the backbone torsion angles phi, psi and omega of tail residues (the same freedoms perturbed in the other steps). The score function used in this phase contained terms for van der Waals repulsion, hydrogen bonding, electrostatics, 2
3 Ramachandran, and residue-pair potentials (which incorporate van der Waals attraction and solvation) (Rohl et al., 2004). After 5,000 perturbing cycles, the lowest-scoring structure seen in the trajectory was recovered. Side-chains were returned to this structure. The crystallographic side-chains were used outside the tail. One of Rosetta's default full atomic representation energy functions, score12, was used for scoring in this phase (Kuhlman et al., 2003) (Smith and Kortemme, 2008). The tail region and any side-chains within 10 Ångstroms of the tail were subjected to a combinatorial repacking wherein a Monte Carlo procedure chose low-energy side-chain conformations from the full set of available rotamers. The side-chains and the tail backbone torsions were then energy minimized. The refinement phase ran for 3,000 cycles of Metropolis Monte Carlo optimization. Most cycles consisted of a random small perturbation. These perturbations were small or shear movements as before, except the perturbation was constrained to within 4 degrees, instead of anywhere in Ramachandran space. This sort of perturbation was immediately followed by a single-pass random-order rotamer optimization of the tail and its neighbors (rotamer trials). After 14 cycles, a minimization of the tail side-chains, tail backbone torsions, and side-chains neighboring the tail was performed. Another 14 cycles after that, a full combinatorial repacking operation was performed before the minimization. At the end of each trajectory, the best structure recorded was reported as output. The protocol as a whole was run for approximately 30,000 trajectories per experiment, which is slightly more than 1,000 processor-days on a 2.3 GHz chip. It is customary to run extremely large numbers of trajectories when doing structure prediction with Rosetta. The random nature of Monte Carlo sampling means that trajectories will sometimes get trapped in poor minima, so many trajectories are needed to sufficiently sample the structure space. Sorting models To search through our dataset for models that addressed the question of how the E2 might bind the E3, we sorted models based on E2-E3 binding energy. This was calculated as the complex score minus the scores of the E2 3
4 and E3-RING (±Nedd8) in isolation. It should be noted that there are two components to this binding energy. There is a constant component represented by the direct, canonical E2-RING interaction. This interaction was optimized by docking as a precursor step but not modified during the main part of the protocol. The second interaction represents binding between the E2 tail and the body of the E3. 4
5 Supplemental References Duda, D.M., Borg, L.A., Scott, D.C., Hunt, H.W., Hammel, M., and Schulman, B.A. (2008). Structural insights into NEDD8 activation of cullin-ring ligases: conformational control of conjugation. Cell 134, Gray, J.J., Moughon, S., Wang, C., Schueler-Furman, O., Kuhlman, B., Rohl, C.A., and Baker, D. (2003). Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol 331, Kim, D.E., Chivian, D., and Baker, D. (2004). Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32, W Kuhlman, B., Dantas, G., Ireton, G.C., Varani, G., Stoddard, B.L., and Baker, D. (2003). Design of a novel globular protein fold with atomic-level accuracy. Science 302, Larkin, M.A., Blackshields, G., Brown, N.P., Chenna, R., McGettigan, P.A., McWilliam, H., Valentin, F., Wallace, I.M., Wilm, A., Lopez, R., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, Rohl, C.A., Strauss, C.E., Misura, K.M., and Baker, D. (2004). Protein structure prediction using Rosetta. Methods Enzymol 383, Saha, A., and Deshaies, R.J. (2008). Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell 32, Smith, C.A., and Kortemme, T. (2008). Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant sidechain prediction. J Mol Biol 380, Zheng, N., Schulman, B.A., Song, L., Miller, J.J., Jeffrey, P.D., Wang, P., Chu, C., Koepp, D.M., Elledge, S.J., Pagano, M., et al. (2002). Structure of the Cul1- Rbx1-Skp1-F boxskp2 SCF ubiquitin ligase complex. Nature 416, Zheng, N., Wang, P., Jeffrey, P.D., and Pavletich, N.P. (2000). Structure of a c- Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell 102,
6 Supplemental Figure Legends Figure S1. Multiple Sequence Alignment of the human Cdc34a, human Cdc34b, and yeast Cdc sequences. While the yeast Cdc34 sequence is 295 amino acids, was included during the alignment procedure (MUSCLE) and resulted in a better alignment than when the full-length sequence was used. Amino acids within the catalytic domain are shaded olive. Amino acids within the acidic tail domain are shaded light-blue. Acidic residues in the tail domain are shaded yellow. Figure S2. The effects of Cul1 neddylation and ubiquitin oxyesterification to Cdc34 on k on are less than 2-fold. All experiments were performed identically to those in Figure 1a. (A) Measurement of k off for Lumio Green labeled WT Cdc34 and neddylated RC CFP complex. k on was estimated from k off and K d was taken from (Saha and Deshaies, 2008). (B) Measurement of k off for WT Cdc34 that was oxyestified to ubiquitin and neddylated RC CFP complex. k on was estimated from k off and K d was taken from (Saha and Deshaies, 2008). (C) Measurement of k off for WT Cdc34 and a full SCF βtrcp complex. Equimolar βtrcp-skp1 was added to the Lumio Cdc34-RC CFP complex. Notice that the value of k off is very nearly the same whether the full SCF complex is assembled or not (27 and 35 sec -1, respectively). The ionic strength of the reaction buffer for all experiments was 50 mm. Figure S3. Binding assays to RC CFP with N-terminally labeled Lumio Green Cdc34 protein show similar results as when the Lumio tag is at the C-terminus. (A) Salt sensitivity of FRET efficiency of RC CFP complex saturated with C- terminally labeled WT Cdc34 (white bars) or RC CFP complex with nearly saturating N-terminally labeled WT Cdc34 (black bars). Notice that N-terminally labeled Cdc34-RC CFP complex is highly salt sensitive, similar to C-terminally labeled Cdc34. (B) Salt sensitivity of Δ221 Cdc34. (C) Titration of N-terminal Lumio-labeled Cdc34 on RC CFP. All experiments were done in duplicate. 6
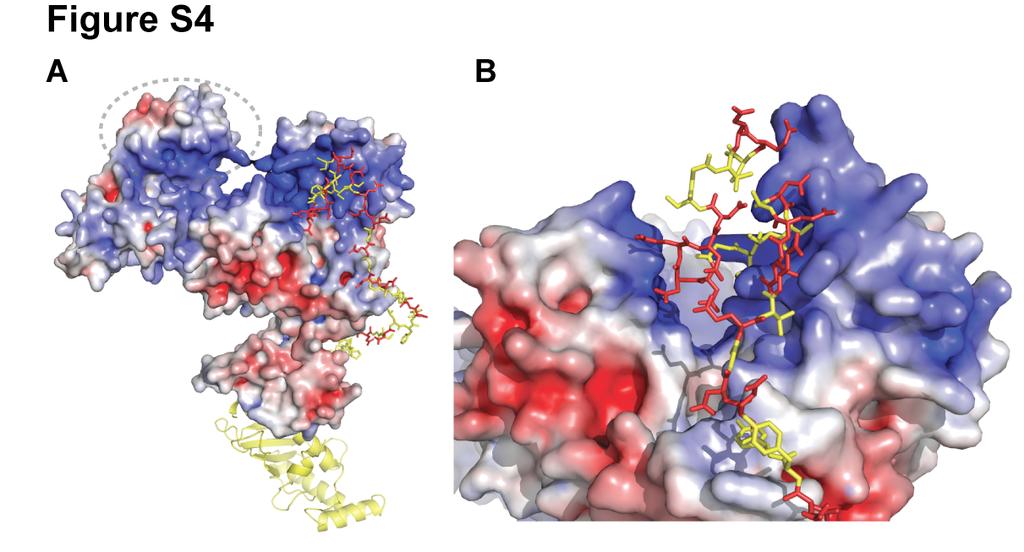
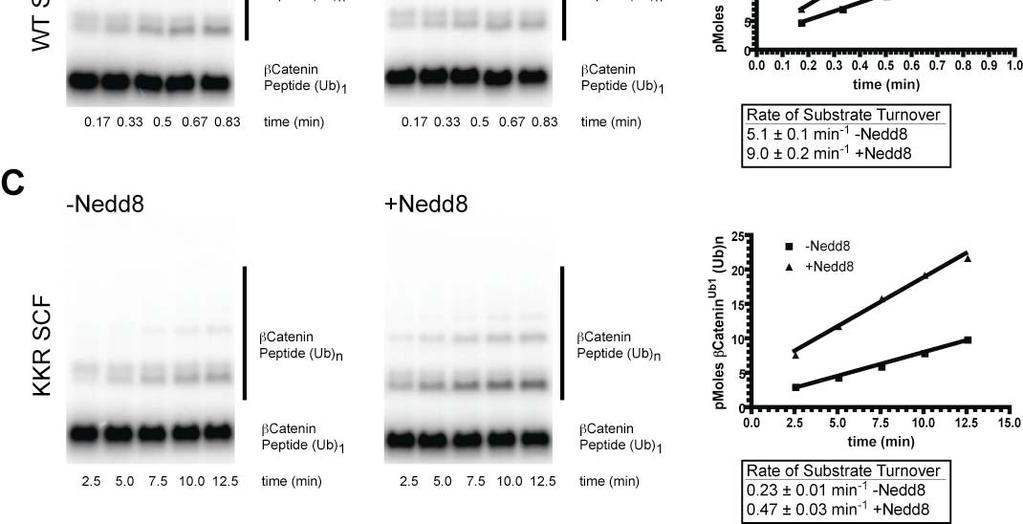
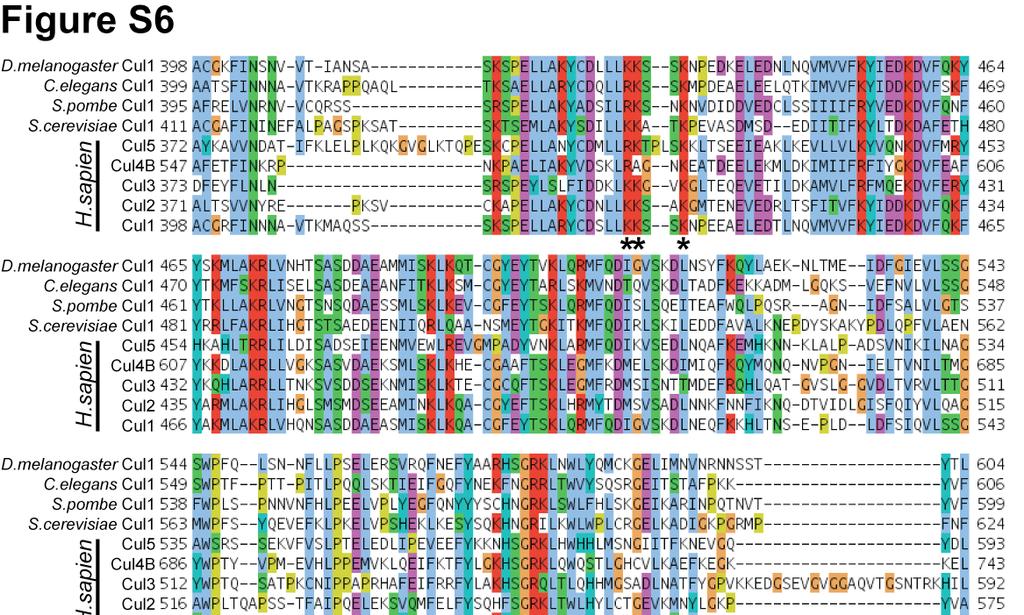
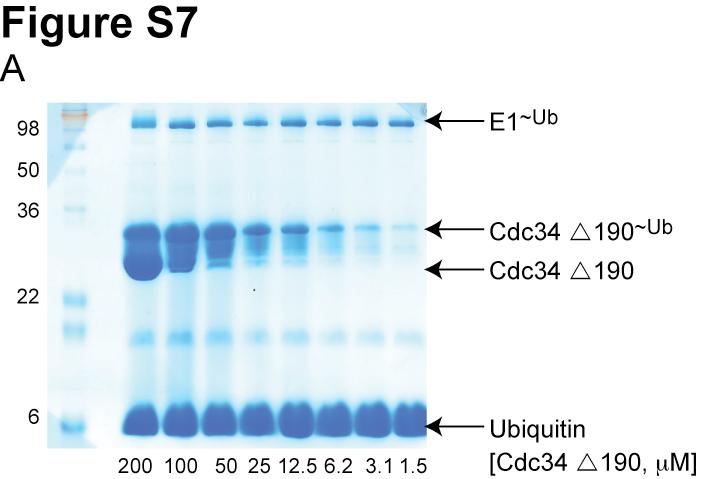
7 Figure S4. (A) Model showing the interaction between the acidic tail and the Cul5 CTD(Nedd8)-Rbx1 structure. Cul5, Rbx1 and Nedd8 are colored according to the electrostatic potential of the surface. The location of Nedd8 in the structure is shown by the dashed gray oval. The catalytic domain of Cdc34 is shown as a yellow ribbon diagram. Cdc34 residues in the tail are yellow balland-sticks, except acidic residues are colored red. (B) Magnified view of (A). Figure S5. Neddylation of KKR SCF results in a similar degree of activation as WT SCF. (A) Time course of the neddylation of WT or KKR RC. Notice that at completion the same amount of product is observed for either WT or KKR RC. (B,C) Ubiquitylation of a mono-ubiquitylated β-catenin peptide by either WT (B) or KKR (C) SCF and 0.5 WT Cdc34. Neddylation stimulated the rate of substrate consumption under multi-turnover conditions by approximately 2-fold for both WT and KKR SCF. Figure S6. Multiple sequence alignment of 9 cullin sequences by MUSCLE. Coloring of conserved residues was performed according to definitions in ClustalX (Larkin et al., 2007). Black asterisks were placed beneath positions in the MSA corresponding to human Cul1 KR claw residues 431, 432, 435, 678, 679 and 681. Figure S7. Cdc34-Δ190 can be fully thioesterified to ubiquitin at concentrations as high as 100. SDS-PAGE in the absence of reducing reagent for reactions containing human E1, various concentrations of human Cdc34-Δ190 (shown), and ubiquitin. The location of apo-e2 and the thioesterified E2-ubiquitin complex are shown. Notice that concentrations as high as 100 E2 can be achieved where little or no apo-e2 is present. Figure S8. Flow diagram illustrating the procedure used for modeling the Cdc34 acidic tail interaction with either Cul1 or neddylated Cul5 CTD. 7
8 Table S1. Constructs, expression, and purification for proteins used in this manuscript. Protein Construct Strain Expression Purification Comments WT hcdc34 a Δ190 hcdc34 NLum d -WT hcdc34 CLum-WT hcdc34 CLum-Δ221 hcdc34 pet11b-hcdc34- His6 pet11b-δ190 hcdc34-his6 pet11b-lumhcdc34-his6 pet11b-hcdc34- Lum-His6 pet11b-δ221 hcdc34-lum-his6 RDB2314 E.coli NiNTA b ->S75 c RDB2315 E.coli NiNTA->S75 RDB2316 E.coli ninta->s75 RDB2317 E.coli ninta->s75 RDB2318 E.coli ninta->s75 C227S hcdc34 RDB2319 E.coli ninta->s75 UbcH5c pet11b-c227shcdc34-his6 pgex-tev- UbcH5c RDB1973 E.coli GST->TEV digest (on beads)->s75 Cul1 NTD Rbx1-Cul1 CTD pal Cul1 NTD, pcool Rbx1 Cul1 CTD RDB2080 RDB2081 E.coli GST- Thrombin- Mono S->S200 e Coexpression Cul1 NTD Rbx1-Cul1 CTD (CFP) pal Cul1 NTD, pcool Rbx1 Cul1 CTD (CFP) RDB2080 RDB2228 E.coli GST- Thrombin- Mono S->S200 Coexpression Cul1 NTD Rbx1-Cul1 CTD K679C pal Cul1 NTD, pcool Rbx1 Cul1 CTD K679C RDB2320 E.coli GST- Thrombin- Mono S->S200 Coexpression Cul1 NTD Rbx1-Cul1 CTD KKR Δ230 ycdc34 f Δ210 ycdc34 βtrcp-skp1 ( ) yscf (Hrt1, Cdc53, Skp1, Cdc4) pal Cul1 NTD, pcool Rbx1 Cul1 CTD KKR pet11b-δ230 ycdc34-his6 pet11b-δ210 ycdc34-his6 RDB2321 E.coli GST- Thrombin- Mono S->S200 RDB2322 E.coli Ni-NTA->S75 RDB2323 E.coli Ni-NTA->S75 Coexpression GST βtrcp-skp1 - Hi5 insect cell GST->Thrombin- Mono Q->S200 Coexpression - Hi5 insect cell Py beads Coexpression a human, b Nickel agarose purification, c Superdex 75 gel filtration column d CCPGCC Lumio tag sequence, e Superdex 200 gel filtration column, f yeast 8
9 Table S2. Reaction conditions for ubiquitylation assays in this manuscript Figure E1 Ub E2 SCF Substrate Competitor 2a he1, 1 2b he1, 1 5a he1, 1 5b he1, 1 5b he1, 1 5c he1, 1 5c he1,1 5d ye1, 1 5d ye1, 1 60 WT Cdc34 (titration series) 150 Δ190 Cdc34 (titration series) 60 WT Cdc34 (titration series) 60 Δ190 Cdc34 (9 ) 60 Δ190 Cdc34 (9 ) 60 Ubc5c (0.75 ) 60 Ubc5c (0.75 ) 300 Δ230 ycdc34 (10 ) 300 Δ210 ycdc34 (10 or 100 ) WT Cul1- N8, Rbx1, Skp1, βtrcp (0.15 ) WT Cul1- N8, Rbx1, Skp1, βtrcp (0.15 ) KKR Cul1, Rbx1, Skp1, βtrcp (0.1 ) WT Cul1, Rbx1, Skp1, βtrcp (0.4 ) KKR Cul1, Rbx1, Skp1, βtrcp (0.4 ) WT Cul1, Rbx1, Skp1, βtrcp (0.2 ) KKR Cul1, Rbx1, Skp1, βtrcp (0.2 ) yscf (0.35 ) yscf (0.35 ) βcatenin Ub peptide (4 ) βcatenin Ub peptide (4 ) βcatenin Ub peptide (3 ) βcatenin Ub peptide (3 ) βcatenin Ub peptide (3 ) βcatenin peptide (5 ) βcatenin peptide (5 ) Cyclin E peptide (30 nm) Cyclin E peptide (30 nm) Cyclin E peptide (20 ) Cyclin E peptide (20 ) 9


10 10
11 11
12 12
13 13
14 14

15 15
16 16
Supporting Online Material for
 www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
www.sciencemag.org/cgi/content/full/309/5742/1868/dc1 Supporting Online Material for Toward High-Resolution de Novo Structure Prediction for Small Proteins Philip Bradley, Kira M. S. Misura, David Baker*
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy
 Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Design of a Novel Globular Protein Fold with Atomic-Level Accuracy Brian Kuhlman, Gautam Dantas, Gregory C. Ireton, Gabriele Varani, Barry L. Stoddard, David Baker Presented by Kate Stafford 4 May 05 Protein
Supporting Information
 Supporting Information Superagonist, Full Agonist, Partial Agonist and Antagonist Actions of Arylguanidines at 5-Hydroxytryptamine-3 (5-HT 3 ) Subunit A Receptors Katie Alix, Shailesh Khatri, Philip D.
Supporting Information Superagonist, Full Agonist, Partial Agonist and Antagonist Actions of Arylguanidines at 5-Hydroxytryptamine-3 (5-HT 3 ) Subunit A Receptors Katie Alix, Shailesh Khatri, Philip D.
HOMOLOGY MODELING. The sequence alignment and template structure are then used to produce a structural model of the target.
 HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
CMPS 6630: Introduction to Computational Biology and Bioinformatics. Tertiary Structure Prediction
 CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 6630: Introduction to Computational Biology and Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the
CMPS 3110: Bioinformatics. Tertiary Structure Prediction
 CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
CMPS 3110: Bioinformatics Tertiary Structure Prediction Tertiary Structure Prediction Why Should Tertiary Structure Prediction Be Possible? Molecules obey the laws of physics! Conformation space is finite
Protein Dynamics. The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron.
 Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Protein Dynamics The space-filling structures of myoglobin and hemoglobin show that there are no pathways for O 2 to reach the heme iron. Below is myoglobin hydrated with 350 water molecules. Only a small
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 Allowed phi-psi angles Red areas are preferred, yellow areas are allowed, and white is avoided 2.3a Hamiltonian
FlexPepDock In a nutshell
 FlexPepDock In a nutshell All Tutorial files are located in http://bit.ly/mxtakv FlexPepdock refinement Step 1 Step 3 - Refinement Step 4 - Selection of models Measure of fit FlexPepdock Ab-initio Step
FlexPepDock In a nutshell All Tutorial files are located in http://bit.ly/mxtakv FlexPepdock refinement Step 1 Step 3 - Refinement Step 4 - Selection of models Measure of fit FlexPepdock Ab-initio Step
THE CRYSTAL STRUCTURE OF THE SGT1-SKP1 COMPLEX: THE LINK BETWEEN
 THE CRYSTAL STRUCTURE OF THE SGT1-SKP1 COMPLEX: THE LINK BETWEEN HSP90 AND BOTH SCF E3 UBIQUITIN LIGASES AND KINETOCHORES Oliver Willhoft, Richard Kerr, Dipali Patel, Wenjuan Zhang, Caezar Al-Jassar, Tina
THE CRYSTAL STRUCTURE OF THE SGT1-SKP1 COMPLEX: THE LINK BETWEEN HSP90 AND BOTH SCF E3 UBIQUITIN LIGASES AND KINETOCHORES Oliver Willhoft, Richard Kerr, Dipali Patel, Wenjuan Zhang, Caezar Al-Jassar, Tina
Protein Structure Prediction, Engineering & Design CHEM 430
 Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Protein Structure Prediction, Engineering & Design CHEM 430 Eero Saarinen The free energy surface of a protein Protein Structure Prediction & Design Full Protein Structure from Sequence - High Alignment
Supplementary figure 1 Application of tmfret in LeuT. (a) To assess the feasibility of using tmfret for distance-dependent measurements in LeuT, a
 To assess the feasibility of using tmfret for distance-dependent measurements in LeuT, a") Supplementary figure 1 Application of tmfret in LeuT. (a) To assess the feasibility of using tmfret for distance-dependent measurements in LeuT, a series of tmfret-pairs comprised of single cysteine mutants
Supplementary figure 1 Application of tmfret in LeuT. (a) To assess the feasibility of using tmfret for distance-dependent measurements in LeuT, a series of tmfret-pairs comprised of single cysteine mutants
Table S1. Overview of used PDZK1 constructs and their binding affinities to peptides. Related to figure 1.
 Table S1. Overview of used PDZK1 constructs and their binding affinities to peptides. Related to figure 1. PDZK1 constru cts Amino acids MW [kda] KD [μm] PEPT2-CT- FITC KD [μm] NHE3-CT- FITC KD [μm] PDZK1-CT-
Table S1. Overview of used PDZK1 constructs and their binding affinities to peptides. Related to figure 1. PDZK1 constru cts Amino acids MW [kda] KD [μm] PEPT2-CT- FITC KD [μm] NHE3-CT- FITC KD [μm] PDZK1-CT-
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Template-Based Modeling of Protein Structure
 Template-Based Modeling of Protein Structure David Constant Biochemistry 218 December 11, 2011 Introduction. Much can be learned about the biology of a protein from its structure. Simply put, structure
Template-Based Modeling of Protein Structure David Constant Biochemistry 218 December 11, 2011 Introduction. Much can be learned about the biology of a protein from its structure. Simply put, structure
Protein structure prediction. CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror
 Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
Protein structure prediction CS/CME/BioE/Biophys/BMI 279 Oct. 10 and 12, 2017 Ron Dror 1 Outline Why predict protein structure? Can we use (pure) physics-based methods? Knowledge-based methods Two major
COMPUTATIONAL PREDICTION OF PROTEIN SMALL MOLECULE INTERFACES USING ROSETTA. Kristian Wallace Kaufmann. Dissertation. Submitted to the Faculty of the
 COMPUTATIONAL PREDICTION OF PROTEIN SMALL MOLECULE INTERFACES USING ROSETTA By Kristian Wallace Kaufmann Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial
COMPUTATIONAL PREDICTION OF PROTEIN SMALL MOLECULE INTERFACES USING ROSETTA By Kristian Wallace Kaufmann Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial
Small Molecules as Rotamers: Generation and Docking in
 Small Molecules as Rotamers: Generation and Docking in ROSETTALIGAND Kristian W. Kaufmann, Jens Meiler Department of Chemistry Vanderbilt University 465 21st Ave. South Nashville, TN 37209 jens.meiler@vanderbilt.edu
Small Molecules as Rotamers: Generation and Docking in ROSETTALIGAND Kristian W. Kaufmann, Jens Meiler Department of Chemistry Vanderbilt University 465 21st Ave. South Nashville, TN 37209 jens.meiler@vanderbilt.edu
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
The protein folding problem consists of two parts:
 Energetics and kinetics of protein folding The protein folding problem consists of two parts: 1)Creating a stable, well-defined structure that is significantly more stable than all other possible structures.
Energetics and kinetics of protein folding The protein folding problem consists of two parts: 1)Creating a stable, well-defined structure that is significantly more stable than all other possible structures.
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University
 Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Alpha-helical Topology and Tertiary Structure Prediction of Globular Proteins Scott R. McAllister Christodoulos A. Floudas Princeton University Department of Chemical Engineering Program of Applied and
Table 1. Crystallographic data collection, phasing and refinement statistics. Native Hg soaked Mn soaked 1 Mn soaked 2
 Table 1. Crystallographic data collection, phasing and refinement statistics Native Hg soaked Mn soaked 1 Mn soaked 2 Data collection Space group P2 1 2 1 2 1 P2 1 2 1 2 1 P2 1 2 1 2 1 P2 1 2 1 2 1 Cell
Table 1. Crystallographic data collection, phasing and refinement statistics Native Hg soaked Mn soaked 1 Mn soaked 2 Data collection Space group P2 1 2 1 2 1 P2 1 2 1 2 1 P2 1 2 1 2 1 P2 1 2 1 2 1 Cell
BSc and MSc Degree Examinations
 Examination Candidate Number: Desk Number: BSc and MSc Degree Examinations 2018-9 Department : BIOLOGY Title of Exam: Molecular Biology and Biochemistry Part I Time Allowed: 1 hour and 30 minutes Marking
Examination Candidate Number: Desk Number: BSc and MSc Degree Examinations 2018-9 Department : BIOLOGY Title of Exam: Molecular Biology and Biochemistry Part I Time Allowed: 1 hour and 30 minutes Marking
Programme Last week s quiz results + Summary Fold recognition Break Exercise: Modelling remote homologues
 Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Programme 8.00-8.20 Last week s quiz results + Summary 8.20-9.00 Fold recognition 9.00-9.15 Break 9.15-11.20 Exercise: Modelling remote homologues 11.20-11.40 Summary & discussion 11.40-12.00 Quiz 1 Feedback
Protein Structure Prediction
 Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Page 1 Protein Structure Prediction Russ B. Altman BMI 214 CS 274 Protein Folding is different from structure prediction --Folding is concerned with the process of taking the 3D shape, usually based on
Protein structure analysis. Risto Laakso 10th January 2005
 Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Protein structure analysis Risto Laakso risto.laakso@hut.fi 10th January 2005 1 1 Summary Various methods of protein structure analysis were examined. Two proteins, 1HLB (Sea cucumber hemoglobin) and 1HLM
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Template Free Protein Structure Modeling Jianlin Cheng, PhD Associate Professor Computer Science Department Informatics Institute University of Missouri, Columbia 2013 Protein Energy Landscape & Free Sampling
Structural Bioinformatics (C3210) Molecular Docking
 Molecular Docking") Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Structural Bioinformatics (C3210) Molecular Docking Molecular Recognition, Molecular Docking Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact
Bioinformatics. Macromolecular structure
 Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Bioinformatics Macromolecular structure Contents Determination of protein structure Structure databases Secondary structure elements (SSE) Tertiary structure Structure analysis Structure alignment Domain
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Bulk behaviour. Alanine. FIG. 1. Chemical structure of the RKLPDA peptide. Numbers on the left mark alpha carbons.
 Bulk behaviour To characterise the conformational behaviour of the peptide, first we looked at the statistics of alpha carbons and the torsion angles. Then they were correlated with positions of positively
Bulk behaviour To characterise the conformational behaviour of the peptide, first we looked at the statistics of alpha carbons and the torsion angles. Then they were correlated with positions of positively
Molecular Dynamics, Monte Carlo and Docking. Lecture 21. Introduction to Bioinformatics MNW2
 Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 If you throw up a stone, it is Physics. If you throw up a stone, it is Physics. If it lands on your head, it is
Molecular Dynamics, Monte Carlo and Docking Lecture 21 Introduction to Bioinformatics MNW2 If you throw up a stone, it is Physics. If you throw up a stone, it is Physics. If it lands on your head, it is
SUPPLEMENTARY FIGURES
 SUPPLEMENTARY FIGURES Supplementary Figure 1 Protein sequence alignment of Vibrionaceae with either a 40-residue insertion or a 44-residue insertion. Identical residues are indicated by red background.
SUPPLEMENTARY FIGURES Supplementary Figure 1 Protein sequence alignment of Vibrionaceae with either a 40-residue insertion or a 44-residue insertion. Identical residues are indicated by red background.
Nature Structural and Molecular Biology: doi: /nsmb Supplementary Figure 1. Definition and assessment of ciap1 constructs.
 Supplementary Figure 1 Definition and assessment of ciap1 constructs. (a) ciap1 constructs used in this study are shown as primary structure schematics with domains colored as in the main text. Mutations
Supplementary Figure 1 Definition and assessment of ciap1 constructs. (a) ciap1 constructs used in this study are shown as primary structure schematics with domains colored as in the main text. Mutations
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation
 Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Polypeptide Folding Using Monte Carlo Sampling, Concerted Rotation, and Continuum Solvation Jakob P. Ulmschneider and William L. Jorgensen J.A.C.S. 2004, 126, 1849-1857 Presented by Laura L. Thomas and
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Table S1. Primers used for the constructions of recombinant GAL1 and λ5 mutants. GAL1-E74A ccgagcagcgggcggctgtctttcc ggaaagacagccgcccgctgctcgg
 SUPPLEMENTAL DATA Table S1. Primers used for the constructions of recombinant GAL1 and λ5 mutants Sense primer (5 to 3 ) Anti-sense primer (5 to 3 ) GAL1 mutants GAL1-E74A ccgagcagcgggcggctgtctttcc ggaaagacagccgcccgctgctcgg
SUPPLEMENTAL DATA Table S1. Primers used for the constructions of recombinant GAL1 and λ5 mutants Sense primer (5 to 3 ) Anti-sense primer (5 to 3 ) GAL1 mutants GAL1-E74A ccgagcagcgggcggctgtctttcc ggaaagacagccgcccgctgctcgg
ALL LECTURES IN SB Introduction
 1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
1. Introduction 2. Molecular Architecture I 3. Molecular Architecture II 4. Molecular Simulation I 5. Molecular Simulation II 6. Bioinformatics I 7. Bioinformatics II 8. Prediction I 9. Prediction II ALL
Supplementary Materials for
 www.sciencesignaling.org/cgi/content/full/5/243/ra68/dc1 Supplementary Materials for Superbinder SH2 Domains Act as Antagonists of Cell Signaling Tomonori Kaneko, Haiming Huang, Xuan Cao, Xing Li, Chengjun
www.sciencesignaling.org/cgi/content/full/5/243/ra68/dc1 Supplementary Materials for Superbinder SH2 Domains Act as Antagonists of Cell Signaling Tomonori Kaneko, Haiming Huang, Xuan Cao, Xing Li, Chengjun
Cryo-EM data collection, refinement and validation statistics
 1 Table S1 Cryo-EM data collection, refinement and validation statistics Data collection and processing CPSF-160 WDR33 (EMDB-7114) (PDB 6BM0) CPSF-160 WDR33 (EMDB-7113) (PDB 6BLY) CPSF-160 WDR33 CPSF-30
1 Table S1 Cryo-EM data collection, refinement and validation statistics Data collection and processing CPSF-160 WDR33 (EMDB-7114) (PDB 6BM0) CPSF-160 WDR33 (EMDB-7113) (PDB 6BLY) CPSF-160 WDR33 CPSF-30
Nature Structural & Molecular Biology: doi: /nsmb Supplementary Figure 1
 Supplementary Figure 1 Identification of the ScDcp2 minimal region interacting with both ScDcp1 and the ScEdc3 LSm domain. Pull-down experiment of untagged ScEdc3 LSm with various ScDcp1-Dcp2-His 6 fragments.
Supplementary Figure 1 Identification of the ScDcp2 minimal region interacting with both ScDcp1 and the ScEdc3 LSm domain. Pull-down experiment of untagged ScEdc3 LSm with various ScDcp1-Dcp2-His 6 fragments.
) P = 1 if exp # " s. + 0 otherwise
 P = 1 if exp # s. + 0 otherwise") Supplementary Material Monte Carlo algorithm procedures. The Monte Carlo conformational search algorithm has been successfully applied by programs dedicated to finding new folds (Jones 2001; Rohl, Strauss,
Supplementary Material Monte Carlo algorithm procedures. The Monte Carlo conformational search algorithm has been successfully applied by programs dedicated to finding new folds (Jones 2001; Rohl, Strauss,
Supplementary Figure 1 Crystal contacts in COP apo structure (PDB code 3S0R)
") Supplementary Figure 1 Crystal contacts in COP apo structure (PDB code 3S0R) Shown in cyan and green are two adjacent tetramers from the crystallographic lattice of COP, forming the only unique inter-tetramer
Supplementary Figure 1 Crystal contacts in COP apo structure (PDB code 3S0R) Shown in cyan and green are two adjacent tetramers from the crystallographic lattice of COP, forming the only unique inter-tetramer
Cks1 CDK1 CDK1 CDK1 CKS1. are ice- lobe. conserved. conserved
 Cks1 d CKS1 Supplementary Figure 1 The -Cks1 crystal lattice. (a) Schematic of the - Cks1 crystal lattice. -Cks1 crystallizes in a lattice that contains c 4 copies of the t - Cks1 dimer in the crystallographic
Cks1 d CKS1 Supplementary Figure 1 The -Cks1 crystal lattice. (a) Schematic of the - Cks1 crystal lattice. -Cks1 crystallizes in a lattice that contains c 4 copies of the t - Cks1 dimer in the crystallographic
CAP 5510 Lecture 3 Protein Structures
 CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
CAP 5510 Lecture 3 Protein Structures Su-Shing Chen Bioinformatics CISE 8/19/2005 Su-Shing Chen, CISE 1 Protein Conformation 8/19/2005 Su-Shing Chen, CISE 2 Protein Conformational Structures Hydrophobicity
Context of the project...3. What is protein design?...3. I The algorithms...3 A Dead-end elimination procedure...4. B Monte-Carlo simulation...
 Laidebeure Stéphane Context of the project...3 What is protein design?...3 I The algorithms...3 A Dead-end elimination procedure...4 B Monte-Carlo simulation...5 II The model...6 A The molecular model...6
Laidebeure Stéphane Context of the project...3 What is protein design?...3 I The algorithms...3 A Dead-end elimination procedure...4 B Monte-Carlo simulation...5 II The model...6 A The molecular model...6
Protein Structure Determination from Pseudocontact Shifts Using ROSETTA
 Supporting Information Protein Structure Determination from Pseudocontact Shifts Using ROSETTA Christophe Schmitz, Robert Vernon, Gottfried Otting, David Baker and Thomas Huber Table S0. Biological Magnetic
Supporting Information Protein Structure Determination from Pseudocontact Shifts Using ROSETTA Christophe Schmitz, Robert Vernon, Gottfried Otting, David Baker and Thomas Huber Table S0. Biological Magnetic
Acta Crystallographica Section D
 Supporting information Acta Crystallographica Section D Volume 70 (2014) Supporting information for article: Structural characterization of the virulence factor Nuclease A from Streptococcus agalactiae
Supporting information Acta Crystallographica Section D Volume 70 (2014) Supporting information for article: Structural characterization of the virulence factor Nuclease A from Streptococcus agalactiae
SUPPLEMENTARY INFORMATION
 Supplementary materials Figure S1 Fusion protein of Sulfolobus solfataricus SRP54 and a signal peptide. a, Expression vector for the fusion protein. The signal peptide of yeast dipeptidyl aminopeptidase
Supplementary materials Figure S1 Fusion protein of Sulfolobus solfataricus SRP54 and a signal peptide. a, Expression vector for the fusion protein. The signal peptide of yeast dipeptidyl aminopeptidase
Homework Problem Set 4 Solutions
 Chemistry 380.37 Dr. Jean M. Standard omework Problem Set 4 Solutions 1. A conformation search is carried out on a system and four low energy stable conformers are obtained. Using the MMFF force field,
Chemistry 380.37 Dr. Jean M. Standard omework Problem Set 4 Solutions 1. A conformation search is carried out on a system and four low energy stable conformers are obtained. Using the MMFF force field,
Homology modeling. Dinesh Gupta ICGEB, New Delhi 1/27/2010 5:59 PM
 Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Homology modeling Dinesh Gupta ICGEB, New Delhi Protein structure prediction Methods: Homology (comparative) modelling Threading Ab-initio Protein Homology modeling Homology modeling is an extrapolation
Supporting Information
 Supporting Information Ottmann et al. 10.1073/pnas.0907587106 Fig. S1. Primary structure alignment of SBT3 with C5 peptidase from Streptococcus pyogenes. The Matchmaker tool in UCSF Chimera (http:// www.cgl.ucsf.edu/chimera)
Supporting Information Ottmann et al. 10.1073/pnas.0907587106 Fig. S1. Primary structure alignment of SBT3 with C5 peptidase from Streptococcus pyogenes. The Matchmaker tool in UCSF Chimera (http:// www.cgl.ucsf.edu/chimera)
Supplementary Information
 Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Supplementary Information Resveratrol Serves as a Protein-Substrate Interaction Stabilizer in Human SIRT1 Activation Xuben Hou,, David Rooklin, Hao Fang *,,, Yingkai Zhang Department of Medicinal Chemistry
Supplementary Materials for
 advances.sciencemag.org/cgi/content/full/3/4/e1600663/dc1 Supplementary Materials for A dynamic hydrophobic core orchestrates allostery in protein kinases Jonggul Kim, Lalima G. Ahuja, Fa-An Chao, Youlin
advances.sciencemag.org/cgi/content/full/3/4/e1600663/dc1 Supplementary Materials for A dynamic hydrophobic core orchestrates allostery in protein kinases Jonggul Kim, Lalima G. Ahuja, Fa-An Chao, Youlin
Template Free Protein Structure Modeling Jianlin Cheng, PhD
 Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Template Free Protein Structure Modeling Jianlin Cheng, PhD Professor Department of EECS Informatics Institute University of Missouri, Columbia 2018 Protein Energy Landscape & Free Sampling http://pubs.acs.org/subscribe/archive/mdd/v03/i09/html/willis.html
Nature Structural & Molecular Biology doi: /nsmb Supplementary Figure 1. CRBN binding assay with thalidomide enantiomers.
 Supplementary Figure 1 CRBN binding assay with thalidomide enantiomers. (a) Competitive elution assay using thalidomide-immobilized beads coupled with racemic thalidomide. Beads were washed three times
Supplementary Figure 1 CRBN binding assay with thalidomide enantiomers. (a) Competitive elution assay using thalidomide-immobilized beads coupled with racemic thalidomide. Beads were washed three times
Modeling for 3D structure prediction
 Modeling for 3D structure prediction What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing
Modeling for 3D structure prediction What is a predicted structure? A structure that is constructed using as the sole source of information data obtained from computer based data-mining. However, mixing
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION
 THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
THE TANGO ALGORITHM: SECONDARY STRUCTURE PROPENSITIES, STATISTICAL MECHANICS APPROXIMATION AND CALIBRATION Calculation of turn and beta intrinsic propensities. A statistical analysis of a protein structure
DISCRETE TUTORIAL. Agustí Emperador. Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING:
 DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
DISCRETE TUTORIAL Agustí Emperador Institute for Research in Biomedicine, Barcelona APPLICATION OF DISCRETE TO FLEXIBLE PROTEIN-PROTEIN DOCKING: STRUCTURAL REFINEMENT OF DOCKING CONFORMATIONS Emperador
Protein Structure. W. M. Grogan, Ph.D. OBJECTIVES
 Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Protein Structure W. M. Grogan, Ph.D. OBJECTIVES 1. Describe the structure and characteristic properties of typical proteins. 2. List and describe the four levels of structure found in proteins. 3. Relate
Abstract. Introduction
 In silico protein design: the implementation of Dead-End Elimination algorithm CS 273 Spring 2005: Project Report Tyrone Anderson 2, Yu Bai1 3, and Caroline E. Moore-Kochlacs 2 1 Biophysics program, 2
In silico protein design: the implementation of Dead-End Elimination algorithm CS 273 Spring 2005: Project Report Tyrone Anderson 2, Yu Bai1 3, and Caroline E. Moore-Kochlacs 2 1 Biophysics program, 2
Docking. GBCB 5874: Problem Solving in GBCB
 Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Docking Benzamidine Docking to Trypsin Relationship to Drug Design Ligand-based design QSAR Pharmacophore modeling Can be done without 3-D structure of protein Receptor/Structure-based design Molecular
Ab-initio protein structure prediction
 Ab-initio protein structure prediction Jaroslaw Pillardy Computational Biology Service Unit Cornell Theory Center, Cornell University Ithaca, NY USA Methods for predicting protein structure 1. Homology
Ab-initio protein structure prediction Jaroslaw Pillardy Computational Biology Service Unit Cornell Theory Center, Cornell University Ithaca, NY USA Methods for predicting protein structure 1. Homology
Serine-7 but not serine-5 phosphorylation primes RNA polymerase II CTD for P-TEFb recognition
 Supplementary Information to Serine-7 but not serine-5 phosphorylation primes RNA polymerase II CTD for P-TEFb recognition Nadine Czudnochowski 1,2, *, Christian A. Bösken 1, * & Matthias Geyer 1 1 Max-Planck-Institut
Supplementary Information to Serine-7 but not serine-5 phosphorylation primes RNA polymerase II CTD for P-TEFb recognition Nadine Czudnochowski 1,2, *, Christian A. Bösken 1, * & Matthias Geyer 1 1 Max-Planck-Institut
From Amino Acids to Proteins - in 4 Easy Steps
 From Amino Acids to Proteins - in 4 Easy Steps Although protein structure appears to be overwhelmingly complex, you can provide your students with a basic understanding of how proteins fold by focusing
From Amino Acids to Proteins - in 4 Easy Steps Although protein structure appears to be overwhelmingly complex, you can provide your students with a basic understanding of how proteins fold by focusing
SUPPLEMENTARY INFORMATION
 doi:10.1038/nature12045 Supplementary Table 1 Data collection and refinement statistics. Native Pt-SAD X-ray source SSRF BL17U SPring-8 BL41XU Wavelength (Å) 0.97947 1.07171 Space group P2 1 2 1 2 1 P2
doi:10.1038/nature12045 Supplementary Table 1 Data collection and refinement statistics. Native Pt-SAD X-ray source SSRF BL17U SPring-8 BL41XU Wavelength (Å) 0.97947 1.07171 Space group P2 1 2 1 2 1 P2
Supplementary Figure 1. Biochemical and sequence alignment analyses the
 Supplementary Figure 1. Biochemical and sequence alignment analyses the interaction of OPTN and TBK1. (a) Analytical gel filtration chromatography analysis of the interaction between TBK1 CTD and OPTN(1-119).
Supplementary Figure 1. Biochemical and sequence alignment analyses the interaction of OPTN and TBK1. (a) Analytical gel filtration chromatography analysis of the interaction between TBK1 CTD and OPTN(1-119).
Supplementary Materials
 Sub-Angstrom Accuracy in Protein Loop Reconstruction by Robotics-Inspired Conformational Sampling Daniel J. Mandell, Evangelos A. Coutsias and Tanja Kortemme Supplementary Materials I. Supplementary Methods
Sub-Angstrom Accuracy in Protein Loop Reconstruction by Robotics-Inspired Conformational Sampling Daniel J. Mandell, Evangelos A. Coutsias and Tanja Kortemme Supplementary Materials I. Supplementary Methods
Nature Structural and Molecular Biology: doi: /nsmb Supplementary Figure 1
 Supplementary Figure 1 Quantitation of the binding of pro53 peptide to sorla Vps10p measured by the AP reporter assay. The graph shows tracings of the typical chromogenic AP reaction observed with AP-pro53
Supplementary Figure 1 Quantitation of the binding of pro53 peptide to sorla Vps10p measured by the AP reporter assay. The graph shows tracings of the typical chromogenic AP reaction observed with AP-pro53
Computational Molecular Modeling
 Computational Molecular Modeling Lecture 1: Structure Models, Properties Chandrajit Bajaj Today s Outline Intro to atoms, bonds, structure, biomolecules, Geometry of Proteins, Nucleic Acids, Ribosomes,
Computational Molecular Modeling Lecture 1: Structure Models, Properties Chandrajit Bajaj Today s Outline Intro to atoms, bonds, structure, biomolecules, Geometry of Proteins, Nucleic Acids, Ribosomes,
Procheck output. Bond angles (Procheck) Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.
 Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics.") Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
Structure verification and validation Bond lengths (Procheck) Introduction to Bioinformatics Iosif Vaisman Email: ivaisman@gmu.edu ----------------------------------------------------------------- Bond
SUPPLEMENTARY INFORMATION
 doi:1.138/nature1737 Supplementary Table 1 variant Description FSEC - 2B12 a FSEC - 6A1 a K d (leucine) c Leucine uptake e K (wild-type like) K (Y18F) K (TS) K (TSY) K288A mutant, lipid facing side chain
doi:1.138/nature1737 Supplementary Table 1 variant Description FSEC - 2B12 a FSEC - 6A1 a K d (leucine) c Leucine uptake e K (wild-type like) K (Y18F) K (TS) K (TSY) K288A mutant, lipid facing side chain
SUPPLEMENTARY INFORMATION
 doi:10.1038/nature11085 Supplementary Tables: Supplementary Table 1. Summary of crystallographic and structure refinement data Structure BRIL-NOP receptor Data collection Number of crystals 23 Space group
doi:10.1038/nature11085 Supplementary Tables: Supplementary Table 1. Summary of crystallographic and structure refinement data Structure BRIL-NOP receptor Data collection Number of crystals 23 Space group
SUPPLEMENTARY INFORMATION
 Supplementary Results DNA binding property of the SRA domain was examined by an electrophoresis mobility shift assay (EMSA) using synthesized 12-bp oligonucleotide duplexes containing unmodified, hemi-methylated,
Supplementary Results DNA binding property of the SRA domain was examined by an electrophoresis mobility shift assay (EMSA) using synthesized 12-bp oligonucleotide duplexes containing unmodified, hemi-methylated,
Protein Folding & Stability. Lecture 11: Margaret A. Daugherty. Fall How do we go from an unfolded polypeptide chain to a
 Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2004 How do we go from an unfolded polypeptide chain to a compact folded protein? (Folding of thioredoxin, F. Richards) Structure - Function
Lecture 11: Protein Folding & Stability Margaret A. Daugherty Fall 2004 How do we go from an unfolded polypeptide chain to a compact folded protein? (Folding of thioredoxin, F. Richards) Structure - Function
SUPPLEMENTARY INFORMATION
 Figure S1. Secondary structure of CAP (in the camp 2 -bound state) 10. α-helices are shown as cylinders and β- strands as arrows. Labeling of secondary structure is indicated. CDB, DBD and the hinge are
Figure S1. Secondary structure of CAP (in the camp 2 -bound state) 10. α-helices are shown as cylinders and β- strands as arrows. Labeling of secondary structure is indicated. CDB, DBD and the hinge are
Supplemental Information. The Mitochondrial Fission Receptor MiD51. Requires ADP as a Cofactor
 Structure, Volume 22 Supplemental Information The Mitochondrial Fission Receptor MiD51 Requires ADP as a Cofactor Oliver C. Losón, Raymond Liu, Michael E. Rome, Shuxia Meng, Jens T. Kaiser, Shu-ou Shan,
Structure, Volume 22 Supplemental Information The Mitochondrial Fission Receptor MiD51 Requires ADP as a Cofactor Oliver C. Losón, Raymond Liu, Michael E. Rome, Shuxia Meng, Jens T. Kaiser, Shu-ou Shan,
Structure and RNA-binding properties. of the Not1 Not2 Not5 module of the yeast Ccr4 Not complex
 Structure and RNA-binding properties of the Not1 Not2 Not5 module of the yeast Ccr4 Not complex Varun Bhaskar 1, Vladimir Roudko 2,3, Jerome Basquin 1, Kundan Sharma 4, Henning Urlaub 4, Bertrand Seraphin
Structure and RNA-binding properties of the Not1 Not2 Not5 module of the yeast Ccr4 Not complex Varun Bhaskar 1, Vladimir Roudko 2,3, Jerome Basquin 1, Kundan Sharma 4, Henning Urlaub 4, Bertrand Seraphin
Protein Modeling Methods. Knowledge. Protein Modeling Methods. Fold Recognition. Knowledge-based methods. Introduction to Bioinformatics
 Protein Modeling Methods Introduction to Bioinformatics Iosif Vaisman Ab initio methods Energy-based methods Knowledge-based methods Email: ivaisman@gmu.edu Protein Modeling Methods Ab initio methods:
Protein Modeling Methods Introduction to Bioinformatics Iosif Vaisman Ab initio methods Energy-based methods Knowledge-based methods Email: ivaisman@gmu.edu Protein Modeling Methods Ab initio methods:
BCMP 201 Protein biochemistry
 BCMP 201 Protein biochemistry BCMP 201 Protein biochemistry with emphasis on the interrelated roles of protein structure, catalytic activity, and macromolecular interactions in biological processes. The
BCMP 201 Protein biochemistry BCMP 201 Protein biochemistry with emphasis on the interrelated roles of protein structure, catalytic activity, and macromolecular interactions in biological processes. The
April, The energy functions include:
 REDUX A collection of Python scripts for torsion angle Monte Carlo protein molecular simulations and analysis The program is based on unified residue peptide model and is designed for more efficient exploration
REDUX A collection of Python scripts for torsion angle Monte Carlo protein molecular simulations and analysis The program is based on unified residue peptide model and is designed for more efficient exploration
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability
 and elementary statistical mechanics. Contributions to protein stability") Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Lecture 2 and 3: Review of forces (ctd.) and elementary statistical mechanics. Contributions to protein stability Part I. Review of forces Covalent bonds Non-covalent Interactions: Van der Waals Interactions
Sensitive NMR Approach for Determining the Binding Mode of Tightly Binding Ligand Molecules to Protein Targets
 Supporting information Sensitive NMR Approach for Determining the Binding Mode of Tightly Binding Ligand Molecules to Protein Targets Wan-Na Chen, Christoph Nitsche, Kala Bharath Pilla, Bim Graham, Thomas
Supporting information Sensitive NMR Approach for Determining the Binding Mode of Tightly Binding Ligand Molecules to Protein Targets Wan-Na Chen, Christoph Nitsche, Kala Bharath Pilla, Bim Graham, Thomas
Tools for Cryo-EM Map Fitting. Paul Emsley MRC Laboratory of Molecular Biology
 Tools for Cryo-EM Map Fitting Paul Emsley MRC Laboratory of Molecular Biology April 2017 Cryo-EM model-building typically need to move more atoms that one does for crystallography the maps are lower resolution
Tools for Cryo-EM Map Fitting Paul Emsley MRC Laboratory of Molecular Biology April 2017 Cryo-EM model-building typically need to move more atoms that one does for crystallography the maps are lower resolution
Syllabus BINF Computational Biology Core Course
 Course Description Syllabus BINF 701-702 Computational Biology Core Course BINF 701/702 is the Computational Biology core course developed at the KU Center for Computational Biology. The course is designed
Course Description Syllabus BINF 701-702 Computational Biology Core Course BINF 701/702 is the Computational Biology core course developed at the KU Center for Computational Biology. The course is designed
Lec.1 Chemistry Of Water
 Lec.1 Chemistry Of Water Biochemistry & Medicine Biochemistry can be defined as the science concerned with the chemical basis of life. Biochemistry can be described as the science concerned with the chemical
Lec.1 Chemistry Of Water Biochemistry & Medicine Biochemistry can be defined as the science concerned with the chemical basis of life. Biochemistry can be described as the science concerned with the chemical
SUPPLEMENTARY INFORMATION
 SUPPLEMENTARY INFORMATION doi:10.1038/nature11539 Supplementary Figure 1 Schematic representation of plant (A) and mammalian (B) P 2B -ATPase domain organization. Actuator (A-), nucleotide binding (N-),
SUPPLEMENTARY INFORMATION doi:10.1038/nature11539 Supplementary Figure 1 Schematic representation of plant (A) and mammalian (B) P 2B -ATPase domain organization. Actuator (A-), nucleotide binding (N-),
Molecular Modeling. Prediction of Protein 3D Structure from Sequence. Vimalkumar Velayudhan. May 21, 2007
 Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
Molecular Modeling Prediction of Protein 3D Structure from Sequence Vimalkumar Velayudhan Jain Institute of Vocational and Advanced Studies May 21, 2007 Vimalkumar Velayudhan Molecular Modeling 1/23 Outline
A: Up regulated proteins B: Down regulated proteins. Susceptible Resistant Susceptible Resistant Resistant Susceptible
 Supplementary Materials: Identification of Biomarkers for Resistance to Fusarium oxysporum f. sp. cubense Infection and in Silico Studies in Musa paradisiaca Cultivar Puttabale through Proteomic Approach
Supplementary Materials: Identification of Biomarkers for Resistance to Fusarium oxysporum f. sp. cubense Infection and in Silico Studies in Musa paradisiaca Cultivar Puttabale through Proteomic Approach
Structure, mechanism and ensemble formation of the Alkylhydroperoxide Reductase subunits. AhpC and AhpF from Escherichia coli
 Structure, mechanism and ensemble formation of the Alkylhydroperoxide Reductase subunits AhpC and AhpF from Escherichia coli Phat Vinh Dip 1,#, Neelagandan Kamariah 2,#, Malathy Sony Subramanian Manimekalai
Structure, mechanism and ensemble formation of the Alkylhydroperoxide Reductase subunits AhpC and AhpF from Escherichia coli Phat Vinh Dip 1,#, Neelagandan Kamariah 2,#, Malathy Sony Subramanian Manimekalai
SUPPLEMENTARY INFORMATION
 doi:10.1038/nature11054 Supplementary Fig. 1 Sequence alignment of Na v Rh with NaChBac, Na v Ab, and eukaryotic Na v and Ca v homologs. Secondary structural elements of Na v Rh are indicated above the
doi:10.1038/nature11054 Supplementary Fig. 1 Sequence alignment of Na v Rh with NaChBac, Na v Ab, and eukaryotic Na v and Ca v homologs. Secondary structural elements of Na v Rh are indicated above the
Supplementary Figures
 1 Supplementary Figures Supplementary Figure 1 Type I FGFR1 inhibitors (a) Chemical structures of a pyrazolylaminopyrimidine inhibitor (henceforth referred to as PAPI; PDB-code of the FGFR1-PAPI complex:
1 Supplementary Figures Supplementary Figure 1 Type I FGFR1 inhibitors (a) Chemical structures of a pyrazolylaminopyrimidine inhibitor (henceforth referred to as PAPI; PDB-code of the FGFR1-PAPI complex:
Today s lecture. Molecular Mechanics and docking. Lecture 22. Introduction to Bioinformatics Docking - ZDOCK. Protein-protein docking
 C N F O N G A V B O N F O M A C S V U Molecular Mechanics and docking Lecture 22 ntroduction to Bioinformatics 2007 oday s lecture 1. Protein interaction and docking a) Zdock method 2. Molecular motion
C N F O N G A V B O N F O M A C S V U Molecular Mechanics and docking Lecture 22 ntroduction to Bioinformatics 2007 oday s lecture 1. Protein interaction and docking a) Zdock method 2. Molecular motion
Protein Structure Basics
 Protein Structure Basics Presented by Alison Fraser, Christine Lee, Pradhuman Jhala, Corban Rivera Importance of Proteins Muscle structure depends on protein-protein interactions Transport across membranes
Protein Structure Basics Presented by Alison Fraser, Christine Lee, Pradhuman Jhala, Corban Rivera Importance of Proteins Muscle structure depends on protein-protein interactions Transport across membranes
SUPPLEMENTARY INFORMATION
 SUPPLEMENTARY INFORMATION doi:10.1038/nature11524 Supplementary discussion Functional analysis of the sugar porter family (SP) signature motifs. As seen in Fig. 5c, single point mutation of the conserved
SUPPLEMENTARY INFORMATION doi:10.1038/nature11524 Supplementary discussion Functional analysis of the sugar porter family (SP) signature motifs. As seen in Fig. 5c, single point mutation of the conserved
Ranjit P. Bahadur Assistant Professor Department of Biotechnology Indian Institute of Technology Kharagpur, India. 1 st November, 2013
 Hydration of protein-rna recognition sites Ranjit P. Bahadur Assistant Professor Department of Biotechnology Indian Institute of Technology Kharagpur, India 1 st November, 2013 Central Dogma of life DNA
Hydration of protein-rna recognition sites Ranjit P. Bahadur Assistant Professor Department of Biotechnology Indian Institute of Technology Kharagpur, India 1 st November, 2013 Central Dogma of life DNA
SUPPLEMENTARY INFORMATION
 doi:10.1038/nature10955 Supplementary Figures Supplementary Figure 1. Electron-density maps and crystallographic dimer structures of the motor domain. (a f) Stereo views of the final electron-density maps
doi:10.1038/nature10955 Supplementary Figures Supplementary Figure 1. Electron-density maps and crystallographic dimer structures of the motor domain. (a f) Stereo views of the final electron-density maps
Rosetta Density-fitting Tutorial Frank DiMaio, January 2010
 Rosetta Density-fitting Tutorial Frank DiMaio, January 2010 This tutorial is intended to guide users to several different ways Rosetta may be used to solve various problems in structure fitting into low-resolution
Rosetta Density-fitting Tutorial Frank DiMaio, January 2010 This tutorial is intended to guide users to several different ways Rosetta may be used to solve various problems in structure fitting into low-resolution
Comparing crystal structure of M.HhaI with and without DNA1, 2 (PDBID:1hmy and PDBID:2hmy),
,") Supporting Information 1. Constructing the starting structure Comparing crystal structure of M.HhaI with and without DNA1, 2 (PDBID:1hmy and PDBID:2hmy), we find that: the RMSD of overall structure and
Supporting Information 1. Constructing the starting structure Comparing crystal structure of M.HhaI with and without DNA1, 2 (PDBID:1hmy and PDBID:2hmy), we find that: the RMSD of overall structure and
Why Do Protein Structures Recur?
 Why Do Protein Structures Recur? Dartmouth Computer Science Technical Report TR2015-775 Rebecca Leong, Gevorg Grigoryan, PhD May 28, 2015 Abstract Protein tertiary structures exhibit an observable degeneracy
Why Do Protein Structures Recur? Dartmouth Computer Science Technical Report TR2015-775 Rebecca Leong, Gevorg Grigoryan, PhD May 28, 2015 Abstract Protein tertiary structures exhibit an observable degeneracy