PC235: 2008 Lecture 5: Quantitation. Arnold Falick
|
|
|
- William Lloyd
- 6 years ago
- Views:
Transcription
1 PC235: 2008 Lecture 5: Quantitation Arnold Falick
2 Summary What you will learn from this lecture: There are many methods to perform quantitation using mass spectrometry (any method worth mentioning has a trendy acronym) Quantitation can be used for studying protein changes, modification changes or as a method for identifying specific results. Quantitation is normally relative (between samples), but can also be absolute. Software for analysis of this type of data is still being developed.
3 Mass Spectrometry is not Inherently Quantitative There are a number of factors that contribute to peak intensity in a mass spectrum Absolute amount of component Amount of component in relation to other components in the sample Ease of ionization of component Components that are of the same concentration may give drastically different signal intensities E.g., if a protein is digested into peptides, different peptides give very different signal intensities (and some peptides may not be detected at all!)
4 Mass Spectrometry is not Inherently Quantitative The same sample can look very different based on the analysis technique tryptic digest in CHCA Same digest in DHB
5 How Can Mass Spectrometry be used for Quantitation? Different types of quantitation can be performed using mass spectrometry Relative quantitation can be performed for chemically similar components present in the same sample Relative quantitation can be performed by comparing peak intensities of the same component in similar samples analyzed sequentially. Comparison to a similar component at a known concentration can be used to estimate absolute quantities. Absolute protein quantities can be crudely estimated without comparison.
6 MS Methods used to Produce Quantitative Data Stable Isotope Labeling: Incorporation of stable isotopes, samples are mixed and then compared to each other. [Isotopes can be incorporated metabolically or chemically] Label-free Quantitation: Samples are run sequentially and either peak intensities or numbers of peptides identified from a protein are compared.
7 Enzymatic Incorporation of Stable Isotopes Serine proteases; e.g., trypsin, will exchange carboxyl terminal oxygens with solution. Incubating a sample in H 2 18 O will cause incorporation of two oxygens, producing a 4Da mass shift. Mix unlabeled and labeled samples together and analyze. Advantages Labels all peptides. Does not affect peptide fragmentation. Can be useful for peptide identification as all C-terminal ions (e.g., y-ions) will give pairs of peaks 4 Da apart.
8 Problems Somewhat difficult to get complete incorporation of both oxygens. 16 O/ 18 O Labeling Label will slowly exchange back once returned to normal water solution. 4 Da is a relatively small mass shift; can get overlap of light and heavy isotope clusters for large peptides. In Summary A quick, easy and cheap quantitation method, but not the most accurate. Wang, J. et al. J Proteom Res (2007)
9 ICAT (Isotope-Coded Affinity Tag) Advantage: Affinity Tag (Biotin) Linker Region Light: 8 1 H Heavy: 8 2 H Cysteine Reactive Group (Iodoacetamide) Can selectively enrich labeled peptides. Problems: Light and heavy reagents do not co-elute in reverse phase, making quantitation of LC-MS data more complicated. Protein must contain Cys The biotin tag causes problems: 1. Difficult to elute quantitatively from streptavidin 2. Tag yields intense fragment ions Fragment biotin rather than peptide Often difficult to identify peptide Gygi, S.P. et al. Nat. Biotechnol. (1999)

10 cicat (cleavable ICAT) 1. Affinity enrich modified peptides. 2. Cleave tag using TFA, removing the biotin. The smaller isotopically labeled tag fragments less than the whole protein, making peptide identification simpler. Light and heavy reagents co-elute, making quantitation easier. Issue Most proteins are identified by a single peptide, requiring high accuracy and reproducibility in measurements to be able to draw conclusions. Hansen, K. C. et al. Mol Cell Proteomics (2003)
11 SILAC (Stable Isotope Labeling of Amino Acids in Culture) In cell culture, replace one (or two) amino acids in the culture medium with 13 C and/or 15 N equivalent. Which amino acid/s should you choose? You want to label as many peptides as possible It needs to be an amino acid that is big enough to give a mass shift of at least 4Da. For tryptic digests, lysine (C6, N2) and/or arginine (C6, N4) are sensible choices. Need to be aware that some amino acids can be synthesized by the cell, or can be converted from one amino acid to another. Safest bet is to keep cells well fed, so there is never a shortage of any amino acid. Ong, S-E. et al. Mol Cell Proteom. (2002)
 is the sole donor of methyl groups for protein (and DNA, RNA) methylation.")
12 SILAC for Modification Quantitation Also possible to label modifications through addition of a labeled version of the donor. S-adenosyl methionine (AdoMet) is the sole donor of methyl groups for protein (and DNA, RNA) methylation. It is synthesized from methionine By adding 13 C 2 H 3 methionine in culture one can label lysine and arginine methylations e.g. in histones. Ong, S-E, Nat Methods (2004) 1 (2)
.")
13 Multiplexing SILAC Three samples can be compared in the same experiment. SILAC Summary Strengths: Can get essentially complete labeling (need about 5 population doublings for complete incorporation). Labeling does not affect fragmentation. Weakness: Requires cell culture. Ong, S-E, Nat Chem Biol (2005) 1 (5)
14 Heavy Methyl Esterification Can methyl esterify side-chain carboxylic acids Used to reduce non-specific binding in IMAC for phosphopeptide enrichment. Can use normal and deuterated methanol, mix samples and compare. Problem Reaction is sometimes difficult to get to completion. 3 Da mass shift is smaller than ideal (get overlap of peptide isotope patterns).
15 Amine-Reactive Labels Deuterated versions of many reagents have been used for labeling peptide N-termini and lysine side-chains. E.g.: Acetic anhydride Propionic anhydride Succinic anhydride Phenyl isocyanate
16 Isotopic Tag for Relative and Absolute Quantitation (itraq) The cleverest and most elegant amine-reactive tagging reagents. Amino-groups are modified (N-terminus, Lys) A set of four reagents, allowing simultaneous comparison of four samples. All reagents attach nominally same mass tag, giving one peak in the MS, but produce different reporter ions in MSMS, allowing relative quantitation. Ross et al. Mol Cell Proteomics (2004) 3, 1154.
 3,")
17 itraq Reagents Ross et al. Mol Cell Proteomics (2004) 3, 1154.

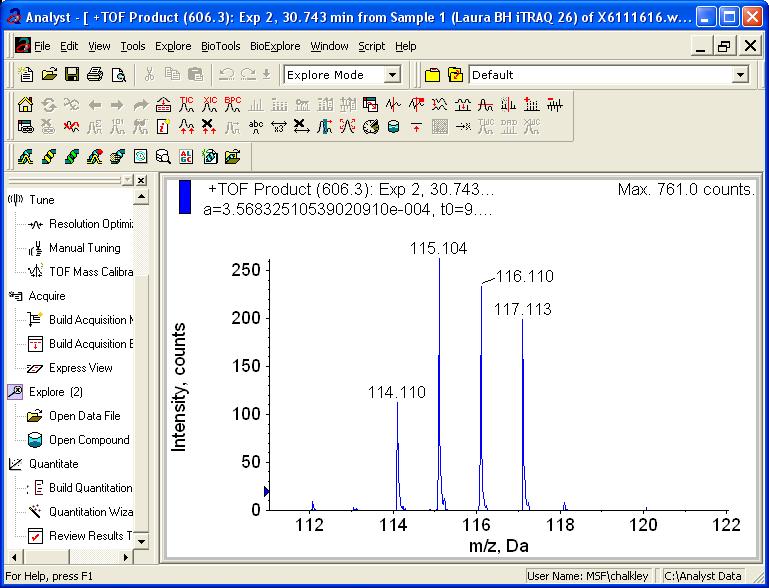
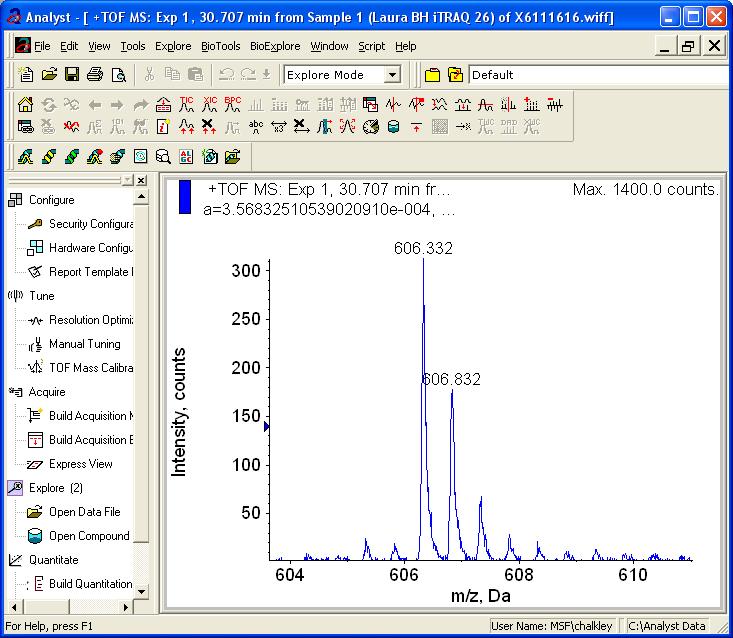
18 Example itraq Data Quantitation MS: Single Peak

19 When four is not enough 8-plex itraq 8-plex version produces reporter ions at m/z 113, 114, 115, 116, 117, 118, 119 and 121 Have to pick m/z that are not normally observed in peptide CID spectra. Choe, L., et al. Proteomics (2007) 7 (issue#20)
20 β-elimination followed by Michael addition of DTT (BEMAD) Primarily aimed at Ser/Thr post-translational modifications Phosphorylation O-Glycosylation (e.g. O-GlcNAcylation) O-Sulfation Alkylated cysteines also undergo reaction. Using 2 H 6 -DTT, can get quantitative information. Isotopic labeling can also be used to identify type of O-linked modification.
21 Absolute Quantitation (AQUA) All mass spectrometry quantitation so far has been relative. What if it is relative to a known amount? Absolute quantitation! For highest accuracy, a calibration curve of known amounts spiked into a sample should be obtained, then from this a peak intensity can be converted into an amount. Peak intensity for a given component is roughly linear with respect to intensity Add isotopic labeled version of peptide of known amount to sample, then compare intensity to unlabeled version in the sample 1. 1 Gerber et al. PNAS (2003) 100 (12)
22 Method of standard addition Sample + 2x std addition signal Sample + std addition Sample conc. Added concentration
23 itraq for Absolute Quantitation
24 Mass Spectrometry-based Protein Quantification Methods At what point are the samples combined? Tissue/ Cells SILAC cicat O 16 /O 18 itraq Internal Standards Protein Peptides MS Analysis
25 Isotopic Differentiation of Interactions as Random or Targeted (I-DIRT) Identifying non-specific interactors isolated as part of an IP/tag pulldown. Tackett et al, J. Prot. Res. (2005) 4, 1752.

26 Localization of Organelle Proteins by Isotope Tagging (LOPIT) Density centrifugation is used for organelle separation. A given organelle will span several fractions. Most fractions contain proteins from more than one organelle. Distribution of proteins between fractions should be the same for those in the same organelle. If separate fractions are labeled with itraq, the reporter peak intensity pattern can be used to identify organellar location. Sadowski, P.G. et al. Nat Protoc (2006)
27 Label-Free Quantitation All approaches so far require purchase of isotopically labeled reagents (can be expensive). What if you want to compare large numbers of samples (10+) What if you can t afford lots of reagents? Peak/Spectral counting Peak area comparison (XIC)
28 Spectral Counting Count the number of peptides identified from a protein in each sample. Do you count repeat identifications of the same peptide? Not accurate at quantifying magnitude of change, but can be used to determine if there is a difference. In general, need a spectral count difference of about 4 peptides in order to be confident of a difference being real. 1 Most proteins in complex mixtures are identified by less than 4 peptides. 1 Old et al. Mol Cell Proteomics (2005)

29 XIC (Extracted Ion Chromatogram) Measure intensity of peak during its elution off HPLC column and into the mass spectrometer. Measure area of peak in XIC. More accurate than selecting peak intensity for one given scan.
30 XIC with Internal Standards When comparing samples for label-free quantitation, how do you adjust for different overall amounts of protein loaded? Add an internal standard to each sample and normalize against this. Adding a protein digest will give multiple internal standards throughout the run. Can normalize to standard eluting at similar time Minimizes effects due to different levels of ion suppression in different samples. Cutillas et al. Mol Cell Proteomics (2007)
31 empai (Exponentially Modified Protein Abundance Index) empai = 10 PAI 1 Where PAI = N observed / N observable What is an observable peptide? Peptides with a precursor mass between Da. There is a roughly linear relationship between log protein concentration and the ratio of observable peptides observed in range of fmoles. If you know how much total protein you analyzed you can derive absolute abundancies. You can observe more peptides than are observable! Ishihama et al. Mol Cell Proteomics (2005)


32 MRM (Multiple Reaction Monitoring) Look for a component of a specific mass that when fragmented forms a fragment of another specific mass. Transition: precursor m/z fragment m/z Very sensitive and specific.
33 MRM Best performed on a triple quadrupole instrument. Scans are very fast, so can perform multiple transition scans on a chromatographic time-scale. If you know retention times, you can schedule different scans at different times during the LC-MS run. It is possible to analyze a few hundred transitions in a one hour run. If you perform MRM analysis of isotopically labeled (light and heavy) peptides can perform quantitation analysis at very high sensitivity. With a calibration curve of amount vs intensity, can approximate absolute concentration. This is an approach that is likely to be powerful for biomarker analysis. Lange et al. Mol Cell Proteomics (2008) [Epub Apr 13]
34 Software for MS Quantitation Analysis Different software is required for different quantitation methods. For most quantitation methods, software needs to be able to read instrument manufacturers raw format. Each manufacturer uses own proprietary format Most quantitation software only works for one type of quantitation on one type of instrument, using one search engine. ProQuant (Applied Biosystems): ICAT, cicat and itraq data acquired on their instruments, searched with their own search engine (Protein Pilot) MSQuant: SILAC data acquired from one of three instrument manufacturers, searched using Mascot Protein Prospector: Any type of isotopic labeling acquired on instruments from two manufacturers.

35 Issues with MS Quantitation Analysis Should you use all data for quantitation? Minimum peak intensity? Peaks near to signal to noise will have much higher variability in quantitation accuracy. Are proteins identified by a single peptide accurately quantified? What if a peptide contains a modification; e.g. oxidized methionine? What if a peptide is observed with and without a missed cleavage? Results are normally reported with a mean and standard deviation. Trinidad et al. Mol Cell Proteomics (2008)
36 Software for Analysis of Label-free Data Quantitation of label-free data is much more difficult from a bioinformatic perspective. When comparing LC-MS runs, elution times are not identical from run-to-run. Mass measurements may vary slightly. Software needs to be able to distort (stretch / squeeze) chromatogram to be able to compare runs.
37 Comparison of Quantitation Accuracy What level of quantitation accuracy can you expect with these methods? Dynamic Range: Most MS methods have a dynamic range accuracy of only about 1 order of magnitude: e.g., cannot distinguish reliably between 20 fold and 100 fold increase. Problem is usually the lower value in the comparison is too close to the signal to noise level. What is the minimum change that can be reliably measured? For label-free methods: 2 fold change For isotopic labeling methods: 30% change
38 Quantitate then MS: Gels Coomassie and Sypro stain densities are relatively linear with respect to amount. Silver stain is less quantitative. Complication: How do you know there is only one major protein component in the band. Greater separation with 2D gels reduces chances of multiple components per spot Multiple components may still be present if starting mixture was complex. 2D gels allow quantitation of a specific-modified version of protein (providing you know what modification state the spot corresponds to!) Will separate different splice variants. Quantitative Westerns: specific, but not very accurate quantitation.

39 Difference Gel Electrophoresis (DIGE) Fluorescent dyes which react with lysine residues. Fluoresce at different wavelengths, so can be differentially visualized. Labeling based on minimal labeling : only 5% of protein is singly labeled. Viswanathan, S. et al. Nat Protocols (2006)

40 Difference Gel Electrophoresis (DIGE) Label alters migration, so majority of protein (unlabeled) does not coelute Cutting out spot for ID is more complicated. Technology sold by GE Healthcare. Can only detect spots using their scanner. Very powerful analysis software available Decyder. Scanner and software cost a small fortune.
41 Conclusions Although not intrinsically quantitative, mass spectrometry can be used for many types of quantitation. Isotopic labeling is most accurate. Label-free approaches are simpler (and cheaper) and more appropriate for comparison of large numbers of samples, but are less accurate. Most quantitation is relative, but if one component is at a known concentration then absolute quantitation can be performed. Software for analyzing this type of data is still being developed.
Chapter 4. strategies for protein quantitation Ⅱ
 Proteomics Chapter 4. strategies for protein quantitation Ⅱ 1 Multiplexed proteomics Multiplexed proteomics is the use of fluorescent stains or probes with different excitation and emission spectra to
Proteomics Chapter 4. strategies for protein quantitation Ⅱ 1 Multiplexed proteomics Multiplexed proteomics is the use of fluorescent stains or probes with different excitation and emission spectra to
NPTEL VIDEO COURSE PROTEOMICS PROF. SANJEEVA SRIVASTAVA
 LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University
 Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Mass Spectrometry and Proteomics - Lecture 5 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously Proteomics Sample prep 144 Lecture 5 Quantitation techniques Search Algorithms Proteomics
Quantitative Proteomics
 Quantitative Proteomics Quantitation AND Mass Spectrometry Condition A Condition B Identify and quantify differently expressed proteins resulting from a change in the environment (stimulus, disease) Lyse
Quantitative Proteomics Quantitation AND Mass Spectrometry Condition A Condition B Identify and quantify differently expressed proteins resulting from a change in the environment (stimulus, disease) Lyse
Quantitative Proteomics
 BSPR workshop 16 th July 2010 Quantitative Proteomics Kathryn Lilley Cambridge Centre for Proteomics Department of Biochemistry University of Cambridge k.s.lilley@bioc.cam.ac.uk www.bio.cam.ac.uk/proteomics/
BSPR workshop 16 th July 2010 Quantitative Proteomics Kathryn Lilley Cambridge Centre for Proteomics Department of Biochemistry University of Cambridge k.s.lilley@bioc.cam.ac.uk www.bio.cam.ac.uk/proteomics/
Comprehensive support for quantitation
 Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Tutorial 1: Setting up your Skyline document
 Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
Atomic masses. Atomic masses of elements. Atomic masses of isotopes. Nominal and exact atomic masses. Example: CO, N 2 ja C 2 H 4
 High-Resolution Mass spectrometry (HR-MS, HRAM-MS) (FT mass spectrometry) MS that enables identifying elemental compositions (empirical formulas) from accurate m/z data 9.05.2017 1 Atomic masses (atomic
High-Resolution Mass spectrometry (HR-MS, HRAM-MS) (FT mass spectrometry) MS that enables identifying elemental compositions (empirical formulas) from accurate m/z data 9.05.2017 1 Atomic masses (atomic
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry
 Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics
 Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Computational Methods for Mass Spectrometry Proteomics
 Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Workshop: SILAC and Alternative Labeling Strategies in Quantitative Proteomics
 Workshop: SILAC and Alternative Labeling Strategies in Quantitative Proteomics SILAC and Stable Isotope Dimethyl-Labeling Approaches in Quantitative Proteomics Ho-Tak Lau, Hyong-Won Suh, Shao-En Ong UW
Workshop: SILAC and Alternative Labeling Strategies in Quantitative Proteomics SILAC and Stable Isotope Dimethyl-Labeling Approaches in Quantitative Proteomics Ho-Tak Lau, Hyong-Won Suh, Shao-En Ong UW
SRM assay generation and data analysis in Skyline
 in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Protein analysis using mass spectrometry
 Protein analysis using mass spectrometry Michael Stadlmeier 2017/12/18 Literature http://www.carellgroup.de/teaching/master 3 What is Proteomics? The proteome is: the entire set of proteins in a given
Protein analysis using mass spectrometry Michael Stadlmeier 2017/12/18 Literature http://www.carellgroup.de/teaching/master 3 What is Proteomics? The proteome is: the entire set of proteins in a given
Aplicació de la proteòmica a la cerca de Biomarcadors proteics Barcelona, 08 de Juny 2010
 Aplicació de la proteòmica a la cerca de Biomarcadors proteics Barcelona, 8 de Juny 21 Eliandre de Oliveira Plataforma de Proteòmica Parc Científic de Barcelona Protein Chemistry Proteomics Hypothesis-free
Aplicació de la proteòmica a la cerca de Biomarcadors proteics Barcelona, 8 de Juny 21 Eliandre de Oliveira Plataforma de Proteòmica Parc Científic de Barcelona Protein Chemistry Proteomics Hypothesis-free
Reagents. Affinity Tag (Biotin) Acid Cleavage Site. Figure 1. Cleavable ICAT Reagent Structure.
 Acid Cleavage Site. Figure 1. Cleavable ICAT Reagent Structure.") DATA SHEET Protein Expression Analysis Reagents Background The ultimate goal of proteomics is to identify and quantify proteins that are relevant to a given biological state; and to unearth networks of
DATA SHEET Protein Expression Analysis Reagents Background The ultimate goal of proteomics is to identify and quantify proteins that are relevant to a given biological state; and to unearth networks of
6 x 5 Ways to Ensure Your LC-MS/MS is Healthy
 6 x 5 Ways to Ensure Your LC-MS/MS is Healthy (Also known as - Tracking Performance with the 6 x 5 LC-MS/MS Peptide Reference Mixture) Mike Rosenblatt, Ph.D. Group Leader Mass Spec Reagents 215. We monitor
6 x 5 Ways to Ensure Your LC-MS/MS is Healthy (Also known as - Tracking Performance with the 6 x 5 LC-MS/MS Peptide Reference Mixture) Mike Rosenblatt, Ph.D. Group Leader Mass Spec Reagents 215. We monitor
SILAC and TMT. IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017
 SILAC and TMT IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017 UHPLC peak chosen at 26.47 min LC Mass at 571.36 chosen for MS/MS MS/MS MS This is a
SILAC and TMT IDeA National Resource for Proteomics Workshop for Graduate Students and Post-docs Renny Lan 5/18/2017 UHPLC peak chosen at 26.47 min LC Mass at 571.36 chosen for MS/MS MS/MS MS This is a
We are IntechOpen, the first native scientific publisher of Open Access books. International authors and editors. Our authors are among the TOP 1%
 We are IntechOpen, the first native scientific publisher of Open Access books 3,350 108,000 1.7 M Open access books available International authors and editors Downloads Our authors are among the 151 Countries
We are IntechOpen, the first native scientific publisher of Open Access books 3,350 108,000 1.7 M Open access books available International authors and editors Downloads Our authors are among the 151 Countries
Methods for proteome analysis of obesity (Adipose tissue)
") Methods for proteome analysis of obesity (Adipose tissue) I. Sample preparation and liquid chromatography-tandem mass spectrometric analysis Instruments, softwares, and materials AB SCIEX Triple TOF 5600
Methods for proteome analysis of obesity (Adipose tissue) I. Sample preparation and liquid chromatography-tandem mass spectrometric analysis Instruments, softwares, and materials AB SCIEX Triple TOF 5600
Protein Identification Using Tandem Mass Spectrometry. Nathan Edwards Informatics Research Applied Biosystems
 Protein Identification Using Tandem Mass Spectrometry Nathan Edwards Informatics Research Applied Biosystems Outline Proteomics context Tandem mass spectrometry Peptide fragmentation Peptide identification
Protein Identification Using Tandem Mass Spectrometry Nathan Edwards Informatics Research Applied Biosystems Outline Proteomics context Tandem mass spectrometry Peptide fragmentation Peptide identification
Key questions of proteomics. Bioinformatics 2. Proteomics. Foundation of proteomics. What proteins are there? Protein digestion
 s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
Quantitative analysis of the proteome
 BMG 744 Proteomics-Mass Spectrometry Quantitative analysis of the proteome Stephen Barnes, PhD sbarnes@uab.edu 1 The ASMS 1996-2007 www.tagate.com/western/wild_west.jpg 2 1 Proteomics Data Standards 2005
BMG 744 Proteomics-Mass Spectrometry Quantitative analysis of the proteome Stephen Barnes, PhD sbarnes@uab.edu 1 The ASMS 1996-2007 www.tagate.com/western/wild_west.jpg 2 1 Proteomics Data Standards 2005
Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent
 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent") Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent concentrations of PcTS (100 µm, blue; 500 µm, green; 1.5 mm,
Figure S1. Interaction of PcTS with αsyn. (a) 1 H- 15 N HSQC NMR spectra of 100 µm αsyn in the absence (0:1, black) and increasing equivalent concentrations of PcTS (100 µm, blue; 500 µm, green; 1.5 mm,
Chem 250 Unit 1 Proteomics by Mass Spectrometry
 Chem 250 Unit 1 Proteomics by Mass Spectrometry Article #1 Quantitative MS for proteomics: teaching a new dog old tricks. MacCoss MJ, Matthews DE., Anal Chem. 2005 Aug 1;77(15):294A-302A. 1. Synopsis 1.1.
Chem 250 Unit 1 Proteomics by Mass Spectrometry Article #1 Quantitative MS for proteomics: teaching a new dog old tricks. MacCoss MJ, Matthews DE., Anal Chem. 2005 Aug 1;77(15):294A-302A. 1. Synopsis 1.1.
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 *
 Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
Amine specific Labeling Reagents for Multiplexed Relative and Absolute Protein Quantitation
 Product Bulletin itraq Reagents itraq Reagents Amine specific Labeling Reagents for Multiplexed Relative and Absolute Protein Quantitation Background Proteomics research includes the characterization of
Product Bulletin itraq Reagents itraq Reagents Amine specific Labeling Reagents for Multiplexed Relative and Absolute Protein Quantitation Background Proteomics research includes the characterization of
Quantitative analysis of the proteome. Proteomics Data Standards
 BMG 744 Proteomics-Mass Spectrometry Quantitative analysis of the proteome Stephen Barnes, PhD sbarnes@uab.edu 1 Proteomics Data Standards 2005 MCP Paris guidelines 2008 HUPO MIAPE(Minimum Information
BMG 744 Proteomics-Mass Spectrometry Quantitative analysis of the proteome Stephen Barnes, PhD sbarnes@uab.edu 1 Proteomics Data Standards 2005 MCP Paris guidelines 2008 HUPO MIAPE(Minimum Information
UCD Conway Institute of Biomolecular & Biomedical Research Graduate Education 2009/2010
 EMERGING PROTEOMIC TECHNOLOGIES - MODULE SCHEDULE & OUTLINE 2010 Course Organiser: Dr. Giuliano Elia Module Co-ordinator: Dr Giuliano Elia Credits: 5 Date & Time Session & Topic Coordinator 14th April
EMERGING PROTEOMIC TECHNOLOGIES - MODULE SCHEDULE & OUTLINE 2010 Course Organiser: Dr. Giuliano Elia Module Co-ordinator: Dr Giuliano Elia Credits: 5 Date & Time Session & Topic Coordinator 14th April
Modeling Mass Spectrometry-Based Protein Analysis
 Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
MS-based proteomics to investigate proteins and their modifications
 MS-based proteomics to investigate proteins and their modifications Francis Impens VIB Proteomics Core October th 217 Overview Mass spectrometry-based proteomics: general workflow Identification of protein
MS-based proteomics to investigate proteins and their modifications Francis Impens VIB Proteomics Core October th 217 Overview Mass spectrometry-based proteomics: general workflow Identification of protein
Chapter 2 What are the Common Mass Spectrometry-Based Analyses Used in Biology?
 Chapter 2 What are the Common Mass Spectrometry-Based Analyses Used in Biology? Abstract Mass spectrometry is used in many field of research, such as biology, chemistry, geology, etc. The focus of this
Chapter 2 What are the Common Mass Spectrometry-Based Analyses Used in Biology? Abstract Mass spectrometry is used in many field of research, such as biology, chemistry, geology, etc. The focus of this
Proteomics. November 13, 2007
 Proteomics November 13, 2007 Acknowledgement Slides presented here have been borrowed from presentations by : Dr. Mark A. Knepper (LKEM, NHLBI, NIH) Dr. Nathan Edwards (Center for Bioinformatics and Computational
Proteomics November 13, 2007 Acknowledgement Slides presented here have been borrowed from presentations by : Dr. Mark A. Knepper (LKEM, NHLBI, NIH) Dr. Nathan Edwards (Center for Bioinformatics and Computational
Nature Methods: doi: /nmeth Supplementary Figure 1. Fragment indexing allows efficient spectra similarity comparisons.
 Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
Supplementary Figure 1 Fragment indexing allows efficient spectra similarity comparisons. The cost and efficiency of spectra similarity calculations can be approximated by the number of fragment comparisons
High-Throughput Protein Quantitation Using Multiple Reaction Monitoring
 High-Throughput Protein Quantitation Using Multiple Reaction Monitoring Application Note Authors Ning Tang, Christine Miller, Joe Roark, Norton Kitagawa and Keith Waddell Agilent Technologies, Inc. Santa
High-Throughput Protein Quantitation Using Multiple Reaction Monitoring Application Note Authors Ning Tang, Christine Miller, Joe Roark, Norton Kitagawa and Keith Waddell Agilent Technologies, Inc. Santa
Proteome-wide label-free quantification with MaxQuant. Jürgen Cox Max Planck Institute of Biochemistry July 2011
 Proteome-wide label-free quantification with MaxQuant Jürgen Cox Max Planck Institute of Biochemistry July 2011 MaxQuant MaxQuant Feature detection Data acquisition Initial Andromeda search Statistics
Proteome-wide label-free quantification with MaxQuant Jürgen Cox Max Planck Institute of Biochemistry July 2011 MaxQuant MaxQuant Feature detection Data acquisition Initial Andromeda search Statistics
PROTEOMICS IN VASCULAR BIOLOGY
 ESC Summerschool 2013 Nice ESC Summer School Cardiovascular Sciences June 18, 2013 PROTEOMICS IN VASCULAR BIOLOGY Prof dr Anton J.G. Horrevoets Molecular Cell Biology and Immunology VU Medical Center Amsterdam
ESC Summerschool 2013 Nice ESC Summer School Cardiovascular Sciences June 18, 2013 PROTEOMICS IN VASCULAR BIOLOGY Prof dr Anton J.G. Horrevoets Molecular Cell Biology and Immunology VU Medical Center Amsterdam
A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen
 994 DOI 10.1002/pmic.200700426 Proteomics 2008, 8, 994 999 TECHNICAL BRIEF A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen John M. Asara 1,
994 DOI 10.1002/pmic.200700426 Proteomics 2008, 8, 994 999 TECHNICAL BRIEF A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen John M. Asara 1,
TOMAHAQ Method Construction
 TOMAHAQ Method Construction Triggered by offset mass accurate-mass high-resolution accurate quantitation (TOMAHAQ) can be performed in the standard method editor of the instrument, without modifications
TOMAHAQ Method Construction Triggered by offset mass accurate-mass high-resolution accurate quantitation (TOMAHAQ) can be performed in the standard method editor of the instrument, without modifications
Comparative and Quantitative Global Proteomics Approaches: An Overview
 Proteomes 2013, 1, 180-218; doi:10.3390/proteomes1030180 Review OPEN ACCESS proteomes ISSN 2227-7382 www.mdpi.com/journal/proteomes Comparative and Quantitative Global Proteomics Approaches: An Overview
Proteomes 2013, 1, 180-218; doi:10.3390/proteomes1030180 Review OPEN ACCESS proteomes ISSN 2227-7382 www.mdpi.com/journal/proteomes Comparative and Quantitative Global Proteomics Approaches: An Overview
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics
 DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics Chih-Chiang Tsou 1,2, Dmitry Avtonomov 2, Brett Larsen 3, Monika Tucholska 3, Hyungwon Choi 4 Anne-Claude Gingras
Rapid Distinction of Leucine and Isoleucine in Monoclonal Antibodies Using Nanoflow. LCMS n. Discovery Attribute Sciences
 Rapid Distinction of Leucine and Isoleucine in Monoclonal Antibodies Using Nanoflow LCMS n Dhanashri Bagal *, Eddie Kast, Ping Cao Discovery Attribute Sciences Amgen, South San Francisco, California, United
Rapid Distinction of Leucine and Isoleucine in Monoclonal Antibodies Using Nanoflow LCMS n Dhanashri Bagal *, Eddie Kast, Ping Cao Discovery Attribute Sciences Amgen, South San Francisco, California, United
Proteolytic 18O-Labeling Strategies for Quantitative Proteomics
 See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/6709639 Proteolytic 18O-Labeling Strategies for Quantitative Proteomics ARTICLE in MASS SPECTROMETRY
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/6709639 Proteolytic 18O-Labeling Strategies for Quantitative Proteomics ARTICLE in MASS SPECTROMETRY
Protocol. Product Use & Liability. Contact us: InfoLine: Order per fax: www:
 Protocol SpikeTides Sets SpikeTides Sets_L heavy SpikeMix SpikeMix_L heavy Peptide Sets for relative quantification of Proteins in Mass Spectrometry Based Assays Contact us: InfoLine: +49-30-6392-7878
Protocol SpikeTides Sets SpikeTides Sets_L heavy SpikeMix SpikeMix_L heavy Peptide Sets for relative quantification of Proteins in Mass Spectrometry Based Assays Contact us: InfoLine: +49-30-6392-7878
TUTORIAL EXERCISES WITH ANSWERS
 TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
Protocol. Product Use & Liability. Contact us: InfoLine: Order per fax: www:
 Protocol SpikeTides Set TAA - light SpikeTides Set TAA_L - heavy Peptide Sets for relative quantification of Tumor Associated Antigens (TAAs) in SRM and MRM Assays Contact us: InfoLine: +49-30-6392-7878
Protocol SpikeTides Set TAA - light SpikeTides Set TAA_L - heavy Peptide Sets for relative quantification of Tumor Associated Antigens (TAAs) in SRM and MRM Assays Contact us: InfoLine: +49-30-6392-7878
Tandem mass spectra were extracted from the Xcalibur data system format. (.RAW) and charge state assignment was performed using in house software
 and charge state assignment was performed using in house software") Supplementary Methods Software Interpretation of Tandem mass spectra Tandem mass spectra were extracted from the Xcalibur data system format (.RAW) and charge state assignment was performed using in house
Supplementary Methods Software Interpretation of Tandem mass spectra Tandem mass spectra were extracted from the Xcalibur data system format (.RAW) and charge state assignment was performed using in house
CHEMICAL COVALENT LABELING AND TANDEM MASS SPECTROMETRY FOR TARGETED PROTEIN CHARACTERIZATION, QUANTIFICATION AND STRUCTURAL ANALYSIS YALI LU
 CHEMICAL COVALENT LABELING AND TANDEM MASS SPECTROMETRY FOR TARGETED PROTEIN CHARACTERIZATION, QUANTIFICATION AND STRUCTURAL ANALYSIS By YALI LU A DISSERTATION Submitted to Michigan State University in
CHEMICAL COVALENT LABELING AND TANDEM MASS SPECTROMETRY FOR TARGETED PROTEIN CHARACTERIZATION, QUANTIFICATION AND STRUCTURAL ANALYSIS By YALI LU A DISSERTATION Submitted to Michigan State University in
Introduction to Proteomics & Bottom-up Proteomics
 Used for MS Short Course at Tsinghua by R. Graham Cooks, Hao Chen, Zheng Ouyang, Andy Tao, Yu Xia and Lingjun Li Introduction to Proteomics & Bottom-up Proteomics W. Andy Tao Purdue University watao@purdue.edu
Used for MS Short Course at Tsinghua by R. Graham Cooks, Hao Chen, Zheng Ouyang, Andy Tao, Yu Xia and Lingjun Li Introduction to Proteomics & Bottom-up Proteomics W. Andy Tao Purdue University watao@purdue.edu
Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were
 Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were developed to allow the analysis of large intact (bigger than
Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were developed to allow the analysis of large intact (bigger than
Peter A. DiMaggio, Jr., Nicolas L. Young, Richard C. Baliban, Benjamin A. Garcia, and Christodoulos A. Floudas. Research
 Research A Mixed Integer Linear Optimization Framework for the Identification and Quantification of Targeted Post-translational Modifications of Highly Modified Proteins Using Multiplexed Electron Transfer
Research A Mixed Integer Linear Optimization Framework for the Identification and Quantification of Targeted Post-translational Modifications of Highly Modified Proteins Using Multiplexed Electron Transfer
Chemical Labeling Strategy for Generation of Internal Standards for Targeted Quantitative Proteomics
 Chemical Labeling Strategy for Generation of Internal Standards for Targeted Quantitative Proteomics mtraq Reagents Triplex Christie Hunter, Brian Williamson, Marjorie Minkoff AB SCIEX, USA The utility
Chemical Labeling Strategy for Generation of Internal Standards for Targeted Quantitative Proteomics mtraq Reagents Triplex Christie Hunter, Brian Williamson, Marjorie Minkoff AB SCIEX, USA The utility
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra. Andrew Keller
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
See for options on how to legitimately share published articles.
 Downloaded via 148.251.232.83 on December 26, 2018 at 05:35:30 (UTC). See https://pubs.acs.org/sharingguidelines for options on how to legitimately share published articles. 294 NLYTICL CHEMISTRY / UGUST
Downloaded via 148.251.232.83 on December 26, 2018 at 05:35:30 (UTC). See https://pubs.acs.org/sharingguidelines for options on how to legitimately share published articles. 294 NLYTICL CHEMISTRY / UGUST
Targeted protein quantification
 Targeted Quantitative Proteomics Targeted protein quantification with high-resolution, accurate-mass MS Highly selective Very sensitive Complex samples HR/AM A more complete quantitative proteomics picture
Targeted Quantitative Proteomics Targeted protein quantification with high-resolution, accurate-mass MS Highly selective Very sensitive Complex samples HR/AM A more complete quantitative proteomics picture
Isobaric Labeling-Based Relative Quantification in Shotgun Proteomics
 This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jpr Isobaric
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. pubs.acs.org/jpr Isobaric
Guide to Peptide Quantitation. Agilent clinical research
 Guide to Peptide Quantitation Agilent clinical research Peptide Quantitation for the Clinical Research Laboratory Peptide quantitation is rapidly growing in clinical research as scientists are translating
Guide to Peptide Quantitation Agilent clinical research Peptide Quantitation for the Clinical Research Laboratory Peptide quantitation is rapidly growing in clinical research as scientists are translating
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry
 Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
SERVA ICPL Kit (Cat.-No )
") INSTRUCTION MANUAL SERVA ICPL Kit (Cat.-No. 39230.01) SERVA Electrophoresis GmbH Carl-Benz-Str. 7 D-69115 Heidelberg Phone +49-6221-138400, Fax +49-6221-1384010 e-mail: info@serva.de http://www.serva.de
INSTRUCTION MANUAL SERVA ICPL Kit (Cat.-No. 39230.01) SERVA Electrophoresis GmbH Carl-Benz-Str. 7 D-69115 Heidelberg Phone +49-6221-138400, Fax +49-6221-1384010 e-mail: info@serva.de http://www.serva.de
In-gel digestion of immunoprecipitated proteins separated by SDS-PAGE
 In-gel digestion of immunoprecipitated proteins separated by SDS-PAGE (Lamond Lab / April 2008)! Perform all the pipetting steps in a laminar flow hood. We routinely do our digestions in our TC room hoods.
In-gel digestion of immunoprecipitated proteins separated by SDS-PAGE (Lamond Lab / April 2008)! Perform all the pipetting steps in a laminar flow hood. We routinely do our digestions in our TC room hoods.
Effective desalting and concentration of in-gel digest samples with Vivapure C18 Micro spin columns prior to MALDI-TOF analysis.
 Introduction The identification of proteins plays an important role in today s pharmaceutical and proteomics research. Commonly used methods for separating proteins from complex samples are 1D or 2D gels.
Introduction The identification of proteins plays an important role in today s pharmaceutical and proteomics research. Commonly used methods for separating proteins from complex samples are 1D or 2D gels.
Rapid and Sensitive Fluorescent Peptide Quantification Using LavaPep
 Rapid and Sensitive Fluorescent Peptide Quantification Using LavaPep Purpose To develop a fast, sensitive and robust fluorescent-based assay, to quantify peptides that is compatible with downstream proteomics
Rapid and Sensitive Fluorescent Peptide Quantification Using LavaPep Purpose To develop a fast, sensitive and robust fluorescent-based assay, to quantify peptides that is compatible with downstream proteomics
Purdue-UAB Botanicals Center for Age- Related Disease
 Purdue-UAB Botanicals Center for Age- Related Disease MALDI-TOF Mass Spectrometry Fingerprinting Technique Landon Wilson MALDI-TOF mass spectrometry is an advanced technique for rapid protein identification
Purdue-UAB Botanicals Center for Age- Related Disease MALDI-TOF Mass Spectrometry Fingerprinting Technique Landon Wilson MALDI-TOF mass spectrometry is an advanced technique for rapid protein identification
Workflow concept. Data goes through the workflow. A Node contains an operation An edge represents data flow The results are brought together in tables
 PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
LECTURE-13. Peptide Mass Fingerprinting HANDOUT. Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of
 LECTURE-13 Peptide Mass Fingerprinting HANDOUT PREAMBLE Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of proteins, drugs and many biological moieties to elucidate
LECTURE-13 Peptide Mass Fingerprinting HANDOUT PREAMBLE Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of proteins, drugs and many biological moieties to elucidate
SeqAn and OpenMS Integration Workshop. Temesgen Dadi, Julianus Pfeuffer, Alexander Fillbrunn The Center for Integrative Bioinformatics (CIBI)
") SeqAn and OpenMS Integration Workshop Temesgen Dadi, Julianus Pfeuffer, Alexander Fillbrunn The Center for Integrative Bioinformatics (CIBI) Mass-spectrometry data analysis in KNIME Julianus Pfeuffer,
SeqAn and OpenMS Integration Workshop Temesgen Dadi, Julianus Pfeuffer, Alexander Fillbrunn The Center for Integrative Bioinformatics (CIBI) Mass-spectrometry data analysis in KNIME Julianus Pfeuffer,
Translational Biomarker Core
 Translational Biomarker Core Instrumentation Thermo Scientific TSQ Quantum Triple Quadrupole Mass Spectrometers. There are two TSQ Quantum Ultra AM instruments available in the TBC. The TSQ Quantum Ultra
Translational Biomarker Core Instrumentation Thermo Scientific TSQ Quantum Triple Quadrupole Mass Spectrometers. There are two TSQ Quantum Ultra AM instruments available in the TBC. The TSQ Quantum Ultra
Advances in quantitative proteomics using stable isotope tags
 Advances in quantitative proteomics using stable isotope tags Mark R. Flory, Timothy J. Griffin, Daniel Martin and Ruedi Aebersold A great deal of current biological and clinical research is directed at
Advances in quantitative proteomics using stable isotope tags Mark R. Flory, Timothy J. Griffin, Daniel Martin and Ruedi Aebersold A great deal of current biological and clinical research is directed at
Towards the Prediction of Protein Abundance from Tandem Mass Spectrometry Data
 Towards the Prediction of Protein Abundance from Tandem Mass Spectrometry Data Anthony J Bonner Han Liu Abstract This paper addresses a central problem of Proteomics: estimating the amounts of each of
Towards the Prediction of Protein Abundance from Tandem Mass Spectrometry Data Anthony J Bonner Han Liu Abstract This paper addresses a central problem of Proteomics: estimating the amounts of each of
Developing Algorithms for the Determination of Relative Abundances of Peptides from LC/MS Data
 Developing Algorithms for the Determination of Relative Abundances of Peptides from LC/MS Data RIPS Team Jake Marcus (Project Manager) Anne Eaton Melanie Kanter Aru Ray Faculty Mentors Shawn Cokus Matteo
Developing Algorithms for the Determination of Relative Abundances of Peptides from LC/MS Data RIPS Team Jake Marcus (Project Manager) Anne Eaton Melanie Kanter Aru Ray Faculty Mentors Shawn Cokus Matteo
Genome wide analysis of protein and mrna half lives reveals dynamic properties of mammalian gene expression
 Genome wide analysis of protein and mrna half lives reveals dynamic properties of mammalian gene expression Matthias Selbach Cell Signaling and Mass Spectrometry Max Delbrück Center for Molecular Medicine
Genome wide analysis of protein and mrna half lives reveals dynamic properties of mammalian gene expression Matthias Selbach Cell Signaling and Mass Spectrometry Max Delbrück Center for Molecular Medicine
Increasing the Multiplexing of Protein Quantitation from 6- to 10-Plex with Reporter Ion Isotopologues
 Increasing the Multiplexing of Protein Quantitation from 6- to 1-Plex with Reporter Ion Isotopologues Rosa Viner, 1 Ryan Bomgarden, 2 Michael Blank, 1 John Rogers 2 1 Thermo Fisher Scientific, San Jose,
Increasing the Multiplexing of Protein Quantitation from 6- to 1-Plex with Reporter Ion Isotopologues Rosa Viner, 1 Ryan Bomgarden, 2 Michael Blank, 1 John Rogers 2 1 Thermo Fisher Scientific, San Jose,
Lecture 15: Realities of Genome Assembly Protein Sequencing
 Lecture 15: Realities of Genome Assembly Protein Sequencing Study Chapter 8.10-8.15 1 Euler s Theorems A graph is balanced if for every vertex the number of incoming edges equals to the number of outgoing
Lecture 15: Realities of Genome Assembly Protein Sequencing Study Chapter 8.10-8.15 1 Euler s Theorems A graph is balanced if for every vertex the number of incoming edges equals to the number of outgoing
Electron Transfer Dissociation of N-linked Glycopeptides from a Recombinant mab Using SYNAPT G2-S HDMS
 Electron Transfer Dissociation of N-linked Glycopeptides from a Recombinant mab Using SYNAPT G2-S HDMS Jonathan P. Williams, Jeffery M. Brown, Stephane Houel, Ying Qing Yu, and Weibin Chen Waters Corporation,
Electron Transfer Dissociation of N-linked Glycopeptides from a Recombinant mab Using SYNAPT G2-S HDMS Jonathan P. Williams, Jeffery M. Brown, Stephane Houel, Ying Qing Yu, and Weibin Chen Waters Corporation,
Proteomics: the first decade and beyond. (2003) Patterson and Aebersold Nat Genet 33 Suppl: from
 Patterson and Aebersold Nat Genet 33 Suppl: from") Advances in mass spectrometry and the generation of large quantities of nucleotide sequence information, combined with computational algorithms that could correlate the two, led to the emergence of proteomics
Advances in mass spectrometry and the generation of large quantities of nucleotide sequence information, combined with computational algorithms that could correlate the two, led to the emergence of proteomics
Peptide Targeted Quantification By High Resolution Mass Spectrometry A Paradigm Shift? Zhiqi Hao Thermo Fisher Scientific San Jose, CA
 Peptide Targeted Quantification By High Resolution Mass Spectrometry A Paradigm Shift? Zhiqi Hao Thermo Fisher Scientific San Jose, CA Proteomics is Turning Quantitative Hmmm.. Which ones are my targets?
Peptide Targeted Quantification By High Resolution Mass Spectrometry A Paradigm Shift? Zhiqi Hao Thermo Fisher Scientific San Jose, CA Proteomics is Turning Quantitative Hmmm.. Which ones are my targets?
The Agilent 6495 Triple Quadrupole LC/MS: Peptide Quantitation Performance
 The Agilent 495 Triple Quadrupole LC/MS: Peptide Quantitation Performance Technical Overview Introduction Sample complexity and the low concentration of certain biomarkers are the major challenges encountered
The Agilent 495 Triple Quadrupole LC/MS: Peptide Quantitation Performance Technical Overview Introduction Sample complexity and the low concentration of certain biomarkers are the major challenges encountered
WADA Technical Document TD2003IDCR
 IDENTIFICATION CRITERIA FOR QUALITATIVE ASSAYS INCORPORATING CHROMATOGRAPHY AND MASS SPECTROMETRY The appropriate analytical characteristics must be documented for a particular assay. The Laboratory must
IDENTIFICATION CRITERIA FOR QUALITATIVE ASSAYS INCORPORATING CHROMATOGRAPHY AND MASS SPECTROMETRY The appropriate analytical characteristics must be documented for a particular assay. The Laboratory must
Qualitative Proteomics (how to obtain high-confidence high-throughput protein identification!)
") Qualitative Proteomics (how to obtain high-confidence high-throughput protein identification!) James A. Mobley, Ph.D. Director of Research in Urology Associate Director of Mass Spectrometry (contact: mobleyja@uab.edu)
Qualitative Proteomics (how to obtain high-confidence high-throughput protein identification!) James A. Mobley, Ph.D. Director of Research in Urology Associate Director of Mass Spectrometry (contact: mobleyja@uab.edu)
Quantitative Analysis of Opioids Using a Triple-Quadrupole GC/MS/MS
 PO-CON1408E Quantitative Analysis of Opioids Using a Pittcon 2014 1090-8P Laura Chambers, Richard Whitney, Ph.D., Nicole M. Lock, Zhuangzhi Max Wang, Ph.D., Clifford M. Taylor; Shimadzu Scientific Instruments,
PO-CON1408E Quantitative Analysis of Opioids Using a Pittcon 2014 1090-8P Laura Chambers, Richard Whitney, Ph.D., Nicole M. Lock, Zhuangzhi Max Wang, Ph.D., Clifford M. Taylor; Shimadzu Scientific Instruments,
Designed for Accuracy. Innovation with Integrity. High resolution quantitative proteomics LC-MS
 Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
Designed for Accuracy High resolution quantitative proteomics Innovation with Integrity LC-MS Setting New Standards in Accuracy The development of mass spectrometry based proteomics approaches has dramatically
Introduction to Proteomics: Fragmentation of protonated peptides and manual sequencing
 Introduction to Proteomics: ragmentation of protonated peptides and manual sequencing Árpád Somogyi CCIC SP SU Summer Workshop S/S hod using ESI Ion rap (Bottom up) 1 An alternative strategy for complex
Introduction to Proteomics: ragmentation of protonated peptides and manual sequencing Árpád Somogyi CCIC SP SU Summer Workshop S/S hod using ESI Ion rap (Bottom up) 1 An alternative strategy for complex
Exam I Answer Key: Summer 2006, Semester C
 1. Which of the following tripeptides would migrate most rapidly towards the negative electrode if electrophoresis is carried out at ph 3.0? a. gly-gly-gly b. glu-glu-asp c. lys-glu-lys d. val-asn-lys
1. Which of the following tripeptides would migrate most rapidly towards the negative electrode if electrophoresis is carried out at ph 3.0? a. gly-gly-gly b. glu-glu-asp c. lys-glu-lys d. val-asn-lys
Identification of Human Hemoglobin Protein Variants Using Electrospray Ionization-Electron Transfer Dissociation Mass Spectrometry
 Identification of Human Hemoglobin Protein Variants Using Electrospray Ionization-Electron Transfer Dissociation Mass Spectrometry Jonathan Williams Waters Corporation, Milford, MA, USA A P P L I C AT
Identification of Human Hemoglobin Protein Variants Using Electrospray Ionization-Electron Transfer Dissociation Mass Spectrometry Jonathan Williams Waters Corporation, Milford, MA, USA A P P L I C AT
Highly Sensitive and Rugged GC/MS/MS Tool
 Highly Sensitive and Rugged GC/MS/MS Tool For Pesticide Multiresidue Analysis in Food Samples Agilent 7 Series Triple Quadrupole GC/MS. The world s first MS/MS designed specifically for GC Analysis Introduction
Highly Sensitive and Rugged GC/MS/MS Tool For Pesticide Multiresidue Analysis in Food Samples Agilent 7 Series Triple Quadrupole GC/MS. The world s first MS/MS designed specifically for GC Analysis Introduction
Optimization and Use of Peptide Mass Measurement Accuracy in Shotgun Proteomics* S
 Research Optimization and Use of Peptide Mass Measurement Accuracy in Shotgun Proteomics* S Wilhelm Haas, Brendan K. Faherty, Scott A. Gerber, Joshua E. Elias, Sean A. Beausoleil, Corey E. Bakalarski,
Research Optimization and Use of Peptide Mass Measurement Accuracy in Shotgun Proteomics* S Wilhelm Haas, Brendan K. Faherty, Scott A. Gerber, Joshua E. Elias, Sean A. Beausoleil, Corey E. Bakalarski,
Yifei Bao. Beatrix. Manor Askenazi
 Detection and Correction of Interference in MS1 Quantitation of Peptides Using their Isotope Distributions Yifei Bao Department of Computer Science Stevens Institute of Technology Beatrix Ueberheide Department
Detection and Correction of Interference in MS1 Quantitation of Peptides Using their Isotope Distributions Yifei Bao Department of Computer Science Stevens Institute of Technology Beatrix Ueberheide Department
WADA Technical Document TD2015IDCR
 MINIMUM CRITERIA FOR CHROMATOGRAPHIC-MASS SPECTROMETRIC CONFIRMATION OF THE IDENTITY OF ANALYTES FOR DOPING CONTROL PURPOSES. The ability of a method to identify an analyte is a function of the entire
MINIMUM CRITERIA FOR CHROMATOGRAPHIC-MASS SPECTROMETRIC CONFIRMATION OF THE IDENTITY OF ANALYTES FOR DOPING CONTROL PURPOSES. The ability of a method to identify an analyte is a function of the entire
Quantitative analysis of small molecules in biological samples. Jeevan Prasain, Ph.D. Department of Pharmacology & Toxicology, UAB.
 Quantitative analysis of small molecules in biological samples 100 Jeevan Prasain, Ph.D. Department of Pharmacology & Toxicology, UAB % 0 300 500 700 900 1100 1300 1500 1700 m/z Class Overview Introduction
Quantitative analysis of small molecules in biological samples 100 Jeevan Prasain, Ph.D. Department of Pharmacology & Toxicology, UAB % 0 300 500 700 900 1100 1300 1500 1700 m/z Class Overview Introduction
Protein separation and characterization
 Address:800 S Wineville Avenue, Ontario, CA 91761,USA Website:www.aladdin-e.com Email USA: tech@aladdin-e.com Email EU: eutech@aladdin-e.com Email Asia Pacific: cntech@aladdin-e.com Protein separation
Address:800 S Wineville Avenue, Ontario, CA 91761,USA Website:www.aladdin-e.com Email USA: tech@aladdin-e.com Email EU: eutech@aladdin-e.com Email Asia Pacific: cntech@aladdin-e.com Protein separation
Overview - MS Proteomics in One Slide. MS masses of peptides. MS/MS fragments of a peptide. Results! Match to sequence database
 Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Overview - MS Proteomics in One Slide Obtain protein Digest into peptides Acquire spectra in mass spectrometer MS masses of peptides MS/MS fragments of a peptide Results! Match to sequence database 2 But
Chapter 5. Complexation of Tholins by 18-crown-6:
 5-1 Chapter 5. Complexation of Tholins by 18-crown-6: Identification of Primary Amines 5.1. Introduction Electrospray ionization (ESI) is an excellent technique for the ionization of complex mixtures,
5-1 Chapter 5. Complexation of Tholins by 18-crown-6: Identification of Primary Amines 5.1. Introduction Electrospray ionization (ESI) is an excellent technique for the ionization of complex mixtures,
Biochemistry 3100 Sample Problems Chemical and Physical Methods
 (1) Describe the function of the following compounds in chemical sequencing, synthesis and modification: (a) Dithiothreitol (c) t-butyloxycarbonyl chloride (b) Dicyclohexyl carbodiimide (d) Cyanogen Bromide
(1) Describe the function of the following compounds in chemical sequencing, synthesis and modification: (a) Dithiothreitol (c) t-butyloxycarbonyl chloride (b) Dicyclohexyl carbodiimide (d) Cyanogen Bromide
Mass Spectrometry Based De Novo Peptide Sequencing Error Correction
 Mass Spectrometry Based De Novo Peptide Sequencing Error Correction by Chenyu Yao A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Mathematics
Mass Spectrometry Based De Novo Peptide Sequencing Error Correction by Chenyu Yao A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Master of Mathematics
HOWTO, example workflow and data files. (Version )
") HOWTO, example workflow and data files. (Version 20 09 2017) 1 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD
HOWTO, example workflow and data files. (Version 20 09 2017) 1 Introduction: SugarQb is a collection of software tools (Nodes) which enable the automated identification of intact glycopeptides from HCD
Biological Mass Spectrometry
 Biochemistry 412 Biological Mass Spectrometry February 13 th, 2007 Proteomics The study of the complete complement of proteins found in an organism Degrees of Freedom for Protein Variability Covalent Modifications
Biochemistry 412 Biological Mass Spectrometry February 13 th, 2007 Proteomics The study of the complete complement of proteins found in an organism Degrees of Freedom for Protein Variability Covalent Modifications
Mass spectrometry based proteomics
 Practice-oriented, student-friendly modernization of the biomedical education for strengthening the international competitiveness of the rural Hungarian universities TÁMOP-4.1.1.C-13/1/KONV-2014-0001 Mass
Practice-oriented, student-friendly modernization of the biomedical education for strengthening the international competitiveness of the rural Hungarian universities TÁMOP-4.1.1.C-13/1/KONV-2014-0001 Mass
Supporting Information. Experimental details
 Electronic Supporting Information Experimental details Chemicals and reagents Pseudoboehmite (78.4 wt% Al 2 O 3 ), phosphoric acid (85 wt%), triethylamine (TEA, 99%), tetrabutyl titanate (IV) (99%) and
Electronic Supporting Information Experimental details Chemicals and reagents Pseudoboehmite (78.4 wt% Al 2 O 3 ), phosphoric acid (85 wt%), triethylamine (TEA, 99%), tetrabutyl titanate (IV) (99%) and