Tutorial 1: Setting up your Skyline document
|
|
|
- Lorena Goodman
- 5 years ago
- Views:
Transcription
1 Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region Change Location Formats English (United States). Open Skyline and select Blank Document at Start Page. Open the Peptide Settings (under the menu Settings ) and go through all tabs to adjust these settings to your current project. Caution! Make sure that you understand every single setting that you set here. Digestion: Enzyme: Select the proteolytic specificity of the enzyme that was used to digest the proteins in your sample. We usually use trypsin, which cleaves C-terminally of lysine and arginine, but not if these are followed by proline: select Trypsin [KR P] Max missed cleavages: Select the number of missed cleavages that you would like to tolerate. We usually work with fully tryptic peptides only: select 0 Background proteome: Here you can build a background proteome from a protein fasta file using the digestion settings defined above. Alternatively, you can directly add an already in-silico digested proteome file (format.protdb). To generate a background proteome according to the digestion settings above click Add (In Background proteome) In the Name field, enter TubercuList_v2-6. Click on Create Define the path ( Skyline folder on USB key) of your new background proteome and enter the file name TubercuList_v2-6 and click Save in the lower right corner of the window. Add File Select the TubercuList_v2-6.fasta file provided in the folder Tutorial-1_Settings. The generated proteome file should contain 3982 proteins. Confirm by clicking OK, then close the Peptide Settings by again clicking OK. Now the background proteome is being generated. You can follow the progress in the bottom lane of the Skyline interface. Depending on the size of the file this may take a few seconds up to several minutes. Tip! We suggest to work with a general folder called Skyline where you save all project-independent spectral libraries, background proteomes, irt databases etc. that you create within Skyline. This helps to find these important files later on when you have many different Skyline projects. Prediction: Re-open the Peptide Settings and select the Prediction tab. Retention time predictor: Select a retention time predictor and use it to create scheduled SRM methods and to support data analysis. We typically work with the SSRCalc predictor or with a custom irt predictor. We will create a scheduled SRM method in a later tutorial, but for now no retention time predictor needs to be activated ("None ). Use measured retention times when present: This option allows working with actually measured retention times (instead of predicted) for those peptides for which this information is included in the Skyline file. For non-scheduled SRM this tab is not functional and we leave the default (activated). Time window: Enter the time window, which you would like to use for your scheduled measurements. For non-scheduled SRM this tab is not functional and we leave the default (2 minutes). 1
2 Drift Time predictor: This option relates to drift times from ion mobility separations which we will not cover in the tutorial. We do not need this option and leave the default None. Use spectral library drift times when present: We leave the default (deactivated). Resolving power: We leave the default (deactivated). Filter: Change to the Filter tab. Enter the peptide amino acid length that you would like to work with. We work with: Min length: 7 Max length: 25 Exclude N-terminal AAs: The N-terminus of a protein might be post-translationally processed (modified and/or cleaved) and might thus not be suited for protein quantification. When working with a synthetic peptide library we typically do not exclude N-terminal amino acids. So herein please enter 0. Exclude potentially ragged ends: Ragged ends are peptides with KK, RR, RK or KR sequences at one or both ends. Such peptides might not be fully cleaved and hence may not be suited for quantification. However, if no alternative peptides are available one might rather quantify with a ragged end peptide then not at all. When working with a synthetic peptide library we typically do not select this option. So herein please leave it deactivated. Exclude peptides containing: Cys, Met, His: These residues are prone to modifications, such as oxidation. NXT/NXS: This is a glycosylation motif. RP/KP: Lys or Arg followed by Pro are sometimes cleaved by trypsin. When working with a synthetic peptide library we typically do not select these options. So herein please leave them deactivated. Auto-select all matching peptides: Needs to be activated if peptides should be automatically selected from a background proteome or a spectral library file. Activate. Library: Change to the Library tab. Libraries: Here you can insert or build spectral libraries containing MS2 spectra. Spectral libraries can be downloaded from public sources or built within Skyline from your own data. Several libraries can be selected at the same time. Be aware that the order in the list matters: the higher up in the list, the higher the priority in case there is an MS2 spectrum for the same peptide in more than one library. For a more detailed library generation protocol please refer to Tutorial 3 - Library. For now we will work with a comprehensive public Mtb library ( which was generated on a QTrap instrument using synthetic peptides (for more information see O. T. Schubert et al., Cell Host Microbe, 2013, 13, 602). This library can be found on your USB key in the project-independent Skyline folder and is called Mtb_DirtyPeptides_QT_filtered_cons.sptxt. Click Edit list Add Name: Mtb_proteome_library Browse Select the sptxt file ( Mtb_DirtyPeptides_QT_filtered_cons.sptxt ) in the folder called Skyline Click OK twice. Activate the new Mtb_proteome_library by checking the box. Confirm by clicking OK. Skyline will now read this library, which takes a few seconds. Again you can follow the progress in the bottom lane of the Skyline interface. Re-open the Peptide Settings and select again the Library tab. Pick peptide matching: Select if peptides should be automatically selected according to the filter settings (defined in the Filter tab before) or according to the library settings defined below. 2

 - TFRatio (SpectraST score for spectrum quality) - Total intensity (sum of all peaks in the MS2 spectrum) For this study we do not need this setting.")
3 For this study we pre-selected the target peptides and we do not need this setting. We keep the default setting ( Library ). Rank peptides by: Here you can define a ranking of all peptides available for a given protein in the library. Depending on which tool you used to create your library, you can select between: - Picked intensity (sum of selected peak intensities in the spectrum for a given peptide) - Spectrum count (sum of all spectra for a given peptide) - TFRatio (SpectraST score for spectrum quality) - Total intensity (sum of all peaks in the MS2 spectrum) For this study we do not need this setting. Limit peptides per protein: Limits the number of automatically selected peptides per protein from the library. For this study we do not need this setting. Modifications: Change to the Modifications tab. Structural modifications: Structural modifications concern chemical modifications of peptides. They can either be static (always present) or variable (sometimes present, sometimes not). By default Carbamidomethyl (C) is activated, which comes from the reduction and alkylation step during sample preparation to avoid formation of disulphide bonds between cysteine residues. Keep this modification. Max variable mods and Max neutral losses: Select the maximal number of variable modifications and neutral losses according to your project. Leave the default setting (3 variable modifications and 1 neutral loss). Isotope label type: Here you can define the isotope label type you plan to work with. Leave the default heavy as label type. Isotope modifications: Here you can define the chemical composition of your isotopic modification. For our case study we will work with stable isotope labelled synthetic reference peptides of which the C-terminal Arg or Lys are heavy labelled. Activate this modification by going to Edit list Add Name: select the following two modifications from the drop-down menu one after another: - Label:13C(6)15N(2) (C-term K) - Label:13C(6)15N(4) (C-term R) After they have been added (by Clicking OK in Edit Isotope Modifications ), then activate both label types by checking the boxes in the Isotope modifications window. Internal standard type: Define which labelling state should be your internal standard Our internal standard will be the spiked-in heavy reference peptides and we thus keep the default setting heavy. 3



4 Quantification: The quantification tab specifies settings required for absolute quantification. We do not use these options for the tutorial and do not change any settings here. Finally: Click OK to confirm all peptide settings. Overview Peptide Settings: 4


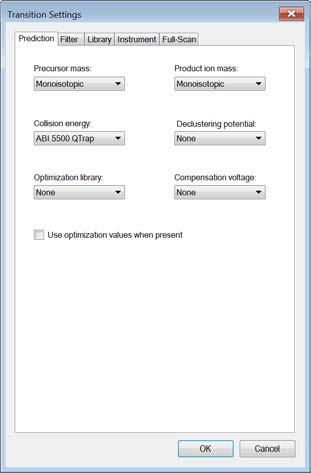
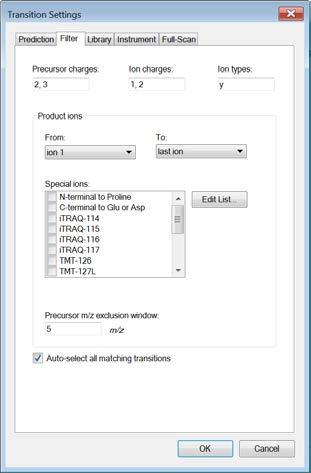
5 Now open the Transition Settings (under the menu Settings ) and go through all tabs to adjust the settings to your current project. Caution! Make again sure that you understand every single setting that you set here. Prediction: Open the Transition Settings and go to the Prediction tab. Precursor mass and Product ion mass: Here you define the basic MS parameters of your data. For both, precursor and product ion mass, we work with the monoisotopic mass. Collision energy: Define how the collision energy that you are going to use should be calculated. Ideally, the optimal collision energy equation is determined for each instrument and we will discuss how to do this in Tutorial 4 - Parameters. If you do not have any information available, use the recommendations by the vendor or by Skyline. Select the ABI 5500 QTrap collision energy. To see the underlying formula of the collision energy calculation select Edit current, go back with Cancel ). Declustering potential: Define the declustering potential that should be applied to your sample when it is injected into the mass spectrometer. This parameter will as well be discussed in Tutorial 4 - Parameters. We do not set a declustering potential. Optimization library: If you have carried out a collision energy optimisation experiment within Skyline, you can directly apply the optimised values. We do not set an optimization library. Use optimisation values when present: We do not activate this option for now. Compensation voltage: We do not activate this option. Filter: Change to the Filter tab. Precursor charges: Define which precursor charge states you would like to consider for your SRM measurements. We usually consider doubly and triply charged peptides, enter 2, 3. Product ion charges: Define which product ion charge states you would like to consider for your SRM measurements. We usually consider singly and doubly charged product ions, enter 1, 2. Ion types: Define which product ion types you would like to consider for your SRM measurement (you can define a,b,c,x,y,z and p ions, p stands for precursor). 5
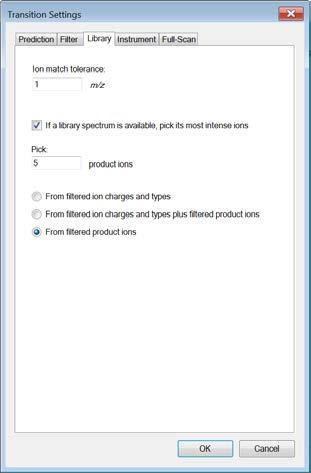
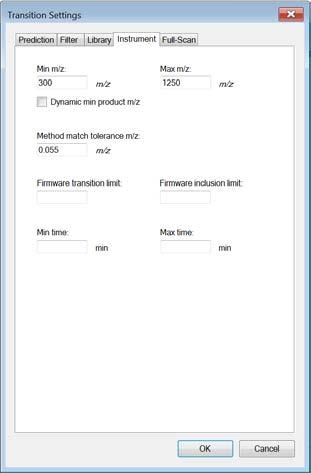
6 In this study we only focus on y-ions, so enter y. Note: We do not consider b-ions in this case study, because compared to y- ions they tend to be less intense and less conserved between instruments. Product ions: In this window you can define a filter to automatically select transitions for all peptides in your Skyline document. For example, to filter for the whole y-ion series, ranging from the first to the last y-ion of a peptide, enter: From: ion 1 and To: last ion. Further criteria to automatically include typically very intense transitions, such as N-terminal to Pro or C-terminal to Glu or Asp, can be selected or customdefined. In our case study we want to consider all y-ions, hence we select From: ion 1 To: last ion. Special ions: Uncheck N-terminal to Proline, as we would like to select transitions based on a library spectrum only. Precursor m/z exclusion window: Here you can exclude a certain mass window around the precursor m/z value for transition selection. In this m/z range transitions are typically very noisy and therefore not suited for identification and quantification. Set the precursor exclusion window to 5 Th (which is +/- 2.5 Th around the precursor). Auto-select all matching transitions: Needs to be activated if transitions should be automatically selected for all peptides based either on the filter settings or the library. Activate. Library: Change to the Library tab. Ion match tolerance: Here you can define the mass accuracy you would like to tolerate for the selection of fragment ions from your spectral library. This depends on the instrument type that was used to acquire the library spectra. Lower values help to get a more specific peak assignment of the spectra, but if the instrument did not have this accuracy you will lose your peaks. Our library has been acquired on a QTrap instrument for which we usually use a mass tolerance of 1.0 m/z. If a library spectrum is available, pick its most intense ions: Here you can specify if the library should be used to guide transition selection and how many transitions per precursor should be considered. Activate and enter 5 product ions From filtered : Here you can choose which settings from the Filter tab you would like to consider for the automatic transition selection. We select From filtered product ions. Instrument: Change to the Instrument tab. Specify the m/z range of your instrument Min m/z: 300 m/z to Max m/z: 1250 m/z (maximum value for QTrap 5500) Dynamic min product m/z: This function is only useful for chromatograms from LTQ MS2 data. Do not activate this option. Method match tolerance: Here you can define the tolerance in mass difference between the theoretical masses calculated by Skyline and the masses given in an imported raw file. If for a given raw file the corresponding method file has been generated with Skyline beforehand, the difference between Skyline and raw file masses will be 0. However, if other mass calculators have been used for method generation and/or due to rounding issues, minor mass differences can occur. We leave the default setting of m/z. Firmware transition limit: In case your instrument of choice has a maximal transition number limit you can enter this here. We do not enter a limit. Firmware inclusion limit: In case you use Skyline for inclusion list generation (e.g. for PRM) you can specify the list limit here. We do not enter a limit. 6
7 Min time, Max time: Here you can limit the part of the HPLC gradient to extract data from (e.g. in SWATH experiments). We leave these settings undefined. Full-Scan: This tab is not needed for SRM measurements, but for MS1 and/or MS2 filtering analysis. For our case study we do not activate this tab Finally: Click OK to confirm all transition settings. Save the Skyline document to folder Tutorial-1_Settings with the name: TarProCourse_Tut1_Settings.sky. 7
8 Overview Transition Settings: 8
9 Exercises 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? 2. What is the chemical formula of the loss occurring in pyro-glutamate formation on N- terminal glutamic acid? 3. How many proteins are in the fasta file and how many are in the Skyline background proteome? What could be the reason for the discrepancy? 4. Chymotrypsin cleaves C-terminal to which amino acids? 5. What is the slope for triply charged precursor ions defined in the collision energy equation of the ABI 5500 QTrap Instrument? 6. Which transition settings do you need to apply to automatically select the complete doublycharged y-ion series of a peptide between 500 and 1000 Da? Caution! Do not save any of the changes you made for the exercises. We would like to thank SystemsX for supporting the Zurich Targeted Proteomics Course
SRM assay generation and data analysis in Skyline
 in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
TUTORIAL EXERCISES WITH ANSWERS
 TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
Skyline Small Molecule Targets
 Skyline Small Molecule Targets The Skyline Targeted Proteomics Environment provides informative visual displays of the raw mass spectrometer data you import into your Skyline documents. Originally developed
Skyline Small Molecule Targets The Skyline Targeted Proteomics Environment provides informative visual displays of the raw mass spectrometer data you import into your Skyline documents. Originally developed
Tutorial 2: Analysis of DIA data in Skyline
 Tutorial 2: Analysis of DIA data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantitation using
Tutorial 2: Analysis of DIA data in Skyline In this tutorial we will learn how to use Skyline to perform targeted post-acquisition analysis for peptide and inferred protein detection and quantitation using
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
MassHunter Software Overview
 MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
MassHunter Software Overview 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks A
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics
 DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
Analyst Software. Peptide and Protein Quantitation Tutorial
 This document is provided to customers who have purchased AB Sciex equipment to use in the operation of such AB Sciex equipment. This document is copyright protected and any reproduction of this document
This document is provided to customers who have purchased AB Sciex equipment to use in the operation of such AB Sciex equipment. This document is copyright protected and any reproduction of this document
Information Dependent Acquisition (IDA) 1
 1") Information Dependent Acquisition (IDA) Information Dependent Acquisition (IDA) enables on the fly acquisition of MS/MS spectra during a chromatographic run. Analyst Software IDA is optimized to generate
Information Dependent Acquisition (IDA) Information Dependent Acquisition (IDA) enables on the fly acquisition of MS/MS spectra during a chromatographic run. Analyst Software IDA is optimized to generate
Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent
 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent") Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent concentrations of PcTS (100 µm, blue; 500 µm, green; 1.5 mm,
Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent concentrations of PcTS (100 µm, blue; 500 µm, green; 1.5 mm,
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 *
 Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
Proteomics. November 13, 2007
 Proteomics November 13, 2007 Acknowledgement Slides presented here have been borrowed from presentations by : Dr. Mark A. Knepper (LKEM, NHLBI, NIH) Dr. Nathan Edwards (Center for Bioinformatics and Computational
Proteomics November 13, 2007 Acknowledgement Slides presented here have been borrowed from presentations by : Dr. Mark A. Knepper (LKEM, NHLBI, NIH) Dr. Nathan Edwards (Center for Bioinformatics and Computational
1. Prepare the MALDI sample plate by spotting an angiotensin standard and the test sample(s).
.") Analysis of a Peptide Sequence from a Proteolytic Digest by MALDI-TOF Post-Source Decay (PSD) and Collision-Induced Dissociation (CID) Standard Operating Procedure Purpose: The following procedure may
Analysis of a Peptide Sequence from a Proteolytic Digest by MALDI-TOF Post-Source Decay (PSD) and Collision-Induced Dissociation (CID) Standard Operating Procedure Purpose: The following procedure may
Protein Identification Using Tandem Mass Spectrometry. Nathan Edwards Informatics Research Applied Biosystems
 Protein Identification Using Tandem Mass Spectrometry Nathan Edwards Informatics Research Applied Biosystems Outline Proteomics context Tandem mass spectrometry Peptide fragmentation Peptide identification
Protein Identification Using Tandem Mass Spectrometry Nathan Edwards Informatics Research Applied Biosystems Outline Proteomics context Tandem mass spectrometry Peptide fragmentation Peptide identification
Tutorial 1: Library Generation from DDA data
 Tutorial 1: Library Generation from DDA data 1. Introduction Before a targeted, peptide-centric DIA analysis can be performed, a spectral library containing peptide-query parameters needs to be generated.
Tutorial 1: Library Generation from DDA data 1. Introduction Before a targeted, peptide-centric DIA analysis can be performed, a spectral library containing peptide-query parameters needs to be generated.
Modeling Mass Spectrometry-Based Protein Analysis
 Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra. Andrew Keller
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
Computational Methods for Mass Spectrometry Proteomics
 Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Protocol. Product Use & Liability. Contact us: InfoLine: Order per fax: www:
 Protocol SpikeTides Set TAA - light SpikeTides Set TAA_L - heavy Peptide Sets for relative quantification of Tumor Associated Antigens (TAAs) in SRM and MRM Assays Contact us: InfoLine: +49-30-6392-7878
Protocol SpikeTides Set TAA - light SpikeTides Set TAA_L - heavy Peptide Sets for relative quantification of Tumor Associated Antigens (TAAs) in SRM and MRM Assays Contact us: InfoLine: +49-30-6392-7878
HOWTO, example workflow and data files. (Version )
") HOWTO, example workflow and data files. (Version 20 09 2017) 1 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD
HOWTO, example workflow and data files. (Version 20 09 2017) 1 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Day 2 October 17, 2006 Andrew Keller Rosetta Bioinformatics, Seattle Outline Need to validate peptide assignments to MS/MS
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Day 2 October 17, 2006 Andrew Keller Rosetta Bioinformatics, Seattle Outline Need to validate peptide assignments to MS/MS
MassHunter TOF/QTOF Users Meeting
 MassHunter TOF/QTOF Users Meeting 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks
MassHunter TOF/QTOF Users Meeting 1 Qualitative Analysis Workflows Workflows in Qualitative Analysis allow the user to only see and work with the areas and dialog boxes they need for their specific tasks
Viewing and Analyzing Proteins, Ligands and their Complexes 2
 2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
Nature Methods: doi: /nmeth Supplementary Figure 1. Fragment indexing allows efficient spectra similarity comparisons.
 Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
Tutorial 3: Building a spectral library in Skyline
 SRM Curse 2013 Tutrial 3 Spectral Library Tutrial 3: Building a spectral library in Skyline Spectral libraries fr SRM methd design and fr data analysis can be either directly added t a Skyline dcument
SRM Curse 2013 Tutrial 3 Spectral Library Tutrial 3: Building a spectral library in Skyline Spectral libraries fr SRM methd design and fr data analysis can be either directly added t a Skyline dcument
NPTEL VIDEO COURSE PROTEOMICS PROF. SANJEEVA SRIVASTAVA
 LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
ProMass Deconvolution User Training. Novatia LLC January, 2013
 ProMass Deconvolution User Training Novatia LLC January, 2013 Overview General info about ProMass Features Basics of how ProMass Deconvolution works Example Spectra Manual Deconvolution with ProMass Deconvolution
ProMass Deconvolution User Training Novatia LLC January, 2013 Overview General info about ProMass Features Basics of how ProMass Deconvolution works Example Spectra Manual Deconvolution with ProMass Deconvolution
Quantitation of High Resolution MS Data Using UNIFI: Acquiring and Processing Full Scan or Tof-MRM (Targeted HRMS) Datasets for Quantitative Assays
 Datasets for Quantitative Assays") : Acquiring and Processing Full Scan or Tof-MRM (Targeted HRMS) Datasets for Quantitative Assays Mark Wrona, Jayne Kirk, and Yun Alelyunas Waters Corporation, Milford, MA, USA APPLICATION BENEFITS Ability
: Acquiring and Processing Full Scan or Tof-MRM (Targeted HRMS) Datasets for Quantitative Assays Mark Wrona, Jayne Kirk, and Yun Alelyunas Waters Corporation, Milford, MA, USA APPLICATION BENEFITS Ability
All Ions MS/MS: Targeted Screening and Quantitation Using Agilent TOF and Q-TOF LC/MS Systems
 All Ions MS/MS: Targeted Screening and Quantitation Using Agilent TOF and Q-TOF LC/MS Systems Technical Overview Introduction All Ions MS/MS is a technique that is available for Agilent high resolution
All Ions MS/MS: Targeted Screening and Quantitation Using Agilent TOF and Q-TOF LC/MS Systems Technical Overview Introduction All Ions MS/MS is a technique that is available for Agilent high resolution
Spectronaut Pulsar. User Manual
 Spectronaut Pulsar User Manual 1 General Information... 6 1.1 Computer System Requirements... 6 1.2 Scope of Spectronaut Software... 6 1.3 Spectronaut Pulsar... 6 1.4 Spectronaut Release Features... 7
Spectronaut Pulsar User Manual 1 General Information... 6 1.1 Computer System Requirements... 6 1.2 Scope of Spectronaut Software... 6 1.3 Spectronaut Pulsar... 6 1.4 Spectronaut Release Features... 7
Protocol. Product Use & Liability. Contact us: InfoLine: Order per fax: www:
 Protocol SpikeTides Sets SpikeTides Sets_L heavy SpikeMix SpikeMix_L heavy Peptide Sets for relative quantification of Proteins in Mass Spectrometry Based Assays Contact us: InfoLine: +49-30-6392-7878
Protocol SpikeTides Sets SpikeTides Sets_L heavy SpikeMix SpikeMix_L heavy Peptide Sets for relative quantification of Proteins in Mass Spectrometry Based Assays Contact us: InfoLine: +49-30-6392-7878
Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were
 Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were developed to allow the analysis of large intact (bigger than
Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were developed to allow the analysis of large intact (bigger than
Last updated: Copyright
 Last updated: 2012-08-20 Copyright 2004-2012 plabel (v2.4) User s Manual by Bioinformatics Group, Institute of Computing Technology, Chinese Academy of Sciences Tel: 86-10-62601016 Email: zhangkun01@ict.ac.cn,
Last updated: 2012-08-20 Copyright 2004-2012 plabel (v2.4) User s Manual by Bioinformatics Group, Institute of Computing Technology, Chinese Academy of Sciences Tel: 86-10-62601016 Email: zhangkun01@ict.ac.cn,
Comprehensive support for quantitation
 Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
High-Throughput Protein Quantitation Using Multiple Reaction Monitoring
 High-Throughput Protein Quantitation Using Multiple Reaction Monitoring Application Note Authors Ning Tang, Christine Miller, Joe Roark, Norton Kitagawa and Keith Waddell Agilent Technologies, Inc. Santa
High-Throughput Protein Quantitation Using Multiple Reaction Monitoring Application Note Authors Ning Tang, Christine Miller, Joe Roark, Norton Kitagawa and Keith Waddell Agilent Technologies, Inc. Santa
Peptide Mass Fingerprinting (PMF) Data Acquisition Using the Voyager DE- PRO Database Search with PMF Data using Ms-Fit
 Data Acquisition Using the Voyager DE- PRO Database Search with PMF Data using Ms-Fit") Peptide Mass Fingerprinting (PMF) Data Acquisition Using the Voyager DE- PRO Introduction page 1 Sample preparation (ZipTip) page 1 Data acquisition using the Voyager DE Pro page 5 Resolution and mass
Peptide Mass Fingerprinting (PMF) Data Acquisition Using the Voyager DE- PRO Introduction page 1 Sample preparation (ZipTip) page 1 Data acquisition using the Voyager DE Pro page 5 Resolution and mass
TOMAHAQ Method Construction
 TOMAHAQ Method Construction Triggered by offset mass accurate-mass high-resolution accurate quantitation (TOMAHAQ) can be performed in the standard method editor of the instrument, without modifications
TOMAHAQ Method Construction Triggered by offset mass accurate-mass high-resolution accurate quantitation (TOMAHAQ) can be performed in the standard method editor of the instrument, without modifications
Mnova Software for Analyzing Reaction Monitoring NMR Spectra
 Mnova Software for Analyzing Reaction Monitoring NMR Spectra Version 10 Chen Peng, PhD, VP of Business Development, US & China Mestrelab Research SL San Diego, CA, USA chen.peng@mestrelab.com 858.736.4563
Mnova Software for Analyzing Reaction Monitoring NMR Spectra Version 10 Chen Peng, PhD, VP of Business Development, US & China Mestrelab Research SL San Diego, CA, USA chen.peng@mestrelab.com 858.736.4563
LECTURE-13. Peptide Mass Fingerprinting HANDOUT. Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of
 LECTURE-13 Peptide Mass Fingerprinting HANDOUT PREAMBLE Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of proteins, drugs and many biological moieties to elucidate
LECTURE-13 Peptide Mass Fingerprinting HANDOUT PREAMBLE Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of proteins, drugs and many biological moieties to elucidate
MS-based proteomics to investigate proteins and their modifications
 MS-based proteomics to investigate proteins and their modifications Francis Impens VIB Proteomics Core October th 217 Overview Mass spectrometry-based proteomics: general workflow Identification of protein
MS-based proteomics to investigate proteins and their modifications Francis Impens VIB Proteomics Core October th 217 Overview Mass spectrometry-based proteomics: general workflow Identification of protein
Tandem mass spectra were extracted from the Xcalibur data system format. (.RAW) and charge state assignment was performed using in house software
 and charge state assignment was performed using in house software") Supplementary Methods Software Interpretation of Tandem mass spectra Tandem mass spectra were extracted from the Xcalibur data system format (.RAW) and charge state assignment was performed using in house
Supplementary Methods Software Interpretation of Tandem mass spectra Tandem mass spectra were extracted from the Xcalibur data system format (.RAW) and charge state assignment was performed using in house
Cerno Application Note Extending the Limits of Mass Spectrometry
 Creation of Accurate Mass Library for NIST Database Search Novel MS calibration has been shown to enable accurate mass and elemental composition determination on quadrupole GC/MS systems for either molecular
Creation of Accurate Mass Library for NIST Database Search Novel MS calibration has been shown to enable accurate mass and elemental composition determination on quadrupole GC/MS systems for either molecular
MS/MS of Peptides Manual Sequencing of Protonated Peptides
 S/S of Peptides anual Sequencing of Protonated Peptides Árpád Somogyi Associate irector CCIC, ass Spectrometry and Proteomics Laboratory SU July 11, 2018 Peptides Product Ion Scan Product ion spectra contain
S/S of Peptides anual Sequencing of Protonated Peptides Árpád Somogyi Associate irector CCIC, ass Spectrometry and Proteomics Laboratory SU July 11, 2018 Peptides Product Ion Scan Product ion spectra contain
Lecture 15: Realities of Genome Assembly Protein Sequencing
 Lecture 15: Realities of Genome Assembly Protein Sequencing Study Chapter 8.10-8.15 1 Euler s Theorems A graph is balanced if for every vertex the number of incoming edges equals to the number of outgoing
Lecture 15: Realities of Genome Assembly Protein Sequencing Study Chapter 8.10-8.15 1 Euler s Theorems A graph is balanced if for every vertex the number of incoming edges equals to the number of outgoing
Agilent MassHunter Quantitative Data Analysis
 Agilent MassHunter Quantitative Data Analysis Presenters: Howard Sanford Stephen Harnos MassHunter Quantitation: Batch and Method Setup Outliers, Data Review, Reporting 1 MassHunter Quantitative Analysis
Agilent MassHunter Quantitative Data Analysis Presenters: Howard Sanford Stephen Harnos MassHunter Quantitation: Batch and Method Setup Outliers, Data Review, Reporting 1 MassHunter Quantitative Analysis
DISCLAIMER. Some General Comments on this Workbook. How to Use This Workbook. Peptide-MS-Calc.xls 1
 Peptide-MS-Calc.xls 1 DISCLAIMER This workbook was written using Excel X on Mac S X. Whilst it will probably work on recent versions of Excel for windows, you may experience some problems. If you benefit
Peptide-MS-Calc.xls 1 DISCLAIMER This workbook was written using Excel X on Mac S X. Whilst it will probably work on recent versions of Excel for windows, you may experience some problems. If you benefit
Purdue-UAB Botanicals Center for Age- Related Disease
 Purdue-UAB Botanicals Center for Age- Related Disease MALDI-TOF Mass Spectrometry Fingerprinting Technique Landon Wilson MALDI-TOF mass spectrometry is an advanced technique for rapid protein identification
Purdue-UAB Botanicals Center for Age- Related Disease MALDI-TOF Mass Spectrometry Fingerprinting Technique Landon Wilson MALDI-TOF mass spectrometry is an advanced technique for rapid protein identification
Analyst Software. Automatic Optimization Tutorial
 This document is provided to customers who have purchased AB Sciex equipment to use in the operation of such AB Sciex equipment. This document is copyright protected and any reproduction of this document
This document is provided to customers who have purchased AB Sciex equipment to use in the operation of such AB Sciex equipment. This document is copyright protected and any reproduction of this document
Ligand Scout Tutorials
 Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
Ligand Scout Tutorials Step : Creating a pharmacophore from a protein-ligand complex. Type ke6 in the upper right area of the screen and press the button Download *+. The protein will be downloaded and
GAS CHROMATOGRAPHY MASS SPECTROMETRY. Pre-Lab Questions
 GAS CHROMATOGRAPHY MASS SPECTROMETRY Pre-Lab Questions Questions to be answered before doing the experiment. The answers are due at the beginning of each experiment without exception (the questions are
GAS CHROMATOGRAPHY MASS SPECTROMETRY Pre-Lab Questions Questions to be answered before doing the experiment. The answers are due at the beginning of each experiment without exception (the questions are
Spectrum-to-Spectrum Searching Using a. Proteome-wide Spectral Library
 MCP Papers in Press. Published on April 30, 2011 as Manuscript M111.007666 Spectrum-to-Spectrum Searching Using a Proteome-wide Spectral Library Chia-Yu Yen, Stephane Houel, Natalie G. Ahn, and William
MCP Papers in Press. Published on April 30, 2011 as Manuscript M111.007666 Spectrum-to-Spectrum Searching Using a Proteome-wide Spectral Library Chia-Yu Yen, Stephane Houel, Natalie G. Ahn, and William
Relative quantification using TMT11plex on a modified Q Exactive HF mass spectrometer
 POSTER NOTE 6558 Relative quantification using TMT11plex on a modified mass spectrometer Authors Tabiwang N. Arrey, 1 Rosa Viner, 2 Ryan D. Bomgarden, 3 Eugen Damoc, 1 Markus Kellmann, 1 Thomas Moehring,
POSTER NOTE 6558 Relative quantification using TMT11plex on a modified mass spectrometer Authors Tabiwang N. Arrey, 1 Rosa Viner, 2 Ryan D. Bomgarden, 3 Eugen Damoc, 1 Markus Kellmann, 1 Thomas Moehring,
Workflow concept. Data goes through the workflow. A Node contains an operation An edge represents data flow The results are brought together in tables
 PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
Agilent All Ions MS/MS
 Agilent All Ions MS/MS Workflow Overview A Determine fragment ions for LC/MS Quant method B Develop final Quant method Develop LC/MS Qualitative Analysis method Process data with Find by Formula Build
Agilent All Ions MS/MS Workflow Overview A Determine fragment ions for LC/MS Quant method B Develop final Quant method Develop LC/MS Qualitative Analysis method Process data with Find by Formula Build
Comparing whole genomes
 BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
BioNumerics Tutorial: Comparing whole genomes 1 Aim The Chromosome Comparison window in BioNumerics has been designed for large-scale comparison of sequences of unlimited length. In this tutorial you will
Portal. User Guide Version 1.0. Contributors
 Portal www.dockthor.lncc.br User Guide Version 1.0 Contributors Diogo A. Marinho, Isabella A. Guedes, Eduardo Krempser, Camila S. de Magalhães, Hélio J. C. Barbosa and Laurent E. Dardenne www.gmmsb.lncc.br
Portal www.dockthor.lncc.br User Guide Version 1.0 Contributors Diogo A. Marinho, Isabella A. Guedes, Eduardo Krempser, Camila S. de Magalhães, Hélio J. C. Barbosa and Laurent E. Dardenne www.gmmsb.lncc.br
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry
 Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University
 Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Introduction to Spark
 1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
1 As you become familiar or continue to explore the Cresset technology and software applications, we encourage you to look through the user manual. This is accessible from the Help menu. However, don t
Live Webinar : How to be more Successful with your ACQUITY QDa Detector?
 Live Webinar : How to be more Successful with your ACQUITY QDa Detector? Q&A Transcript ---------------- Q - How do you generate multiple charges reproductively? A - If you use the same settings on the
Live Webinar : How to be more Successful with your ACQUITY QDa Detector? Q&A Transcript ---------------- Q - How do you generate multiple charges reproductively? A - If you use the same settings on the
PC235: 2008 Lecture 5: Quantitation. Arnold Falick
 PC235: 2008 Lecture 5: Quantitation Arnold Falick falickam@berkeley.edu Summary What you will learn from this lecture: There are many methods to perform quantitation using mass spectrometry (any method
PC235: 2008 Lecture 5: Quantitation Arnold Falick falickam@berkeley.edu Summary What you will learn from this lecture: There are many methods to perform quantitation using mass spectrometry (any method
Jasco V-670 absorption spectrometer
 Laser Spectroscopy Labs Jasco V-670 absorption spectrometer Operation instructions 1. Turn ON the power switch on the right side of the spectrophotometer. It takes about 5 minutes for the light source
Laser Spectroscopy Labs Jasco V-670 absorption spectrometer Operation instructions 1. Turn ON the power switch on the right side of the spectrophotometer. It takes about 5 minutes for the light source
SIERRA ANALYTICS, INC. Version Polymerix Software User Manual
 SIERRA ANALYTICS, INC. Version 3.0.0 Polymerix Software User Manual V E R S I O N 3.0.0 M A R C H 2013 Polymerix Software User Manual Copyright 2010 to 2013 Sierra Analytics, Inc. 5815 Stoddard Road, Suite
SIERRA ANALYTICS, INC. Version 3.0.0 Polymerix Software User Manual V E R S I O N 3.0.0 M A R C H 2013 Polymerix Software User Manual Copyright 2010 to 2013 Sierra Analytics, Inc. 5815 Stoddard Road, Suite
The Power of LC MALDI: Identification of Proteins by LC MALDI MS/MS Using the Applied Biosystems 4700 Proteomics Analyzer with TOF/TOF Optics
 APPLICATION NOTE TOF MS The Power of LC MALDI: Identification of Proteins by LC MALDI MS/MS Using the Applied Biosystems 4700 Proteomics Analyzer with TOF/TOF Optics Purpose The Applied Biosystems 4700
APPLICATION NOTE TOF MS The Power of LC MALDI: Identification of Proteins by LC MALDI MS/MS Using the Applied Biosystems 4700 Proteomics Analyzer with TOF/TOF Optics Purpose The Applied Biosystems 4700
The Pitfalls of Peaklist Generation Software Performance on Database Searches
 Proceedings of the 56th ASMS Conference on Mass Spectrometry and Allied Topics, Denver, CO, June 1-5, 2008 The Pitfalls of Peaklist Generation Software Performance on Database Searches Aenoch J. Lynn,
Proceedings of the 56th ASMS Conference on Mass Spectrometry and Allied Topics, Denver, CO, June 1-5, 2008 The Pitfalls of Peaklist Generation Software Performance on Database Searches Aenoch J. Lynn,
MS-MS Analysis Programs
 MS-MS Analysis Programs Basic Process Genome - Gives AA sequences of proteins Use this to predict spectra Compare data to prediction Determine degree of correctness Make assignment Did we see the protein?
MS-MS Analysis Programs Basic Process Genome - Gives AA sequences of proteins Use this to predict spectra Compare data to prediction Determine degree of correctness Make assignment Did we see the protein?
Performing Peptide Bioanalysis Using High Resolution Mass Spectrometry with Target Enhancement MRM Acquisition
 Performing Peptide Bioanalysis Using High Resolution Mass Spectrometry with Target Enhancement MRM Acquisition Yun Wang Alelyunas, Mark D. Wrona, and Nick Tomczyk Waters Corporation, Milford, MA, USA GOAL
Performing Peptide Bioanalysis Using High Resolution Mass Spectrometry with Target Enhancement MRM Acquisition Yun Wang Alelyunas, Mark D. Wrona, and Nick Tomczyk Waters Corporation, Milford, MA, USA GOAL
HR/AM Targeted Peptide Quantification on a Q Exactive MS: A Unique Combination of High Selectivity, High Sensitivity, and High Throughput
 HR/AM Targeted Peptide Quantification on a Q Exactive MS: A Unique Combination of High Selectivity, High Sensitivity, and High Throughput Yi Zhang 1, Zhiqi Hao 1, Markus Kellmann 2 and Andreas FR. Huhmer
HR/AM Targeted Peptide Quantification on a Q Exactive MS: A Unique Combination of High Selectivity, High Sensitivity, and High Throughput Yi Zhang 1, Zhiqi Hao 1, Markus Kellmann 2 and Andreas FR. Huhmer
Agilent MassHunter Quantitative Data Analysis
 Agilent MassHunter Quantitative Data Analysis Presenters: Howard Sanford Stephen Harnos MassHunter Quantitation: Batch Table, Compound Information Setup, Calibration Curve and Globals Settings 1 MassHunter
Agilent MassHunter Quantitative Data Analysis Presenters: Howard Sanford Stephen Harnos MassHunter Quantitation: Batch Table, Compound Information Setup, Calibration Curve and Globals Settings 1 MassHunter
Overview - MS Proteomics in One Slide. MS masses of peptides. MS/MS fragments of a peptide. Results! Match to sequence database
 Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Self-assembling covalent organic frameworks functionalized. magnetic graphene hydrophilic biocomposite as an ultrasensitive
 Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2017 Electronic Supporting Information for: Self-assembling covalent organic frameworks functionalized
Electronic Supplementary Material (ESI) for Nanoscale. This journal is The Royal Society of Chemistry 2017 Electronic Supporting Information for: Self-assembling covalent organic frameworks functionalized
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry
 Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
NMR Assignments using NMRView II: Sequential Assignments
 NMR Assignments using NMRView II: Sequential Assignments DO THE FOLLOWING, IF YOU HAVE NOT ALREADY DONE SO: For Mac OS X, you should have a subdirectory nmrview. At UGA this is /Users/bcmb8190/nmrview.
NMR Assignments using NMRView II: Sequential Assignments DO THE FOLLOWING, IF YOU HAVE NOT ALREADY DONE SO: For Mac OS X, you should have a subdirectory nmrview. At UGA this is /Users/bcmb8190/nmrview.
WADA Technical Document TD2003IDCR
 IDENTIFICATION CRITERIA FOR QUALITATIVE ASSAYS INCORPORATING CHROMATOGRAPHY AND MASS SPECTROMETRY The appropriate analytical characteristics must be documented for a particular assay. The Laboratory must
IDENTIFICATION CRITERIA FOR QUALITATIVE ASSAYS INCORPORATING CHROMATOGRAPHY AND MASS SPECTROMETRY The appropriate analytical characteristics must be documented for a particular assay. The Laboratory must
The Agilent 6495 Triple Quadrupole LC/MS: Peptide Quantitation Performance
 The Agilent 495 Triple Quadrupole LC/MS: Peptide Quantitation Performance Technical Overview Introduction Sample complexity and the low concentration of certain biomarkers are the major challenges encountered
The Agilent 495 Triple Quadrupole LC/MS: Peptide Quantitation Performance Technical Overview Introduction Sample complexity and the low concentration of certain biomarkers are the major challenges encountered
ICM-Chemist-Pro How-To Guide. Version 3.6-1h Last Updated 12/29/2009
 ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
ICM-Chemist-Pro How-To Guide Version 3.6-1h Last Updated 12/29/2009 ICM-Chemist-Pro ICM 3D LIGAND EDITOR: SETUP 1. Read in a ligand molecule or PDB file. How to setup the ligand in the ICM 3D Ligand Editor.
Site-specific Identification of Lysine Acetylation Stoichiometries in Mammalian Cells
 Supplementary Information Site-specific Identification of Lysine Acetylation Stoichiometries in Mammalian Cells Tong Zhou 1, 2, Ying-hua Chung 1, 2, Jianji Chen 1, Yue Chen 1 1. Department of Biochemistry,
Supplementary Information Site-specific Identification of Lysine Acetylation Stoichiometries in Mammalian Cells Tong Zhou 1, 2, Ying-hua Chung 1, 2, Jianji Chen 1, Yue Chen 1 1. Department of Biochemistry,
X!TandemPipeline (Myosine Anabolisée) validating, filtering and grouping MSMS identifications
 validating, filtering and grouping MSMS identifications") X!TandemPipeline 3.3.3 (Myosine Anabolisée) validating, filtering and grouping MSMS identifications Olivier Langella and Benoit Valot langella@moulon.inra.fr; valot@moulon.inra.fr PAPPSO - http://pappso.inra.fr/
X!TandemPipeline 3.3.3 (Myosine Anabolisée) validating, filtering and grouping MSMS identifications Olivier Langella and Benoit Valot langella@moulon.inra.fr; valot@moulon.inra.fr PAPPSO - http://pappso.inra.fr/
OECD QSAR Toolbox v.4.1. Tutorial illustrating new options for grouping with metabolism
 OECD QSAR Toolbox v.4.1 Tutorial illustrating new options for grouping with metabolism Outlook Background Objectives Specific Aims The exercise Workflow 2 Background Grouping with metabolism is a procedure
OECD QSAR Toolbox v.4.1 Tutorial illustrating new options for grouping with metabolism Outlook Background Objectives Specific Aims The exercise Workflow 2 Background Grouping with metabolism is a procedure
Separation of Large and Small Peptides by Supercritical Fluid Chromatography and Detection by Mass Spectrometry
 Separation of Large and Small Peptides by Supercritical Fluid Chromatography and Detection by Mass Spectrometry Application Note Biologics and Biosimilars Author Edgar Naegele Agilent Technologies, Inc.
Separation of Large and Small Peptides by Supercritical Fluid Chromatography and Detection by Mass Spectrometry Application Note Biologics and Biosimilars Author Edgar Naegele Agilent Technologies, Inc.
Quantitative Proteomics
 Quantitative Proteomics Quantitation AND Mass Spectrometry Condition A Condition B Identify and quantify differently expressed proteins resulting from a change in the environment (stimulus, disease) Lyse
Quantitative Proteomics Quantitation AND Mass Spectrometry Condition A Condition B Identify and quantify differently expressed proteins resulting from a change in the environment (stimulus, disease) Lyse
Peter A. DiMaggio, Jr., Nicolas L. Young, Richard C. Baliban, Benjamin A. Garcia, and Christodoulos A. Floudas. Research
 Research A Mixed Integer Linear Optimization Framework for the Identification and Quantification of Targeted Post-translational Modifications of Highly Modified Proteins Using Multiplexed Electron Transfer
Research A Mixed Integer Linear Optimization Framework for the Identification and Quantification of Targeted Post-translational Modifications of Highly Modified Proteins Using Multiplexed Electron Transfer
Rapid Distinction of Leucine and Isoleucine in Monoclonal Antibodies Using Nanoflow. LCMS n. Discovery Attribute Sciences
 Rapid Distinction of Leucine and Isoleucine in Monoclonal Antibodies Using Nanoflow LCMS n Dhanashri Bagal *, Eddie Kast, Ping Cao Discovery Attribute Sciences Amgen, South San Francisco, California, United
Rapid Distinction of Leucine and Isoleucine in Monoclonal Antibodies Using Nanoflow LCMS n Dhanashri Bagal *, Eddie Kast, Ping Cao Discovery Attribute Sciences Amgen, South San Francisco, California, United
Designed for Accuracy. Innovation with Integrity. High resolution quantitative proteomics LC-MS
 Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
Key questions of proteomics. Bioinformatics 2. Proteomics. Foundation of proteomics. What proteins are there? Protein digestion
 s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
CycloBranch. Tutorials
 CycloBranch Tutorials Outline Tutorial 1: Does a peptide have a cycle? Tutorial 2: How to determine a tag? Tutorial 3: How to determine a complete sequence? Tutorial 4: How to determine a branched sequence?
CycloBranch Tutorials Outline Tutorial 1: Does a peptide have a cycle? Tutorial 2: How to determine a tag? Tutorial 3: How to determine a complete sequence? Tutorial 4: How to determine a branched sequence?
Improved Throughput and Reproducibility for Targeted Protein Quantification Using a New High-Performance Triple Quadrupole Mass Spectrometer
 Improved Throughput and Reproducibility for Targeted Protein Quantification Using a New High-Performance Triple Quadrupole Mass Spectrometer Reiko Kiyonami, Mary Blackburn, Andreas FR Hühme: Thermo Fisher
Improved Throughput and Reproducibility for Targeted Protein Quantification Using a New High-Performance Triple Quadrupole Mass Spectrometer Reiko Kiyonami, Mary Blackburn, Andreas FR Hühme: Thermo Fisher
Introduction to Structure Preparation and Visualization
 Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Introduction to Structure Preparation and Visualization Created with: Release 2018-4 Prerequisites: Release 2018-2 or higher Access to the internet Categories: Molecular Visualization, Structure-Based
Introduction to Proteomics: Fragmentation of protonated peptides and manual sequencing
 Introduction to Proteomics: ragmentation of protonated peptides and manual sequencing Árpád Somogyi CCIC SP SU Summer Workshop S/S hod using ESI Ion rap (Bottom up) 1 An alternative strategy for complex
Introduction to Proteomics: ragmentation of protonated peptides and manual sequencing Árpád Somogyi CCIC SP SU Summer Workshop S/S hod using ESI Ion rap (Bottom up) 1 An alternative strategy for complex
MetWorks Metabolite Identification Software
 m a s s s p e c t r o m e t r y MetWorks Metabolite Identification Software Enabling Confident Analysis of Metabolism Data Part of Thermo Fisher Scientific MetWorks Software for the Confident Analysis
m a s s s p e c t r o m e t r y MetWorks Metabolite Identification Software Enabling Confident Analysis of Metabolism Data Part of Thermo Fisher Scientific MetWorks Software for the Confident Analysis
Protein Fragment Search Program ver Overview: Contents:
 Protein Fragment Search Program ver 1.1.1 Developed by: BioPhysics Laboratory, Faculty of Life and Environmental Science, Shimane University 1060 Nishikawatsu-cho, Matsue-shi, Shimane, 690-8504, Japan
Protein Fragment Search Program ver 1.1.1 Developed by: BioPhysics Laboratory, Faculty of Life and Environmental Science, Shimane University 1060 Nishikawatsu-cho, Matsue-shi, Shimane, 690-8504, Japan
Other Methods for Generating Ions 1. MALDI matrix assisted laser desorption ionization MS 2. Spray ionization techniques 3. Fast atom bombardment 4.
 Other Methods for Generating Ions 1. MALDI matrix assisted laser desorption ionization MS 2. Spray ionization techniques 3. Fast atom bombardment 4. Field Desorption 5. MS MS techniques Matrix assisted
Other Methods for Generating Ions 1. MALDI matrix assisted laser desorption ionization MS 2. Spray ionization techniques 3. Fast atom bombardment 4. Field Desorption 5. MS MS techniques Matrix assisted
Uncontrolled Copy. SOP-060 Chromatography, Tune Evaluation and Troubleshooting. Table of Contents. 1. Principle... 3
 Table of Contents 1. Principle... 3 2. Mass Spectrometer Evaluation... 3 3. GC NPD Evaluation... 5 4. Acid Neutral test injection evaluation (GC/MS/FID)... 6 5. Discrepancies... 6 6. Troubleshooting...
Table of Contents 1. Principle... 3 2. Mass Spectrometer Evaluation... 3 3. GC NPD Evaluation... 5 4. Acid Neutral test injection evaluation (GC/MS/FID)... 6 5. Discrepancies... 6 6. Troubleshooting...
The Theory of HPLC. Quantitative and Qualitative HPLC
 The Theory of HPLC Quantitative and Qualitative HPLC i Wherever you see this symbol, it is important to access the on-line course as there is interactive material that cannot be fully shown in this reference
The Theory of HPLC Quantitative and Qualitative HPLC i Wherever you see this symbol, it is important to access the on-line course as there is interactive material that cannot be fully shown in this reference
Examples of Protein Modeling. Protein Modeling. Primary Structure. Protein Structure Description. Protein Sequence Sources. Importing Sequences to MOE
 Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
Examples of Protein Modeling Protein Modeling Visualization Examination of an experimental structure to gain insight about a research question Dynamics To examine the dynamics of protein structures To
De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra. Xiaowen Liu
 De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra Xiaowen Liu Department of BioHealth Informatics, Department of Computer and Information Sciences, Indiana University-Purdue
De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra Xiaowen Liu Department of BioHealth Informatics, Department of Computer and Information Sciences, Indiana University-Purdue
Improved Validation of Peptide MS/MS Assignments. Using Spectral Intensity Prediction
 MCP Papers in Press. Published on October 2, 2006 as Manuscript M600320-MCP200 Improved Validation of Peptide MS/MS Assignments Using Spectral Intensity Prediction Shaojun Sun 1, Karen Meyer-Arendt 2,
MCP Papers in Press. Published on October 2, 2006 as Manuscript M600320-MCP200 Improved Validation of Peptide MS/MS Assignments Using Spectral Intensity Prediction Shaojun Sun 1, Karen Meyer-Arendt 2,
WindNinja Tutorial 3: Point Initialization
 WindNinja Tutorial 3: Point Initialization 6/27/2018 Introduction Welcome to WindNinja Tutorial 3: Point Initialization. This tutorial will step you through the process of downloading weather station data
WindNinja Tutorial 3: Point Initialization 6/27/2018 Introduction Welcome to WindNinja Tutorial 3: Point Initialization. This tutorial will step you through the process of downloading weather station data
AB SCIEX SelexION Technology Used to Improve Mass Spectral Library Searching Scores by Removal of Isobaric Interferences
 AB SCIEX SelexION Technology Used to Improve Mass Spectral Library Searching s by Removal of Isobaric Interferences Differential Mobility Used as a Tool to Address Selectivity Challenges Adrian M. Taylor
AB SCIEX SelexION Technology Used to Improve Mass Spectral Library Searching s by Removal of Isobaric Interferences Differential Mobility Used as a Tool to Address Selectivity Challenges Adrian M. Taylor