Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were
|
|
|
- Martha McKenzie
- 5 years ago
- Views:
Transcription
 molecule.")
1 Mass spectrometry has been used a lot in biology since the late 1950 s. However it really came into play in the late 1980 s once methods were developed to allow the analysis of large intact (bigger than 1,000 Daltons) molecule. Two soft ionization techniques, Electrospray and Matrix Assisted Laser Desorption led to a huge jump in popularity as did the development of much more compact (bench top rather than whole laboratory) mass spectrometers. 1!
 where they can be manipulated by electrostatic and/or magnetic fields and separated.")
2 Mass spectrometry is dependant on the ability to turn the analyte of interest into individual but intact, charged molecules in the gas phase. These are released into a low pressure area (a vacuum from 10-3 to torr) where they can be manipulated by electrostatic and/or magnetic fields and separated. The force fields require the molecule to be charged, neutral molecules cannot be manipulated and are lost from the system. 2!

3 Here the spectrum of a small molecule caffeine, shows basically one main peak and a few larger but much less intense peaks. 3!
, S33 (0.8), S34 (4.2%) These atoms are common in biological systems. Since the heavy isotopes are rare, this is not significant for most small molecules.")
4 Isotope distributions in nature: Carbon C12 (98.9%), C13 (1.1%), C14 (small) Hydrogen H1 (99.98%), Deuterium (0.015%), Tritium (small) Oxygen O16 (99.8%), O17 (0.04%), O18 (0.2%) Sulphur S32 (95.0%), S33 (0.8), S34 (4.2%) These atoms are common in biological systems. Since the heavy isotopes are rare, this is not significant for most small molecules. Once one starts to look at biological molecules like peptides, the mass changes can be significant. For example, insulin has a mass of over 6,000 and thus a 1% shift by each of carbon, oxygen, hydrogen etc. can spread the mass from the lightest molecule (all C12, H1, O16 etc) to the heaviest (all C13, D2, O18 etc) over a range of mass units. However some elements such as bromine have almost equally distributed isotopes (Br % and Br %) which give rise to spectra with all peaks appearing as doublets. The effect of the isotope distribution on the shape of the spectrum (sometimes called the isotope envelope) becomes much more pronounced when analysing larger biomolecules. Here the various masses are shown for the peptide hormone glucagon. 4!

5 Here is a spectrum of pure substance P, a peptide. Since a mass spectrometer always measures m/z the mass to charge ratio, two peaks are found. Here we see two main peaks, the doubly and singly charged ions. 5!
 divided by the")
6 In order to find out what you are looking at, i.e. is it singly, doubly etc charged, one looks at the details of the isotope distribution. Since isotopes are always one mass unit apart, if the peaks are one unit apart, the ion is singly charge since the mass difference (1 mass unit) divided by the charge (1+) is 1. 6!

7 If the peaks are 0.5 mass units apart, they are doubly charged. Remember m/ z,that the mass difference is 1 unit between isotopes and if the charge is 2 then the the mass change is 1/2 = 0.5 7!

8 An electric field accelerates the ions to a high speed. After this, they are directed into a magnetic field which applies a force to each ion perpendicular to the plane defined by the particles' direction of travel and the magnetic field lines. This force deflects the ions (makes them curve instead of traveling in a straight line) to varying degrees depending on their mass-to-charge ratio. Lighter ions get deflected more than the heavier ions. This is due to Newton's second law of motion. The acceleration of a particle is inversely proportional to its mass. Therefore, the magnetic field deflects the lighter ions more than it does the heavier ions. The detector measures the deflection of each resulting ion beam. From this measurement, the mass-to-charge ratios of all the ions produced in the source can be determined. 8!
9 All of these mass spectrometers have many things in common. Firstly they possess an Ion Source, that produces ions, an Analyzer that sorts them in some way by their masses, and a Detector that measures the relative intensities of different masses. The underlying principle of all mass spectrometers is that the paths of gas phase ions in electric and magnetic fields are dependent on their mass-to-charge ratios which is used by the analyzer to distinguish the ions from one another. 9!

10 The simplest type of mass spectrometer involves a single mass separation stage. The ions that are passed from the source into the mass analyzer give a simple read out of the intact molecular ions (assuming the ionization method is soft enough) 10!

11 As interest grew in analyzing the structure of molecules, more complex mass spectrometers were developed with two mass separation stages. The first stage allows the selection of unique molecules by creating a single mass window to filter away other molecular species. The isolated molecule can then be broken into smaller components by a variety of techniques and the resultant fragment ions can be analysed in the second mass seperation stage. This is called tandem mass spectrometry or MS/MS since it originally was carried out using two mass spectrometers joined together in tandem. 11!

12 Fragmentation of gas-phase ions is essential to tandem mass spectrometry and occurs between different stages of mass analysis. There are many methods used to fragment the ions and can result in different types of fragmentation and thus different information about the structure and composition of the molecule. There are a number of different tandem MS experiments, which each have their own applications and offer their own information. An instrument equipped for tandem MS can still be used to run MS experiments. Tandem MS can be done in either time or space. Tandem MS in space involves the physical separation of the instrument components (QqQ or QTOF), tandem MS in time involves the use of an ion trap. Post-source fragmentation is most often what is being used in a tandem mass spectrometry experiment. Energy can also be added to the, usually already vibrationally excited, ions through post-source collisions with neutral atoms or molecules, the absorption of radiation, or the transfer or capture of an electron by a multiply charged ion. Collision-induced dissociation (CID), also called collisionally activated dissociation (CAD), involves the collision of an ion with a neutral atom or molecule in the gas phase and subsequent dissociation of the ion. In mass spectrometry, collision-induced dissociation (CID), referred to by some as collisionally activated dissociation (CAD), is a mechanism by which to fragment molecular ions in the gas phase. The molecular ions are usually accelerated by some electrical potential to high kinetic energy in the vacuum of a mass spectrometer and then allowed to collide with neutral gas molecules (often helium, nitrogen or argon). In the collision some of the kinetic energy is converted into internal energy which results in bond breakage and the fragmentation of the molecular ion into smaller fragments. These fragment ions can then be analyzed by a mass spectrometer. In peptide analysis, CID cleaves randomly along the peptide backbone producing b and y ions (see later section 12!

13 Two types of MS/MS experiments can be carried out depending on the instrument type being used. The first approach developed was tandem in space, in which the parent molecule of interest is fragmented in one part of the instrument before being moved to a second part for the analysis of the daughter (fragment) ions. 13!

14 The alternative to tandem in space, is the type of experiment that is carried out in an ion trap; tandem in time. Here the isolation of the parent molecule and the analysis of the daughter ions produced by fragmentation occur in the same part of the instrument. The two processes are merely separated by time, the parent isolation occurs first, then the fragmentation and finally the daughter analysis is carried out in the same part of the trap. 14!

15 Genomics began with the goal of sequencing entire genomes. To accomplish this task, two different sequencing approaches were developed. These methods can be thought of in the following way: Imagine that you have the complete works of an author, written in a language that you studied in school, but never became fluent in. Moreover, the books are in such bad shape that if you open them, they disintegrate. You have two alternatives. You can remove one page at a time, preserve it and decipher it. Or you can open all the books at once and then pick up the fragments of paper and use the words on them to figure out how they fit together. The page-by-page approach to sequencing the human genome was used by the public genome-sequencing consortium. This group first figured out how all the pages fit together and then deciphered all the words on each page. Finally, it assembled the pages back together to produce the whole genome. The advantage of this approach is that it is very precise. The disadvantage is that it takes a long time. The biotechnology company Celera used the other method, called whole genome shotgun sequencing, in its competing effort to sequence the human genome. This method is equivalent to figuring out what s written on all the fragments of paper from all of the volumes and then figuring out how they piece together. To do this procedure effectively requires starting with several copies of each volume so that overlaps among the fragments can be found. The number of original copies is referred to as coverage. To produce a high-quality sequence by this method usually requires eight- to tenfold coverage. The disadvantage of this method is that you rarely get the whole sequence to line up. The advantage is that the portion of the sequence that does line up is acquired much more rapidly than via the pageby-page method. 15!

16 Proteins can be identified in simple mixtures by digesting them with an enzyme and then measuring the masses of the peptides formed. The set of masses is called the peptide fingerprint. A database is made containing all the proteins in the species genome and the masses of all the peptides from each protein produced by a certain enzyme are calculated. Thus each protein has a theoretical peptide fingerprint. The experimental fingerprint is then compared to all the theoretical fingerprints and the best match is calculated. This should be the correct identity of the unknown protein. 16!

17 Fingerprints are generated by using specific proteases. These are ones that cut after known amino-acids and hence one can predict theoretically which peptides will be formed.trypsin is the most commonly used protease in proteomics studies since it cuts after arginine and lysine and on average generates peptides that are around 12 amino acids long on average. This is ideal for ESI MS/MS analysis. 17!

18 A specific enzyme, here trypsin, is used to cut the protein into peptides. Trypsin cuts after arginine (R) and lysine (K) and the masses of the peptides can then be calculated. The experimentally determined masses are then searched against the theoretical masses in the fingerprint database to try and find the best match between the two sets of masses. The result of the search is returned as a list of matches according a probability of this not occuring at random. 18!

19 Here the output results from database search using the popular Mascot program are shown. A graph is shown to aid visualization. The green box indicates an area where the probability of a hit being correct is less than the significance threshold set, usually The red bars outside the box indicate proteins that are likely hits. 19!

20 The first time a peptide match to a query (one spectrum) appears in the report, it is shown in bold face. Whenever the top ranking peptide match appears, it is shown in red. This means that protein hits with peptide matches that are both bold and red are the most likely assignments. These hits represent the highest scoring protein that contains one or more top ranking peptide matches. 20!

21 The concept of shotgun proteomics is shown above. Instead of separating the proteins, the entire cell extract is digested with proteases and then the complex mixture is separated. The peptides are eluted from the final separation method, usually reversed-phase chromatography directly into the mass spectrometer where they are automatically subjected to MS/MS analysis. The peptides are identified in a similar way to how proteins are identified. Maybe 10 peptides are entering the mass spectrometer. The MS picks automatically the most intense, isolates it (throwing away the other 9 peptides) and then smashes it into pieces. The mass of the peptide is used to search the database to find all peptides with the same mass. The fragmentation spectra of all these peptides are then calculated and compared to the experimental fragments observed. The best matching peptide sequence is then selected. 21!

22 Here an automatic RP-HPLC-MS/MS run is shown. The mass spectrometer first accumulates a normal MS scan. It finds the 10 most intense peaks. It uses a mass window of around 10 to prevent picking all the isotopes in a intense peak envelope. The mass spectrometer then sequentially performs MS/MS on each of the ten peaks and then returns to MS mode. The ten peaks are then placed in an exclusion list which tells the mass spectrometer to ignore these masses for the next 5 minutes to ensure they have all eluted and are not repeatedly analysed. The next ten most intense peaks are then determined and scheduled for MS/MS: 22!

23 The experimental data is generated by the automatic accumulation of MS/MS spectra of tryptic peptides from the multi-dimensional peptide separation. A list of intact peptide masses, each with a list of their fragment masses is generated. In a manner analogous to protein fingerprinting, a theoretical in silico list of the masses of all the tryptic peptides predicted for a specific genome together with their predicted fragment ions is generated. In a first pass, the best theoretical 1000 matching peptide intact massesis generated for each experimental parent mass. Then a cross-correlation analysis is done between the experimental MS/MS spectrum and every theoretical spectrum. The crosscorrelation indicates which is the best matching spectrum and again the probability of the match not occurring at random is calculated. 23!
24 This shows the MASCOT output for such search. The green area shows insignificant matches and the red boxes indicate significant protein identifications. 24!
25 25!

26 26!
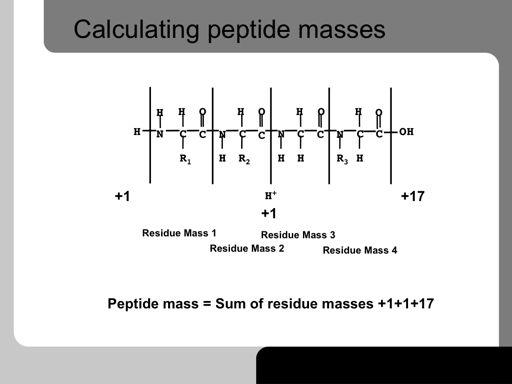
27 27!
28 28!
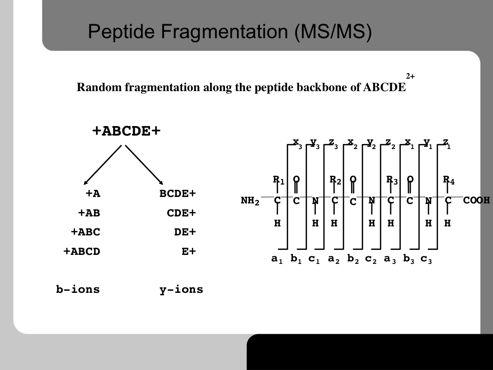
29 29!
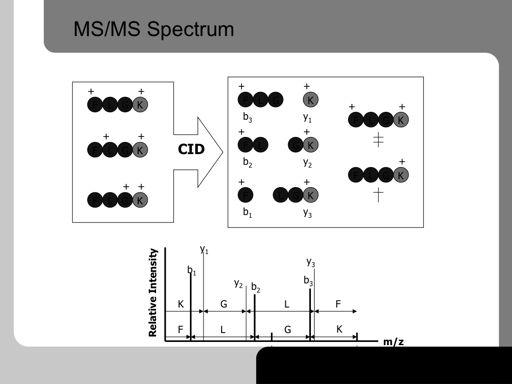
30 30!
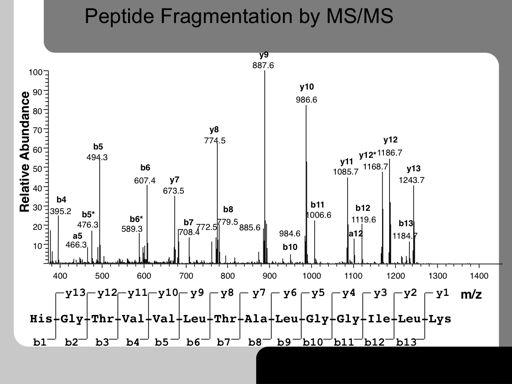
31 31!

32 32!
33 This shows the MASCOT output for such search. The green area shows insignificant matches and the red boxes indicate significant protein identifications. 33!
34 34

35 35

36 36
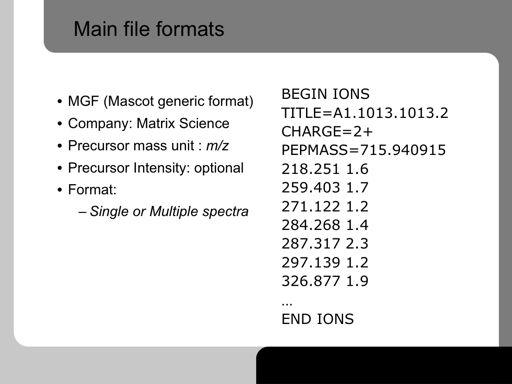
37 37

38 Nominal values: important: we have discret space, unit: 1 m/z 38!
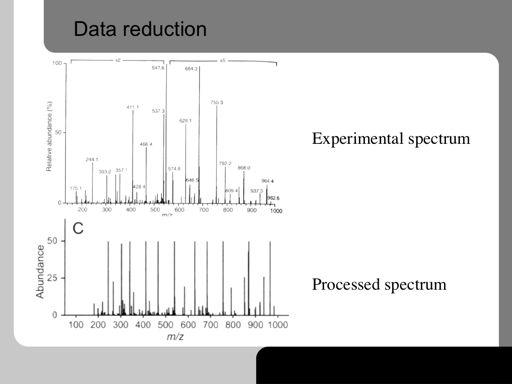
39 39!

40 40!
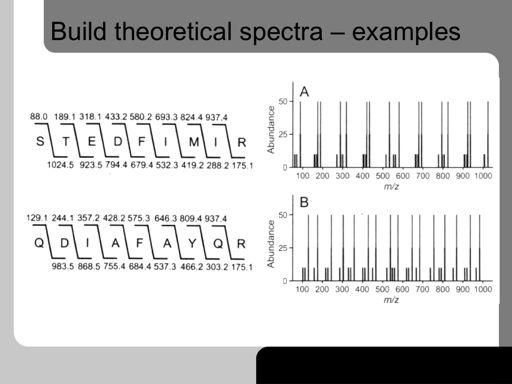
41 41!
42 42!
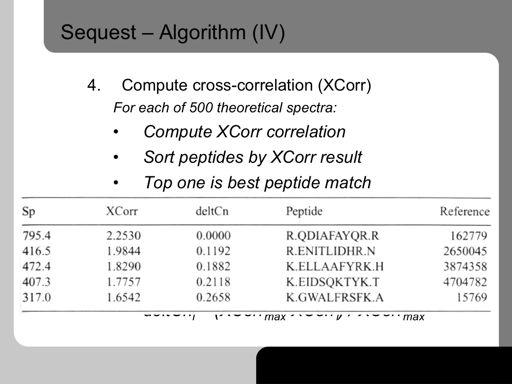
43 43!

44 44!

45 45!

46 46!
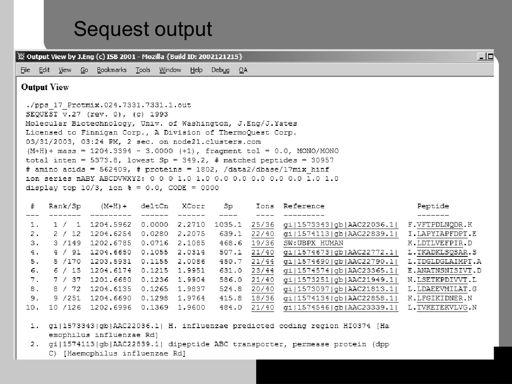
47 47!
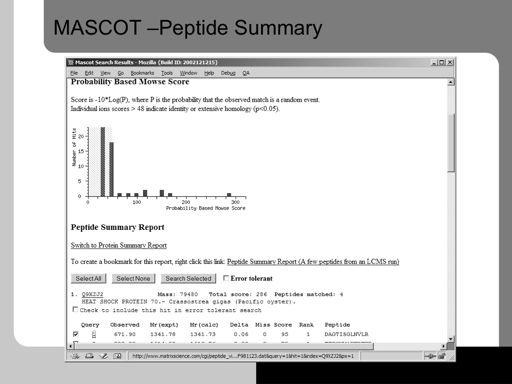
48 48!
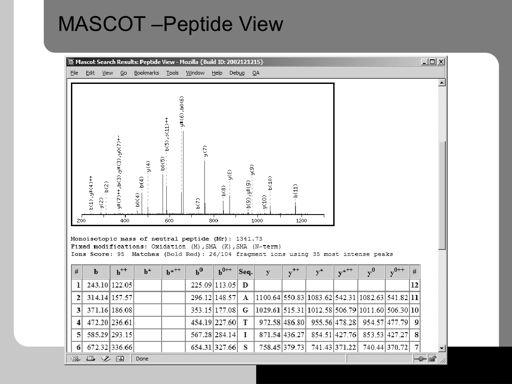
49 49!
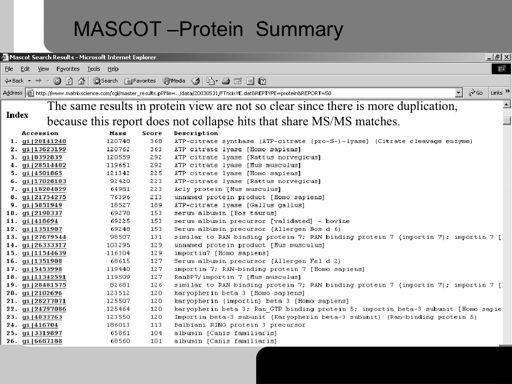
50 50!

51 51!

52 In bioinformatics, Basic Local Alignment Search Tool, or BLAST, is an algorithm for comparing primary biological sequence information, such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences. A BLAST search enables a researcher to compare a query sequence with a library or database of sequences, and identify library sequences that resemble the query sequence above a certain threshold. BLAST searches for high scoring sequence alignments between the query sequence and sequences in the database using a heuristic approach that approximates the Smith- Waterman algorithm. The exhaustive Smith-Waterman approach is too slow for searching large genomic databases such as GenBank. Therefore, the BLAST algorithm uses a heuristic approach that is less accurate than the Smith- Waterman but over 50 times faster. The speed and relatively good accuracy of BLAST are the key technical innovation of the BLAST programs. The BLAST algorithm can be conceptually divided into three stages. In the first stage, BLAST searches for exact matches of a small fixed length W between the query and sequences in the database. For example, given the sequences AGTTAC and ACTTAG and a word length W = 3, BLAST would identify the matching substring TTA that is common to both sequences. These exact matches are known as seeds. By default, W = 11 is used for nucleic seeds. In the second stage, BLAST tries to extend the match in both directions, starting at the seed. The ungapped alignment process extends the initial seed match of length W in each direction in an attempt to boost the alignment score. If a high-scoring un-gapped alignment is found, the database sequence passes on to the third stage. 52!
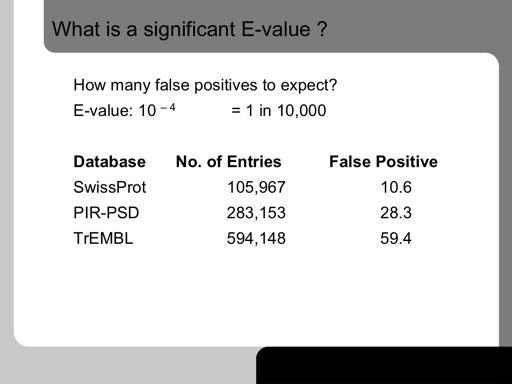
53 53!

54 54!

55 55!

56 56!

57 57!

58 58!

59 59!
60 60!
61 61!
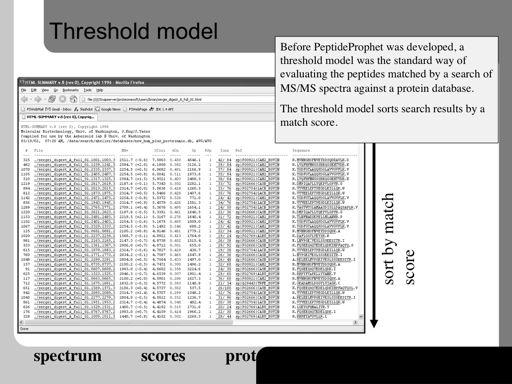
62 62!

63 63!

64 64!

65 65!

66 66!
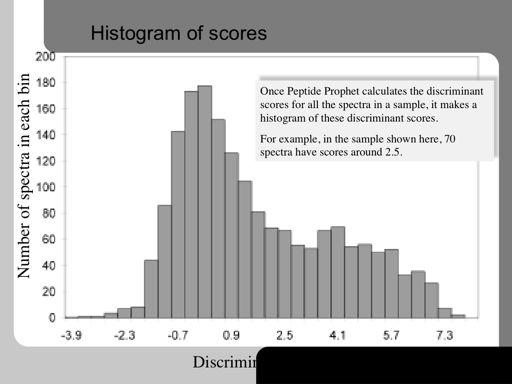
67 67!
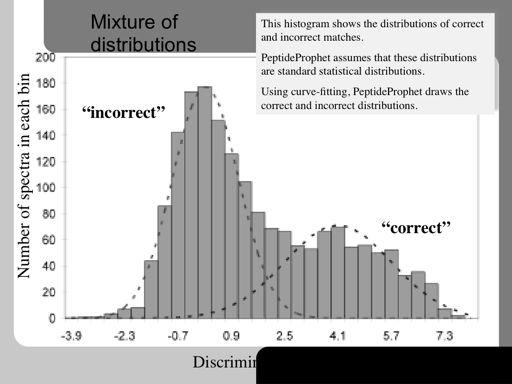
68 68!
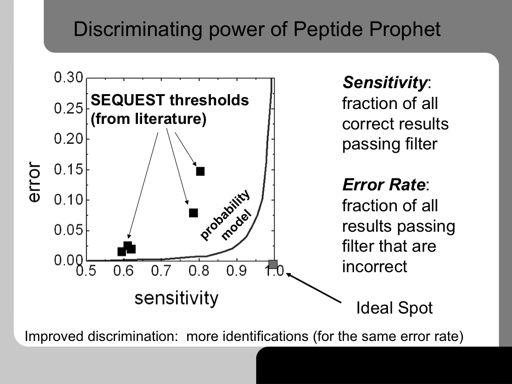
69 69!
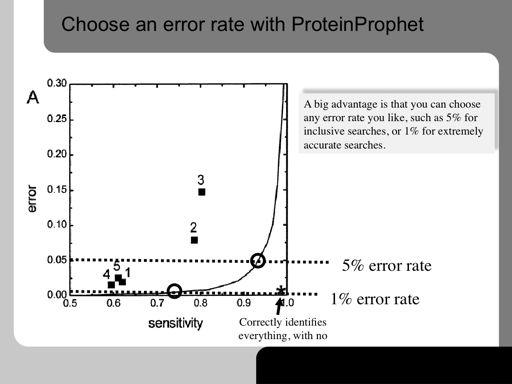
70 70!
71 71!

72 72!
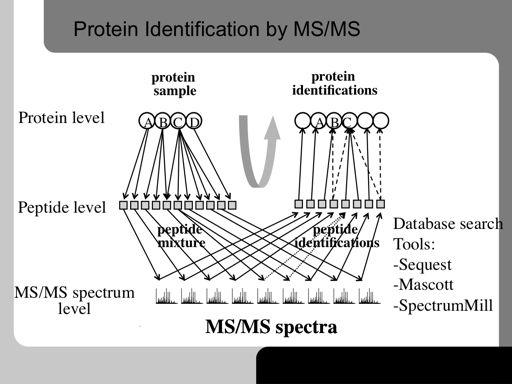
73 73!

74 74!

75 75!

76 76!

77 77!
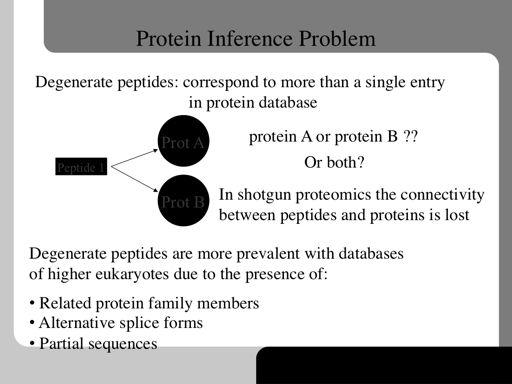
78 78!
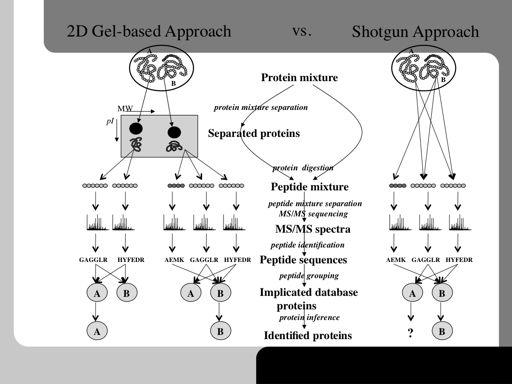
79 79!
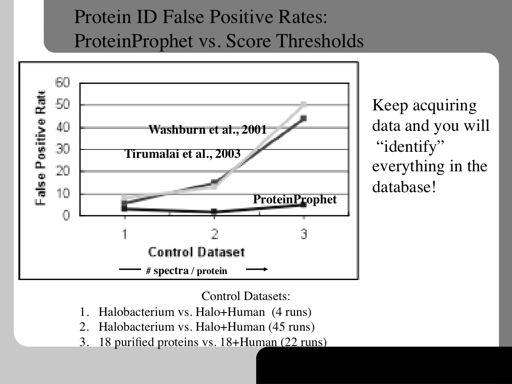
80 80!
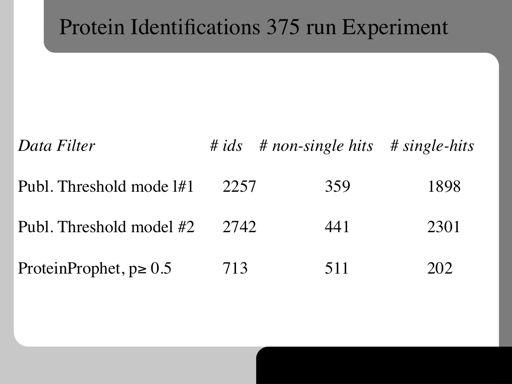
81 81!
Lecture 8: Mass Spectrometry
 intensity Lecture 8: Mass Spectrometry Relative abundance m/z 1 Ethylbenzene CH 2 CH 3 + m/z = 106 CH 2 + m/z = 91 C 8 H 10 MW = 106 CH + m/z = 77 + 2 2 What information can be obtained from a MS spectrum?
intensity Lecture 8: Mass Spectrometry Relative abundance m/z 1 Ethylbenzene CH 2 CH 3 + m/z = 106 CH 2 + m/z = 91 C 8 H 10 MW = 106 CH + m/z = 77 + 2 2 What information can be obtained from a MS spectrum?
Lecture 8: Mass Spectrometry
 intensity Lecture 8: Mass Spectrometry Relative abundance m/z 1 Ethylbenzene experiment CH 2 CH 3 + m/z = 106 CH 2 + m/z = 91 C 8 H 10 MW = 106 CH + m/z = 77 + 2 2 What information can we get from MS spectrum?
intensity Lecture 8: Mass Spectrometry Relative abundance m/z 1 Ethylbenzene experiment CH 2 CH 3 + m/z = 106 CH 2 + m/z = 91 C 8 H 10 MW = 106 CH + m/z = 77 + 2 2 What information can we get from MS spectrum?
TANDEM MASS SPECTROSCOPY
 TANDEM MASS SPECTROSCOPY 1 MASS SPECTROMETER TYPES OF MASS SPECTROMETER PRINCIPLE TANDEM MASS SPECTROMETER INSTRUMENTATION QUADRAPOLE MASS ANALYZER TRIPLE QUADRAPOLE MASS ANALYZER TIME OF FLIGHT MASS ANALYSER
TANDEM MASS SPECTROSCOPY 1 MASS SPECTROMETER TYPES OF MASS SPECTROMETER PRINCIPLE TANDEM MASS SPECTROMETER INSTRUMENTATION QUADRAPOLE MASS ANALYZER TRIPLE QUADRAPOLE MASS ANALYZER TIME OF FLIGHT MASS ANALYSER
(Refer Slide Time 00:09) (Refer Slide Time 00:13)
 (Refer Slide Time 00:13)") (Refer Slide Time 00:09) Mass Spectrometry Based Proteomics Professor Sanjeeva Srivastava Department of Biosciences and Bioengineering Indian Institute of Technology, Bombay Mod 02 Lecture Number 09 (Refer
(Refer Slide Time 00:09) Mass Spectrometry Based Proteomics Professor Sanjeeva Srivastava Department of Biosciences and Bioengineering Indian Institute of Technology, Bombay Mod 02 Lecture Number 09 (Refer
Introduction to Mass Spectrometry (MS)
") Introduction to Mass Spectrometry (MS) MS Mass Spectrometry (MS) This is a very powerful analytical tool that can provide information on both molecular mass and molecular structure. Molecules come in all
Introduction to Mass Spectrometry (MS) MS Mass Spectrometry (MS) This is a very powerful analytical tool that can provide information on both molecular mass and molecular structure. Molecules come in all
Fundamentals of Mass Spectrometry. Fundamentals of Mass Spectrometry. Learning Objective. Proteomics
 Mass spectrometry (MS) is the technique for protein identification and analysis by production of charged molecular species in vacuum, and their separation by magnetic and electric fields based on mass
Mass spectrometry (MS) is the technique for protein identification and analysis by production of charged molecular species in vacuum, and their separation by magnetic and electric fields based on mass
Computational Methods for Mass Spectrometry Proteomics
 Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
Computational Methods for Mass Spectrometry Proteomics Eidhammer, Ingvar ISBN-13: 9780470512975 Table of Contents Preface. Acknowledgements. 1 Protein, Proteome, and Proteomics. 1.1 Primary goals for studying
CHROMATOGRAPHY AND MASS SPECTROMETER
 22 CHROMATOGRAPHY AND MASS SPECTROMETER 22.1 INTRODUCTION We know that the biochemistry or biological chemistry deals with the study of molecules present in organisms. These molecules are called as biomolecules
22 CHROMATOGRAPHY AND MASS SPECTROMETER 22.1 INTRODUCTION We know that the biochemistry or biological chemistry deals with the study of molecules present in organisms. These molecules are called as biomolecules
LECTURE-13. Peptide Mass Fingerprinting HANDOUT. Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of
 LECTURE-13 Peptide Mass Fingerprinting HANDOUT PREAMBLE Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of proteins, drugs and many biological moieties to elucidate
LECTURE-13 Peptide Mass Fingerprinting HANDOUT PREAMBLE Mass spectrometry is an indispensable tool for qualitative and quantitative analysis of proteins, drugs and many biological moieties to elucidate
Other Methods for Generating Ions 1. MALDI matrix assisted laser desorption ionization MS 2. Spray ionization techniques 3. Fast atom bombardment 4.
 Other Methods for Generating Ions 1. MALDI matrix assisted laser desorption ionization MS 2. Spray ionization techniques 3. Fast atom bombardment 4. Field Desorption 5. MS MS techniques Matrix assisted
Other Methods for Generating Ions 1. MALDI matrix assisted laser desorption ionization MS 2. Spray ionization techniques 3. Fast atom bombardment 4. Field Desorption 5. MS MS techniques Matrix assisted
MS/MS .LQGVRI0606([SHULPHQWV
 0DVV6SHFWURPHWHUV Tandem Mass Spectrometry (MS/MS) :KDWLV0606" Mass spectrometers are commonly combined with separation devices such as gas chromatographs (GC) and liquid chromatographs (LC). The GC or
0DVV6SHFWURPHWHUV Tandem Mass Spectrometry (MS/MS) :KDWLV0606" Mass spectrometers are commonly combined with separation devices such as gas chromatographs (GC) and liquid chromatographs (LC). The GC or
MS-based proteomics to investigate proteins and their modifications
 MS-based proteomics to investigate proteins and their modifications Francis Impens VIB Proteomics Core October th 217 Overview Mass spectrometry-based proteomics: general workflow Identification of protein
MS-based proteomics to investigate proteins and their modifications Francis Impens VIB Proteomics Core October th 217 Overview Mass spectrometry-based proteomics: general workflow Identification of protein
BST 226 Statistical Methods for Bioinformatics David M. Rocke. January 22, 2014 BST 226 Statistical Methods for Bioinformatics 1
 BST 226 Statistical Methods for Bioinformatics David M. Rocke January 22, 2014 BST 226 Statistical Methods for Bioinformatics 1 Mass Spectrometry Mass spectrometry (mass spec, MS) comprises a set of instrumental
BST 226 Statistical Methods for Bioinformatics David M. Rocke January 22, 2014 BST 226 Statistical Methods for Bioinformatics 1 Mass Spectrometry Mass spectrometry (mass spec, MS) comprises a set of instrumental
Instrumental Analysis. Mass Spectrometry. Lecturer:! Somsak Sirichai
 303351 Instrumental Analysis Mass Spectrometry Lecturer:! Somsak Sirichai Mass Spectrometry What is Mass spectrometry (MS)? An analytic method that employs ionization and mass analysis of compounds in
303351 Instrumental Analysis Mass Spectrometry Lecturer:! Somsak Sirichai Mass Spectrometry What is Mass spectrometry (MS)? An analytic method that employs ionization and mass analysis of compounds in
Atomic masses. Atomic masses of elements. Atomic masses of isotopes. Nominal and exact atomic masses. Example: CO, N 2 ja C 2 H 4
 High-Resolution Mass spectrometry (HR-MS, HRAM-MS) (FT mass spectrometry) MS that enables identifying elemental compositions (empirical formulas) from accurate m/z data 9.05.2017 1 Atomic masses (atomic
High-Resolution Mass spectrometry (HR-MS, HRAM-MS) (FT mass spectrometry) MS that enables identifying elemental compositions (empirical formulas) from accurate m/z data 9.05.2017 1 Atomic masses (atomic
sample was a solution that was evaporated in the spectrometer (such as with ESI-MS) ions such as H +, Na +, K +, or NH 4
 ions such as H +, Na +, K +, or NH 4") Introduction to Spectroscopy V: Mass Spectrometry Basic Theory: Unlike other forms of spectroscopy used in structure elucidation of organic molecules mass spectrometry does not involve absorption/emission
Introduction to Spectroscopy V: Mass Spectrometry Basic Theory: Unlike other forms of spectroscopy used in structure elucidation of organic molecules mass spectrometry does not involve absorption/emission
Tutorial 1: Setting up your Skyline document
 Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
Tutorial 1: Setting up your Skyline document Caution! For using Skyline the number formats of your computer have to be set to English (United States). Open the Control Panel Clock, Language, and Region
LECTURE-11. Hybrid MS Configurations HANDOUT. As discussed in our previous lecture, mass spectrometry is by far the most versatile
 LECTURE-11 Hybrid MS Configurations HANDOUT PREAMBLE As discussed in our previous lecture, mass spectrometry is by far the most versatile technique used in proteomics. We had also discussed some of the
LECTURE-11 Hybrid MS Configurations HANDOUT PREAMBLE As discussed in our previous lecture, mass spectrometry is by far the most versatile technique used in proteomics. We had also discussed some of the
Chemistry 311: Topic 3 - Mass Spectrometry
 Mass Spectroscopy: A technique used to measure the mass-to-charge ratio of molecules and atoms. Often characteristic ions produced by an induced unimolecular dissociation of a molecule are measured. These
Mass Spectroscopy: A technique used to measure the mass-to-charge ratio of molecules and atoms. Often characteristic ions produced by an induced unimolecular dissociation of a molecule are measured. These
Protein Structure Analysis and Verification. Course S Basics for Biosystems of the Cell exercise work. Maija Nevala, BIO, 67485U 16.1.
 Protein Structure Analysis and Verification Course S-114.2500 Basics for Biosystems of the Cell exercise work Maija Nevala, BIO, 67485U 16.1.2008 1. Preface When faced with an unknown protein, scientists
Protein Structure Analysis and Verification Course S-114.2500 Basics for Biosystems of the Cell exercise work Maija Nevala, BIO, 67485U 16.1.2008 1. Preface When faced with an unknown protein, scientists
Proteomics. November 13, 2007
 Proteomics November 13, 2007 Acknowledgement Slides presented here have been borrowed from presentations by : Dr. Mark A. Knepper (LKEM, NHLBI, NIH) Dr. Nathan Edwards (Center for Bioinformatics and Computational
Proteomics November 13, 2007 Acknowledgement Slides presented here have been borrowed from presentations by : Dr. Mark A. Knepper (LKEM, NHLBI, NIH) Dr. Nathan Edwards (Center for Bioinformatics and Computational
Harris: Quantitative Chemical Analysis, Eight Edition
 Harris: Quantitative Chemical Analysis, Eight Edition CHAPTER 21: MASS SPECTROMETRY CHAPTER 21: Opener 21.0 Mass Spectrometry Mass Spectrometry provides information about 1) The elemental composition of
Harris: Quantitative Chemical Analysis, Eight Edition CHAPTER 21: MASS SPECTROMETRY CHAPTER 21: Opener 21.0 Mass Spectrometry Mass Spectrometry provides information about 1) The elemental composition of
Key questions of proteomics. Bioinformatics 2. Proteomics. Foundation of proteomics. What proteins are there? Protein digestion
 s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
s s Key questions of proteomics What proteins are there? Bioinformatics 2 Lecture 2 roteomics How much is there of each of the proteins? - Absolute quantitation - Stoichiometry What (modification/splice)
Powerful Scan Modes of QTRAP System Technology
 Powerful Scan Modes of QTRAP System Technology Unique Hybrid Triple Quadrupole Linear Ion Trap Technology Provides Powerful Workflows to Answer Complex Questions with No Compromises While there are many
Powerful Scan Modes of QTRAP System Technology Unique Hybrid Triple Quadrupole Linear Ion Trap Technology Provides Powerful Workflows to Answer Complex Questions with No Compromises While there are many
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot RPPA Immunohistochemistry
Tandem MS = MS / MS. ESI-MS give information on the mass of a molecule but none on the structure
 Tandem MS = MS / MS ESI-MS give information on the mass of a molecule but none on the structure In tandem MS (MSMS) (pseudo-)molecular ions are selected in MS1 and fragmented by collision with gas. collision
Tandem MS = MS / MS ESI-MS give information on the mass of a molecule but none on the structure In tandem MS (MSMS) (pseudo-)molecular ions are selected in MS1 and fragmented by collision with gas. collision
Qualitative Proteomics (how to obtain high-confidence high-throughput protein identification!)
") Qualitative Proteomics (how to obtain high-confidence high-throughput protein identification!) James A. Mobley, Ph.D. Director of Research in Urology Associate Director of Mass Spectrometry (contact: mobleyja@uab.edu)
Qualitative Proteomics (how to obtain high-confidence high-throughput protein identification!) James A. Mobley, Ph.D. Director of Research in Urology Associate Director of Mass Spectrometry (contact: mobleyja@uab.edu)
MASS ANALYSER. Mass analysers - separate the ions according to their mass-to-charge ratio. sample. Vacuum pumps
 ION ANALYZERS MASS ANALYSER sample Vacuum pumps Mass analysers - separate the ions according to their mass-to-charge ratio MASS ANALYSER Separate the ions according to their mass-to-charge ratio in space
ION ANALYZERS MASS ANALYSER sample Vacuum pumps Mass analysers - separate the ions according to their mass-to-charge ratio MASS ANALYSER Separate the ions according to their mass-to-charge ratio in space
SRM assay generation and data analysis in Skyline
 in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
in Skyline Preparation 1. Download the example data from www.srmcourse.ch/eupa.html (3 raw files, 1 csv file, 1 sptxt file). 2. The number formats of your computer have to be set to English (United States).
Types of Analyzers: Quadrupole: mass filter -part1
 16 Types of Analyzers: Sector or double focusing: magnetic and electric Time-of-flight (TOF) Quadrupole (mass filter) Linear ion trap Quadrupole Ion Trap (3D trap) FTICR fourier transform ion cyclotron
16 Types of Analyzers: Sector or double focusing: magnetic and electric Time-of-flight (TOF) Quadrupole (mass filter) Linear ion trap Quadrupole Ion Trap (3D trap) FTICR fourier transform ion cyclotron
Protein Quantitation II: Multiple Reaction Monitoring. Kelly Ruggles New York University
 Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Protein Quantitation II: Multiple Reaction Monitoring Kelly Ruggles kelly@fenyolab.org New York University Traditional Affinity-based proteomics Use antibodies to quantify proteins Western Blot Immunohistochemistry
Introduction to the Q Trap LC/MS/MS System
 www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +1.847.913.0777 for Refurbished & Certified Lab Equipment ABI Q Trap LC/MS/MS Introduction to the Q Trap LC/MS/MS System The Q Trap
www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +1.847.913.0777 for Refurbished & Certified Lab Equipment ABI Q Trap LC/MS/MS Introduction to the Q Trap LC/MS/MS System The Q Trap
De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra. Xiaowen Liu
 De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra Xiaowen Liu Department of BioHealth Informatics, Department of Computer and Information Sciences, Indiana University-Purdue
De novo Protein Sequencing by Combining Top-Down and Bottom-Up Tandem Mass Spectra Xiaowen Liu Department of BioHealth Informatics, Department of Computer and Information Sciences, Indiana University-Purdue
Mass Spectrometry in MCAL
 Mass Spectrometry in MCAL Two systems: GC-MS, LC-MS GC seperates small, volatile, non-polar material MS is detection devise (Agilent 320-MS TQ Mass Spectrometer) Full scan monitoring SIM single ion monitoring
Mass Spectrometry in MCAL Two systems: GC-MS, LC-MS GC seperates small, volatile, non-polar material MS is detection devise (Agilent 320-MS TQ Mass Spectrometer) Full scan monitoring SIM single ion monitoring
Lecture 15: Realities of Genome Assembly Protein Sequencing
 Lecture 15: Realities of Genome Assembly Protein Sequencing Study Chapter 8.10-8.15 1 Euler s Theorems A graph is balanced if for every vertex the number of incoming edges equals to the number of outgoing
Lecture 15: Realities of Genome Assembly Protein Sequencing Study Chapter 8.10-8.15 1 Euler s Theorems A graph is balanced if for every vertex the number of incoming edges equals to the number of outgoing
Mass Spectrometry and Proteomics - Lecture 2 - Matthias Trost Newcastle University
 Mass Spectrometry and Proteomics - Lecture 2 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously: Resolution and other basics MALDI Electrospray 40 Lecture 2 Mass analysers Detectors
Mass Spectrometry and Proteomics - Lecture 2 - Matthias Trost Newcastle University matthias.trost@ncl.ac.uk Previously: Resolution and other basics MALDI Electrospray 40 Lecture 2 Mass analysers Detectors
MASS SPECTROMETRY. Topics
 MASS SPECTROMETRY MALDI-TOF AND ESI-MS Topics Principle of Mass Spectrometry MALDI-TOF Determination of Mw of Proteins Structural Information by MS: Primary Sequence of a Protein 1 A. Principles Ionization:
MASS SPECTROMETRY MALDI-TOF AND ESI-MS Topics Principle of Mass Spectrometry MALDI-TOF Determination of Mw of Proteins Structural Information by MS: Primary Sequence of a Protein 1 A. Principles Ionization:
NPTEL VIDEO COURSE PROTEOMICS PROF. SANJEEVA SRIVASTAVA
 LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
LECTURE-25 Quantitative proteomics: itraq and TMT TRANSCRIPT Welcome to the proteomics course. Today we will talk about quantitative proteomics and discuss about itraq and TMT techniques. The quantitative
Welcome to Organic Chemistry II
 Welcome to Organic Chemistry II Erika Bryant, Ph.D. erika.bryant@hccs.edu Class Syllabus 3 CHAPTER 12: STRUCTURE DETERMINATION 4 What is this solution Soda Tea Coffee??? 5 What is this solution Soda Tea
Welcome to Organic Chemistry II Erika Bryant, Ph.D. erika.bryant@hccs.edu Class Syllabus 3 CHAPTER 12: STRUCTURE DETERMINATION 4 What is this solution Soda Tea Coffee??? 5 What is this solution Soda Tea
Mass Spectrometry. Hyphenated Techniques GC-MS LC-MS and MS-MS
 Mass Spectrometry Hyphenated Techniques GC-MS LC-MS and MS-MS Reasons for Using Chromatography with MS Mixture analysis by MS alone is difficult Fragmentation from ionization (EI or CI) Fragments from
Mass Spectrometry Hyphenated Techniques GC-MS LC-MS and MS-MS Reasons for Using Chromatography with MS Mixture analysis by MS alone is difficult Fragmentation from ionization (EI or CI) Fragments from
MASS SPECTROSCOPY (MS)
") MASS SPECTOSCOPY (MS) Castor seeds icin (toxic protein) INTODUCTION Does not involve absorption of electromagnetic radiation. It is a spectroscopic technique, by virtue of its use in structure elucidation.
MASS SPECTOSCOPY (MS) Castor seeds icin (toxic protein) INTODUCTION Does not involve absorption of electromagnetic radiation. It is a spectroscopic technique, by virtue of its use in structure elucidation.
Biological Mass Spectrometry
 Biochemistry 412 Biological Mass Spectrometry February 13 th, 2007 Proteomics The study of the complete complement of proteins found in an organism Degrees of Freedom for Protein Variability Covalent Modifications
Biochemistry 412 Biological Mass Spectrometry February 13 th, 2007 Proteomics The study of the complete complement of proteins found in an organism Degrees of Freedom for Protein Variability Covalent Modifications
Comprehensive support for quantitation
 Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
Comprehensive support for quantitation One of the major new features in the current release of Mascot is support for quantitation. This is still work in progress. Our goal is to support all of the popular
An ion source performs the following two functions:
 Ionization The Ion Source An ion source performs the following two functions: 1) converts sample atoms or molecules to ionized particles (ions) in the gas phase (sometimes the task of introducing the atoms
Ionization The Ion Source An ion source performs the following two functions: 1) converts sample atoms or molecules to ionized particles (ions) in the gas phase (sometimes the task of introducing the atoms
Mixture Mode for Peptide Mass Fingerprinting ASMS 2003
 Mixture Mode for Peptide Mass Fingerprinting ASMS 2003 1 Mixture Mode: New in Mascot 1.9 All peptide mass fingerprint searches now test for the possibility that the sample is a mixture of proteins. Mascot
Mixture Mode for Peptide Mass Fingerprinting ASMS 2003 1 Mixture Mode: New in Mascot 1.9 All peptide mass fingerprint searches now test for the possibility that the sample is a mixture of proteins. Mascot
Introduction to LC-MS
 Wednesday April 5, 2017 10am Introduction to LC-MS Amy Patton, MS Laboratory Manager, Pinpoint Testing, LLC Little Rock, AR DESCRIPTION: Amy Patton, laboratory manager for Pinpoint Testing, will begin
Wednesday April 5, 2017 10am Introduction to LC-MS Amy Patton, MS Laboratory Manager, Pinpoint Testing, LLC Little Rock, AR DESCRIPTION: Amy Patton, laboratory manager for Pinpoint Testing, will begin
Bioinformatics and BLAST
 Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Bioinformatics and BLAST Overview Recap of last time Similarity discussion Algorithms: Needleman-Wunsch Smith-Waterman BLAST Implementation issues and current research Recap from Last Time Genome consists
Mass Spectrometry. General Principles
 General Principles Mass Spectrometer: Converts molecules to ions Separates ions (usually positively charged) on the basis of their mass/charge (m/z) ratio Quantifies how many units of each ion are formed
General Principles Mass Spectrometer: Converts molecules to ions Separates ions (usually positively charged) on the basis of their mass/charge (m/z) ratio Quantifies how many units of each ion are formed
Protein Sequencing and Identification by Mass Spectrometry
 Protein Sequencing and Identification by Mass Spectrometry Tandem Mass Spectrometry De Novo Peptide Sequencing Spectrum Graph Protein Identification via Database Search Identifying Post Translationally
Protein Sequencing and Identification by Mass Spectrometry Tandem Mass Spectrometry De Novo Peptide Sequencing Spectrum Graph Protein Identification via Database Search Identifying Post Translationally
Electrophiles are attracted to the π bond Addition sees a π bond replaced with a σ bond There are many different types of addition reactions:
 Nucleophiles and Electrophiles Nucleophiles are the atoms that donates the electron pairs and is added to the molecules (In the example above this is the CN) Electrophiles are the atoms that seek electron
Nucleophiles and Electrophiles Nucleophiles are the atoms that donates the electron pairs and is added to the molecules (In the example above this is the CN) Electrophiles are the atoms that seek electron
ChemActivity L2: Mass Spectrometry
 ChemActivity L2: Mass Spectrometry (How can we determine the mass and molecular formula of an unknown compound?) This activity is designed to be completed in a 1 ½-hour laboratory session or two classroom
ChemActivity L2: Mass Spectrometry (How can we determine the mass and molecular formula of an unknown compound?) This activity is designed to be completed in a 1 ½-hour laboratory session or two classroom
Propose a structure for an alcohol, C4H10O, that has the following
 Propose a structure for an alcohol, C4H10O, that has the following 13CNMR spectral data: Broadband _ decoupled 13CNMR: 19.0, 31.7, 69.5 б DEPT _90: 31.7 б DEPT _ 135: positive peak at 19.0 & 31.7 б, negative
Propose a structure for an alcohol, C4H10O, that has the following 13CNMR spectral data: Broadband _ decoupled 13CNMR: 19.0, 31.7, 69.5 б DEPT _90: 31.7 б DEPT _ 135: positive peak at 19.0 & 31.7 б, negative
for the Novice Mass Spectrometry (^>, John Greaves and John Roboz yc**' CRC Press J Taylor & Francis Group Boca Raton London New York
 Mass Spectrometry for the Novice John Greaves and John Roboz (^>, yc**' CRC Press J Taylor & Francis Group Boca Raton London New York CRC Press is an imprint of the Taylor & Francis Croup, an informa business
Mass Spectrometry for the Novice John Greaves and John Roboz (^>, yc**' CRC Press J Taylor & Francis Group Boca Raton London New York CRC Press is an imprint of the Taylor & Francis Croup, an informa business
Mass Spectrometry - Background
 Mass Spectrometry - Background In mass spectrometry, a substance is bombarded with an electron beam having sufficient energy to fragment the molecule. The positive fragments which are produced (cations
Mass Spectrometry - Background In mass spectrometry, a substance is bombarded with an electron beam having sufficient energy to fragment the molecule. The positive fragments which are produced (cations
The Power of LC MALDI: Identification of Proteins by LC MALDI MS/MS Using the Applied Biosystems 4700 Proteomics Analyzer with TOF/TOF Optics
 APPLICATION NOTE TOF MS The Power of LC MALDI: Identification of Proteins by LC MALDI MS/MS Using the Applied Biosystems 4700 Proteomics Analyzer with TOF/TOF Optics Purpose The Applied Biosystems 4700
APPLICATION NOTE TOF MS The Power of LC MALDI: Identification of Proteins by LC MALDI MS/MS Using the Applied Biosystems 4700 Proteomics Analyzer with TOF/TOF Optics Purpose The Applied Biosystems 4700
Mass spectrometry and elemental analysis
 Mass spectrometry and elemental analysis A schematic representation of a single-focusing mass spectrometer with an electron-impact (EI) ionization source. M: + e _ M +. + 2e _ Ionization and fragmentation
Mass spectrometry and elemental analysis A schematic representation of a single-focusing mass spectrometer with an electron-impact (EI) ionization source. M: + e _ M +. + 2e _ Ionization and fragmentation
Mass spectrometry.
 Mass spectrometry Mass spectrometry provides qualitative and quantitative information about the atomic and molecular composition of inorganic and organic materials. The mass spectrometer produces charged
Mass spectrometry Mass spectrometry provides qualitative and quantitative information about the atomic and molecular composition of inorganic and organic materials. The mass spectrometer produces charged
Mass Analyzers. Principles of the three most common types magnetic sector, quadrupole and time of flight - will be discussed herein.
 Mass Analyzers After the production of ions in ion sources, the next critical step in mass spectrometry is to separate these gas phase ions according to their mass-to-charge ratio (m/z). Ions are extracted
Mass Analyzers After the production of ions in ion sources, the next critical step in mass spectrometry is to separate these gas phase ions according to their mass-to-charge ratio (m/z). Ions are extracted
WADA Technical Document TD2015IDCR
 MINIMUM CRITERIA FOR CHROMATOGRAPHIC-MASS SPECTROMETRIC CONFIRMATION OF THE IDENTITY OF ANALYTES FOR DOPING CONTROL PURPOSES. The ability of a method to identify an analyte is a function of the entire
MINIMUM CRITERIA FOR CHROMATOGRAPHIC-MASS SPECTROMETRIC CONFIRMATION OF THE IDENTITY OF ANALYTES FOR DOPING CONTROL PURPOSES. The ability of a method to identify an analyte is a function of the entire
Protein analysis using mass spectrometry
 Protein analysis using mass spectrometry Michael Stadlmeier 2017/12/18 Literature http://www.carellgroup.de/teaching/master 3 What is Proteomics? The proteome is: the entire set of proteins in a given
Protein analysis using mass spectrometry Michael Stadlmeier 2017/12/18 Literature http://www.carellgroup.de/teaching/master 3 What is Proteomics? The proteome is: the entire set of proteins in a given
Accurate, High-Throughput Protein Identification Using the Q TRAP LC/MS/MS System and Pro ID Software
 www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +1.847.913.0777 for Refurbished & Certified Lab Equipment ABI Q Trap Pro LC/MS/MS Accurate, High-Throughput Protein Identification
www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +1.847.913.0777 for Refurbished & Certified Lab Equipment ABI Q Trap Pro LC/MS/MS Accurate, High-Throughput Protein Identification
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry
 Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
Effective Strategies for Improving Peptide Identification with Tandem Mass Spectrometry by Xi Han A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree
CEE 772 Lecture #27 12/10/2014. CEE 772: Instrumental Methods in Environmental Analysis
 Updated: 10 December 2014 Print version CEE 772: Instrumental Methods in Environmental Analysis Lecture #21 Mass Spectrometry: Mass Filters & Spectrometers (Skoog, Chapt. 20, pp.511 524) (Harris, Chapt.
Updated: 10 December 2014 Print version CEE 772: Instrumental Methods in Environmental Analysis Lecture #21 Mass Spectrometry: Mass Filters & Spectrometers (Skoog, Chapt. 20, pp.511 524) (Harris, Chapt.
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry
 Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
Quantitation of a target protein in crude samples using targeted peptide quantification by Mass Spectrometry Jon Hao, Rong Ye, and Mason Tao Poochon Scientific, Frederick, Maryland 21701 Abstract Background:
SPECTRA LIBRARY ASSISTED DE NOVO PEPTIDE SEQUENCING FOR HCD AND ETD SPECTRA PAIRS
 SPECTRA LIBRARY ASSISTED DE NOVO PEPTIDE SEQUENCING FOR HCD AND ETD SPECTRA PAIRS 1 Yan Yan Department of Computer Science University of Western Ontario, Canada OUTLINE Background Tandem mass spectrometry
SPECTRA LIBRARY ASSISTED DE NOVO PEPTIDE SEQUENCING FOR HCD AND ETD SPECTRA PAIRS 1 Yan Yan Department of Computer Science University of Western Ontario, Canada OUTLINE Background Tandem mass spectrometry
CEE 772: Instrumental Methods in Environmental Analysis
 Updated: 10 December 2014 Print version CEE 772: Instrumental Methods in Environmental Analysis Lecture #21 Mass Spectrometry: Mass Filters & Spectrometers (Skoog, Chapt. 20, pp.511-524) (Harris, Chapt.
Updated: 10 December 2014 Print version CEE 772: Instrumental Methods in Environmental Analysis Lecture #21 Mass Spectrometry: Mass Filters & Spectrometers (Skoog, Chapt. 20, pp.511-524) (Harris, Chapt.
TUTORIAL EXERCISES WITH ANSWERS
 TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
TUTORIAL EXERCISES WITH ANSWERS Tutorial 1 Settings 1. What is the exact monoisotopic mass difference for peptides carrying a 13 C (and NO additional 15 N) labelled C-terminal lysine residue? a. 6.020129
Chapter 5. Complexation of Tholins by 18-crown-6:
 5-1 Chapter 5. Complexation of Tholins by 18-crown-6: Identification of Primary Amines 5.1. Introduction Electrospray ionization (ESI) is an excellent technique for the ionization of complex mixtures,
5-1 Chapter 5. Complexation of Tholins by 18-crown-6: Identification of Primary Amines 5.1. Introduction Electrospray ionization (ESI) is an excellent technique for the ionization of complex mixtures,
Chemistry Instrumental Analysis Lecture 37. Chem 4631
 Chemistry 4631 Instrumental Analysis Lecture 37 Most analytes separated by HPLC are thermally stable and non-volatile (liquids) (unlike in GC) so not ionized easily by EI or CI techniques. MS must be at
Chemistry 4631 Instrumental Analysis Lecture 37 Most analytes separated by HPLC are thermally stable and non-volatile (liquids) (unlike in GC) so not ionized easily by EI or CI techniques. MS must be at
LC-MS Based Metabolomics
 LC-MS Based Metabolomics Analysing the METABOLOME 1. Metabolite Extraction 2. Metabolite detection (with or without separation) 3. Data analysis Metabolite Detection GC-MS: Naturally volatile or made volatile
LC-MS Based Metabolomics Analysing the METABOLOME 1. Metabolite Extraction 2. Metabolite detection (with or without separation) 3. Data analysis Metabolite Detection GC-MS: Naturally volatile or made volatile
Identification of proteins by enzyme digestion, mass
 Method for Screening Peptide Fragment Ion Mass Spectra Prior to Database Searching Roger E. Moore, Mary K. Young, and Terry D. Lee Beckman Research Institute of the City of Hope, Duarte, California, USA
Method for Screening Peptide Fragment Ion Mass Spectra Prior to Database Searching Roger E. Moore, Mary K. Young, and Terry D. Lee Beckman Research Institute of the City of Hope, Duarte, California, USA
What is Tandem Mass Spectrometry? (MS/MS)
") What is Tandem Mass Spectrometry? (MS/MS) Activate MS1 MS2 Gas Surface Photons Electrons http://www.iupac.org/goldbook/t06250.pdf Major difference between MS and MS/MS: two analysis steps Ionization Analysis
What is Tandem Mass Spectrometry? (MS/MS) Activate MS1 MS2 Gas Surface Photons Electrons http://www.iupac.org/goldbook/t06250.pdf Major difference between MS and MS/MS: two analysis steps Ionization Analysis
Chemistry Instrumental Analysis Lecture 34. Chem 4631
 Chemistry 4631 Instrumental Analysis Lecture 34 From molecular to elemental analysis there are three major techniques used for elemental analysis: Optical spectrometry Mass spectrometry X-ray spectrometry
Chemistry 4631 Instrumental Analysis Lecture 34 From molecular to elemental analysis there are three major techniques used for elemental analysis: Optical spectrometry Mass spectrometry X-ray spectrometry
THE MODERN VIEW OF ATOMIC STRUCTURE
 44 CHAPTER 2 Atoms, Molecules, and Ions GO FIGURE What is the charge on the particles that form the beam? Experiment Interpretation Incoming a particles Beam of a particles Source of a particles Nucleus
44 CHAPTER 2 Atoms, Molecules, and Ions GO FIGURE What is the charge on the particles that form the beam? Experiment Interpretation Incoming a particles Beam of a particles Source of a particles Nucleus
Modeling Mass Spectrometry-Based Protein Analysis
 Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
Chapter 8 Jan Eriksson and David Fenyö Abstract The success of mass spectrometry based proteomics depends on efficient methods for data analysis. These methods require a detailed understanding of the information
Thermo Scientific LTQ Orbitrap Velos Hybrid FT Mass Spectrometer
 IET International Equipment Trading Ltd. www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +847.913.0777 for Refurbished & Certified Lab Equipment Thermo Scientific LTQ Orbitrap Velos
IET International Equipment Trading Ltd. www.ietltd.com Proudly serving laboratories worldwide since 1979 CALL +847.913.0777 for Refurbished & Certified Lab Equipment Thermo Scientific LTQ Orbitrap Velos
Towards the Prediction of Protein Abundance from Tandem Mass Spectrometry Data
 Towards the Prediction of Protein Abundance from Tandem Mass Spectrometry Data Anthony J Bonner Han Liu Abstract This paper addresses a central problem of Proteomics: estimating the amounts of each of
Towards the Prediction of Protein Abundance from Tandem Mass Spectrometry Data Anthony J Bonner Han Liu Abstract This paper addresses a central problem of Proteomics: estimating the amounts of each of
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra. Andrew Keller
 PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
PeptideProphet: Validation of Peptide Assignments to MS/MS Spectra Andrew Keller Outline Need to validate peptide assignments to MS/MS spectra Statistical approach to validation Running PeptideProphet
Analytical Technologies in Biotechnology Prof. Dr. Ashwani K. Sharma Department of Biotechnology Indian Institute of Technology, Roorkee
 Analytical Technologies in Biotechnology Prof. Dr. Ashwani K. Sharma Department of Biotechnology Indian Institute of Technology, Roorkee Module - 6 Spectroscopic Techniques Lecture - 6 Atomic Spectroscopy
Analytical Technologies in Biotechnology Prof. Dr. Ashwani K. Sharma Department of Biotechnology Indian Institute of Technology, Roorkee Module - 6 Spectroscopic Techniques Lecture - 6 Atomic Spectroscopy
Chem 250 Unit 1 Proteomics by Mass Spectrometry
 Chem 250 Unit 1 Proteomics by Mass Spectrometry Article #1 Quantitative MS for proteomics: teaching a new dog old tricks. MacCoss MJ, Matthews DE., Anal Chem. 2005 Aug 1;77(15):294A-302A. 1. Synopsis 1.1.
Chem 250 Unit 1 Proteomics by Mass Spectrometry Article #1 Quantitative MS for proteomics: teaching a new dog old tricks. MacCoss MJ, Matthews DE., Anal Chem. 2005 Aug 1;77(15):294A-302A. 1. Synopsis 1.1.
2. Separate the ions based on their mass to charge (m/e) ratio. 3. Measure the relative abundance of the ions that are produced
 ratio. 3. Measure the relative abundance of the ions that are produced") I. Mass spectrometry: capable of providing both quantitative and qualitative information about samples as small as 100 pg (!) and with molar masses in the 10 4-10 5 kdalton range A. The mass spectrometer
I. Mass spectrometry: capable of providing both quantitative and qualitative information about samples as small as 100 pg (!) and with molar masses in the 10 4-10 5 kdalton range A. The mass spectrometer
ZAHID IQBAL WARRAICH
 Q1 Chromatography is an important analytical technique in chemistry. There is a number of techniques under the general heading of chromatography. (a) Paper and gas chromatography rely on partition to separate
Q1 Chromatography is an important analytical technique in chemistry. There is a number of techniques under the general heading of chromatography. (a) Paper and gas chromatography rely on partition to separate
i. This is the best evidence for the fact that electrons in an atom surround the nucleus in certain allowed energy levels or orbitals ii.
 Atomic Structure I. The Atom A. Atomic theory: Devised in 1807 by John Dalton, states that all matter is made up of a small number of different kinds of atoms that are indivisible and indestructible but
Atomic Structure I. The Atom A. Atomic theory: Devised in 1807 by John Dalton, states that all matter is made up of a small number of different kinds of atoms that are indivisible and indestructible but
1.1 Atomic structure
 1.1 Atomic structure History of the atom The model of the atom has changed as our observations of its behavior and properties have increased. A model is used to explain observations. The model changes
1.1 Atomic structure History of the atom The model of the atom has changed as our observations of its behavior and properties have increased. A model is used to explain observations. The model changes
Mass Spectrometry (MS)
") Kevin Burgess, February 20, 2017 1 Mass Spectrometry (MS) from chapter(s) in the recommended text A. Introduction Kevin Burgess, February 20, 2017 2 B. Components f Mass Spectrometers mass-to-charge. molecular
Kevin Burgess, February 20, 2017 1 Mass Spectrometry (MS) from chapter(s) in the recommended text A. Introduction Kevin Burgess, February 20, 2017 2 B. Components f Mass Spectrometers mass-to-charge. molecular
Identification of Human Hemoglobin Protein Variants Using Electrospray Ionization-Electron Transfer Dissociation Mass Spectrometry
 Identification of Human Hemoglobin Protein Variants Using Electrospray Ionization-Electron Transfer Dissociation Mass Spectrometry Jonathan Williams Waters Corporation, Milford, MA, USA A P P L I C AT
Identification of Human Hemoglobin Protein Variants Using Electrospray Ionization-Electron Transfer Dissociation Mass Spectrometry Jonathan Williams Waters Corporation, Milford, MA, USA A P P L I C AT
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics
 Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
Isotopic-Labeling and Mass Spectrometry-Based Quantitative Proteomics Xiao-jun Li, Ph.D. Current address: Homestead Clinical Day 4 October 19, 2006 Protein Quantification LC-MS/MS Data XLink mzxml file
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 *
 Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
Improved 6- Plex TMT Quantification Throughput Using a Linear Ion Trap HCD MS 3 Scan Jane M. Liu, 1,2 * Michael J. Sweredoski, 2 Sonja Hess 2 * 1 Department of Chemistry, Pomona College, Claremont, California
MS Goals and Applications. MS Goals and Applications
 MS Goals and Applications 3 Several variations on a theme, three common steps Form gas-phase ions choice of ionization method depends on sample identity and information required Separate ions on basis
MS Goals and Applications 3 Several variations on a theme, three common steps Form gas-phase ions choice of ionization method depends on sample identity and information required Separate ions on basis
BENG 183 Trey Ideker. Protein Sequencing
 BENG 183 Trey Ideker Protein Sequencing The following slides borrowed from Hong Li s Biochemistry Course: www.sb.fsu.edu/~hongli/4053notes Introduction to Proteins Proteins are of vital importance to biological
BENG 183 Trey Ideker Protein Sequencing The following slides borrowed from Hong Li s Biochemistry Course: www.sb.fsu.edu/~hongli/4053notes Introduction to Proteins Proteins are of vital importance to biological
WADA Technical Document TD2003IDCR
 IDENTIFICATION CRITERIA FOR QUALITATIVE ASSAYS INCORPORATING CHROMATOGRAPHY AND MASS SPECTROMETRY The appropriate analytical characteristics must be documented for a particular assay. The Laboratory must
IDENTIFICATION CRITERIA FOR QUALITATIVE ASSAYS INCORPORATING CHROMATOGRAPHY AND MASS SPECTROMETRY The appropriate analytical characteristics must be documented for a particular assay. The Laboratory must
Information Dependent Acquisition (IDA) 1
 1") Information Dependent Acquisition (IDA) Information Dependent Acquisition (IDA) enables on the fly acquisition of MS/MS spectra during a chromatographic run. Analyst Software IDA is optimized to generate
Information Dependent Acquisition (IDA) Information Dependent Acquisition (IDA) enables on the fly acquisition of MS/MS spectra during a chromatographic run. Analyst Software IDA is optimized to generate
Workflow concept. Data goes through the workflow. A Node contains an operation An edge represents data flow The results are brought together in tables
 PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
PROTEOME DISCOVERER Workflow concept Data goes through the workflow Spectra Peptides Quantitation A Node contains an operation An edge represents data flow The results are brought together in tables Protein
Tandem mass spectra were extracted from the Xcalibur data system format. (.RAW) and charge state assignment was performed using in house software
 and charge state assignment was performed using in house software") Supplementary Methods Software Interpretation of Tandem mass spectra Tandem mass spectra were extracted from the Xcalibur data system format (.RAW) and charge state assignment was performed using in house
Supplementary Methods Software Interpretation of Tandem mass spectra Tandem mass spectra were extracted from the Xcalibur data system format (.RAW) and charge state assignment was performed using in house
Gases. Pressure is formally defined as the force exerted on a surface per unit area:
 Gases Pressure is formally defined as the force exerted on a surface per unit area: Force is measure in Newtons Area is measured in m 2 and it refers to the Area the particle/object is touching (From the
Gases Pressure is formally defined as the force exerted on a surface per unit area: Force is measure in Newtons Area is measured in m 2 and it refers to the Area the particle/object is touching (From the
Protein Identification Using Tandem Mass Spectrometry. Nathan Edwards Informatics Research Applied Biosystems
 Protein Identification Using Tandem Mass Spectrometry Nathan Edwards Informatics Research Applied Biosystems Outline Proteomics context Tandem mass spectrometry Peptide fragmentation Peptide identification
Protein Identification Using Tandem Mass Spectrometry Nathan Edwards Informatics Research Applied Biosystems Outline Proteomics context Tandem mass spectrometry Peptide fragmentation Peptide identification
Interazioni di ioni con elettroni (ECD, ETD) e fotoni (Ion spectroscopy) Gianluca Giorgi. via Aldo Moro Siena
 e fotoni (Ion spectroscopy) Gianluca Giorgi. via Aldo Moro Siena") Interazioni di ioni con elettroni (ECD, ETD) e fotoni (Ion spectroscopy) Gianluca Giorgi Università degli Studi di Siena Dipartimento di Biotecnologie, Chimica e Farmacia via Aldo Moro 53100 Siena e-mail:
Interazioni di ioni con elettroni (ECD, ETD) e fotoni (Ion spectroscopy) Gianluca Giorgi Università degli Studi di Siena Dipartimento di Biotecnologie, Chimica e Farmacia via Aldo Moro 53100 Siena e-mail:
Algorithms in Bioinformatics FOUR Pairwise Sequence Alignment. Pairwise Sequence Alignment. Convention: DNA Sequences 5. Sequence Alignment
 Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
Algorithms in Bioinformatics FOUR Sami Khuri Department of Computer Science San José State University Pairwise Sequence Alignment Homology Similarity Global string alignment Local string alignment Dot
BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer Systems Biology Exp. Methods
 BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 14. Systems Biology Exp. Methods Overview Transcriptomics Basics of microarrays Comparative analysis Interactomics:
BIOINF 4120 Bioinformatics 2 - Structures and Systems - Oliver Kohlbacher Summer 2013 14. Systems Biology Exp. Methods Overview Transcriptomics Basics of microarrays Comparative analysis Interactomics:
Parallel Algorithms For Real-Time Peptide-Spectrum Matching
 Parallel Algorithms For Real-Time Peptide-Spectrum Matching A Thesis Submitted to the College of Graduate Studies and Research in Partial Fulfillment of the Requirements for the degree of Master of Science
Parallel Algorithms For Real-Time Peptide-Spectrum Matching A Thesis Submitted to the College of Graduate Studies and Research in Partial Fulfillment of the Requirements for the degree of Master of Science