An Integrated Approach for the Assessment of Chromosomal Abnormalities
|
|
|
- Brook Leonard
- 5 years ago
- Views:
Transcription
1 An Integrated Approach for the Assessment of Chromosomal Abnormalities Department of Biostatistics Johns Hopkins Bloomberg School of Public Health June 26, 2007
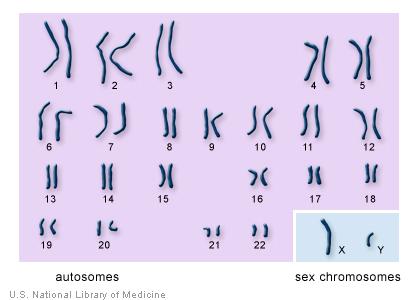
2 Karyotypes
3 Karyotypes General Cytogenetics Information

4 FISH Courtesy of the Pevsner Laboratory

5 SNP chip data
6 Estimation 1 By SNP: Estimate genotype and copy number for each SNP. 2 Within a sample: Borrow strength between SNPs to infer regions of LOH and copy number changes. 3 Between samples: Comparison between normal and disease populations to find chromosomal alterations associated with disease.
7 Deletion

8 Amplification

9 Uniparental Isodisomy

10 Cancer samples

11 Mosaicism

12 SNPchip S4 classes and methods
13 The structure of the data we observe At each SNP, we observe a noisy measure of the true copy number and genotype (and possibly also measures of confidence in those estimates).
14 Another HMM...? Novel (and we believe, important) HMM features: 1 Model the observation sequence of genotype calls and copy number jointly (Vanilla) 2 Integrate confidence estimates of the genotype calls and copy number estimates (ICE)
15 The Vanilla HMM components Observations ĈN and ĜT Hidden states Initial state probability distribution Transition probabilities Emission probabilities
16 Hidden states
17 Transition probabilities Following suggestions in the literature, we model the transition probabilities as a function of the distance d between SNPs. Specifically, let θ(d) 1 e 2d denote the probability that SNP i is not informative (I c ) for SNP at i + 1. For example: τ =P { (d) = P { = P { P i+1 i, d } i+1, I i, d } + P i+1, I c i, d i+1 I, i, d } P { I i, d } + i+1 I c, i, d P I c i, d = P { } θ(d).
18 Emission probabilities We assume conditional independence between copy number estimates and the genotype calls. For example: f( c CN, c GT ) = f( c CN ) f( c GT ) n o n o = f ccn f cgt = β n ccn o β n cgt o.
19 Vanilla HMM 5.0 A D B C E Amp LOH vanilla Norm Del
20 More information The confidence in genotype calls can differ substantially between SNPs! 4 2 sense antisense
21 Integrating confidence estimates for genotype calls Let S cgt be the confidence score for the genotype estimate. We can estimate from Hapmap the following densities: n o n o n f SĤOM ĤOM, HOM, f SĤOM ĤOM, HET, f S HET d HET, d o n HOM, f S HET d HET, d o HET. Note: n o f SĤOM ĤOM, n f S HET d HET, d o n o f SĤOM ĤOM, HOM n f S HET d HET, d o HOM.
22 Emission Probabilities - Loss Recall that f( c CN, c GT ) = f( c CN ) f( c GT ) n o n o = f ccn f cgt = β n ccn o β n cgt o. If the state for a particular SNP is Loss, we have β n cgt, S cgt o = f n o n cgt f S cgt GT, c o.
23 Emission Probabilities - Retention For retention, the true genotype can be HET or HOM: β n cgt, Sd GT o n o n = f cgt f S GT d GT, c o n o n = f cgt f S GT d, HOM GT, c o n + f S d GT, HET GT, c o n o n = f cgt f S GT d HOM, GT, c o n f HOM GT, c o n + f S GT d HET, GT, c o n f HET GT, c o n o n = f cgt f S GT d HOM, GT c o n f HOM GT, c o n + f S GT d HET, GT c o n f HET GT, c o
24 Vanilla ICE comparison 5.0 A D B C E Amp LOH vanilla Norm Del Amp LOH ICE Norm Del Bioconductor package: ICE Amp LOH A D vanilla B vanilla E Norm Del Amp LOH A vanilla D vanilla ICE B ICE E Norm Del ICE ICE
25 A HapMap sample LOH 2 normal AA/BB AB Mb
26 Many HapMap samples
27 SNP Trio

28 SNP Trio
29 SNP Trio
30 HMM for SNP Trio chromosome 10 chromosome 22 BPI BPI UPI F UPI F UPI M UPI M MI D MI D MI S non BPI MI S non BPI BPI BPI position (Mb) position (Mb)
31 Acknowledgments Rob Scharpf Giovanni Parmigiani Rafael Irizarry Benilton Carvalho, Wenyi Wang Jonathan Pevsner Nate Miller, Eli Roberson, Jason Ting
32 References Carvalho B, Bengtsson H, Speed TP, Irizarry RA (2007) Exploration, normalization, and genotype calls of high-density oligonucleotide SNP array data. Biostatistics, 8(2): Scharpf RB, Ting JC, Pevsner J, Ruczinski I (2007). SNPchip: R classes and methods for SNP array data. Bioinformatics, 23(5): Scharpf RB, Parmigiani G, Ruczinski I (2007). A hidden markov model for joint estimation of genotype and copy number in high-throughput SNP chips. JHU Biostatistics Working papers, #136. Ting JC, Ye Y, Thomas GH, Ruczinski I, Pevsner J (2006). Analysis and visualization of chromosomal abnormalities in SNP data with SNPscan. BMC Bioinformatics, 7(1):25. Ting JC, Roberson ED, Miller N, et al, Ruczinski I, Thomas GH, Pevsner J (2007). Visualization of uniparental inheritance, Mendelian inconsistencies, deletions and parent of origin effects in single nucleotide polymorphism trio data with SNPtrio. Human Mutation, (in press). Wang W, Caravalho B, Miller N, Pevsner J, Chakravarti A, Irizarry RA (2006) Estimating genome-wide copy number using allele specific mixture models. JHU Biostatistics Working papers, #122.
An Integrated Approach for the Assessment of Chromosomal Abnormalities
 An Integrated Approach for the Assessment of Chromosomal Abnormalities Department of Biostatistics Johns Hopkins Bloomberg School of Public Health June 6, 2007 Karyotypes Mitosis and Meiosis Meiosis Meiosis
An Integrated Approach for the Assessment of Chromosomal Abnormalities Department of Biostatistics Johns Hopkins Bloomberg School of Public Health June 6, 2007 Karyotypes Mitosis and Meiosis Meiosis Meiosis
Hidden Markov Models for the Assessment of Chromosomal Alterations using High-throughput SNP Arrays
 Hidden Markov Models for the Assessment of Chromosomal Alterations using High-throughput SNP Arrays Department of Biostatistics Johns Hopkins Bloomberg School of Public Health November 18, 2008 Acknowledgments
Hidden Markov Models for the Assessment of Chromosomal Alterations using High-throughput SNP Arrays Department of Biostatistics Johns Hopkins Bloomberg School of Public Health November 18, 2008 Acknowledgments
SNP Association Studies with Case-Parent Trios
 SNP Association Studies with Case-Parent Trios Department of Biostatistics Johns Hopkins Bloomberg School of Public Health July, 00 Acknowledgments Collaborators: Qing Li, Rob Scharpf, Holger Schwender,
SNP Association Studies with Case-Parent Trios Department of Biostatistics Johns Hopkins Bloomberg School of Public Health July, 00 Acknowledgments Collaborators: Qing Li, Rob Scharpf, Holger Schwender,
CSci 8980: Advanced Topics in Graphical Models Analysis of Genetic Variation
 CSci 8980: Advanced Topics in Graphical Models Analysis of Genetic Variation Instructor: Arindam Banerjee November 26, 2007 Genetic Polymorphism Single nucleotide polymorphism (SNP) Genetic Polymorphism
CSci 8980: Advanced Topics in Graphical Models Analysis of Genetic Variation Instructor: Arindam Banerjee November 26, 2007 Genetic Polymorphism Single nucleotide polymorphism (SNP) Genetic Polymorphism
Learning ancestral genetic processes using nonparametric Bayesian models
 Learning ancestral genetic processes using nonparametric Bayesian models Kyung-Ah Sohn October 31, 2011 Committee Members: Eric P. Xing, Chair Zoubin Ghahramani Russell Schwartz Kathryn Roeder Matthew
Learning ancestral genetic processes using nonparametric Bayesian models Kyung-Ah Sohn October 31, 2011 Committee Members: Eric P. Xing, Chair Zoubin Ghahramani Russell Schwartz Kathryn Roeder Matthew
Genotype Imputation. Biostatistics 666
 Genotype Imputation Biostatistics 666 Previously Hidden Markov Models for Relative Pairs Linkage analysis using affected sibling pairs Estimation of pairwise relationships Identity-by-Descent Relatives
Genotype Imputation Biostatistics 666 Previously Hidden Markov Models for Relative Pairs Linkage analysis using affected sibling pairs Estimation of pairwise relationships Identity-by-Descent Relatives
Genotype Imputation. Class Discussion for January 19, 2016
 Genotype Imputation Class Discussion for January 19, 2016 Intuition Patterns of genetic variation in one individual guide our interpretation of the genomes of other individuals Imputation uses previously
Genotype Imputation Class Discussion for January 19, 2016 Intuition Patterns of genetic variation in one individual guide our interpretation of the genomes of other individuals Imputation uses previously
Multiple Change-Point Detection and Analysis of Chromosome Copy Number Variations
 Multiple Change-Point Detection and Analysis of Chromosome Copy Number Variations Yale School of Public Health Joint work with Ning Hao, Yue S. Niu presented @Tsinghua University Outline 1 The Problem
Multiple Change-Point Detection and Analysis of Chromosome Copy Number Variations Yale School of Public Health Joint work with Ning Hao, Yue S. Niu presented @Tsinghua University Outline 1 The Problem
Expected complete data log-likelihood and EM
 Expected complete data log-likelihood and EM In our EM algorithm, the expected complete data log-likelihood Q is a function of a set of model parameters τ, ie M Qτ = log fb m, r m, g m z m, l m, τ p mz
Expected complete data log-likelihood and EM In our EM algorithm, the expected complete data log-likelihood Q is a function of a set of model parameters τ, ie M Qτ = log fb m, r m, g m z m, l m, τ p mz
1 Springer. Nan M. Laird Christoph Lange. The Fundamentals of Modern Statistical Genetics
 1 Springer Nan M. Laird Christoph Lange The Fundamentals of Modern Statistical Genetics 1 Introduction to Statistical Genetics and Background in Molecular Genetics 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
1 Springer Nan M. Laird Christoph Lange The Fundamentals of Modern Statistical Genetics 1 Introduction to Statistical Genetics and Background in Molecular Genetics 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
The Lander-Green Algorithm. Biostatistics 666 Lecture 22
 The Lander-Green Algorithm Biostatistics 666 Lecture Last Lecture Relationship Inferrence Likelihood of genotype data Adapt calculation to different relationships Siblings Half-Siblings Unrelated individuals
The Lander-Green Algorithm Biostatistics 666 Lecture Last Lecture Relationship Inferrence Likelihood of genotype data Adapt calculation to different relationships Siblings Half-Siblings Unrelated individuals
Some New Methods for Family-Based Association Studies
 Some New Methods for Family-Based Association Studies Ingo Ruczinski Department of Biostatistics Johns Hopkins Bloomberg School of Public Health April 8, 20 http: //biostat.jhsph.edu/ iruczins/ Topics
Some New Methods for Family-Based Association Studies Ingo Ruczinski Department of Biostatistics Johns Hopkins Bloomberg School of Public Health April 8, 20 http: //biostat.jhsph.edu/ iruczins/ Topics
Curriculum Links. AQA GCE Biology. AS level
 Curriculum Links AQA GCE Biology Unit 2 BIOL2 The variety of living organisms 3.2.1 Living organisms vary and this variation is influenced by genetic and environmental factors Causes of variation 3.2.2
Curriculum Links AQA GCE Biology Unit 2 BIOL2 The variety of living organisms 3.2.1 Living organisms vary and this variation is influenced by genetic and environmental factors Causes of variation 3.2.2
2. Map genetic distance between markers
 Chapter 5. Linkage Analysis Linkage is an important tool for the mapping of genetic loci and a method for mapping disease loci. With the availability of numerous DNA markers throughout the human genome,
Chapter 5. Linkage Analysis Linkage is an important tool for the mapping of genetic loci and a method for mapping disease loci. With the availability of numerous DNA markers throughout the human genome,
GENETICS - CLUTCH CH.1 INTRODUCTION TO GENETICS.
 !! www.clutchprep.com CONCEPT: HISTORY OF GENETICS The earliest use of genetics was through of plants and animals (8000-1000 B.C.) Selective breeding (artificial selection) is the process of breeding organisms
!! www.clutchprep.com CONCEPT: HISTORY OF GENETICS The earliest use of genetics was through of plants and animals (8000-1000 B.C.) Selective breeding (artificial selection) is the process of breeding organisms
SNP Association Studies with Case-Parent Trios
 SNP Association Studies with Case-Parent Trios Department of Biostatistics Johns Hopkins Bloomberg School of Public Health September 3, 2009 Population-based Association Studies Balding (2006). Nature
SNP Association Studies with Case-Parent Trios Department of Biostatistics Johns Hopkins Bloomberg School of Public Health September 3, 2009 Population-based Association Studies Balding (2006). Nature
Learning Your Identity and Disease from Research Papers: Information Leaks in Genome-Wide Association Study
 Learning Your Identity and Disease from Research Papers: Information Leaks in Genome-Wide Association Study Rui Wang, Yong Li, XiaoFeng Wang, Haixu Tang and Xiaoyong Zhou Indiana University at Bloomington
Learning Your Identity and Disease from Research Papers: Information Leaks in Genome-Wide Association Study Rui Wang, Yong Li, XiaoFeng Wang, Haixu Tang and Xiaoyong Zhou Indiana University at Bloomington
NONPARAMETRIC ADJUSTMENT FOR MEASUREMENT ERROR IN TIME TO EVENT DATA: APPLICATION TO RISK PREDICTION MODELS
 BIRS 2016 1 NONPARAMETRIC ADJUSTMENT FOR MEASUREMENT ERROR IN TIME TO EVENT DATA: APPLICATION TO RISK PREDICTION MODELS Malka Gorfine Tel Aviv University, Israel Joint work with Danielle Braun and Giovanni
BIRS 2016 1 NONPARAMETRIC ADJUSTMENT FOR MEASUREMENT ERROR IN TIME TO EVENT DATA: APPLICATION TO RISK PREDICTION MODELS Malka Gorfine Tel Aviv University, Israel Joint work with Danielle Braun and Giovanni
SNP-SNP Interactions in Case-Parent Trios
 Detection of SNP-SNP Interactions in Case-Parent Trios Department of Biostatistics Johns Hopkins Bloomberg School of Public Health June 2, 2009 Karyotypes http://ghr.nlm.nih.gov/ Single Nucleotide Polymphisms
Detection of SNP-SNP Interactions in Case-Parent Trios Department of Biostatistics Johns Hopkins Bloomberg School of Public Health June 2, 2009 Karyotypes http://ghr.nlm.nih.gov/ Single Nucleotide Polymphisms
RECONSTRUCTING DNA COPY NUMBER BY JOINT SEGMENTATION OF MULTIPLE SEQUENCES. Zhongyang Zhang Kenneth Lange Chiara Sabatti
 RECONSTRUCTING DNA COPY NUMBER BY JOINT SEGMENTATION OF MULTIPLE SEQUENCES By Zhongyang Zhang Kenneth Lange Chiara Sabatti Technical Report 261 March 2012 Division of Biostatistics STANFORD UNIVERSITY
RECONSTRUCTING DNA COPY NUMBER BY JOINT SEGMENTATION OF MULTIPLE SEQUENCES By Zhongyang Zhang Kenneth Lange Chiara Sabatti Technical Report 261 March 2012 Division of Biostatistics STANFORD UNIVERSITY
UNIT 8 BIOLOGY: Meiosis and Heredity Page 148
 UNIT 8 BIOLOGY: Meiosis and Heredity Page 148 CP: CHAPTER 6, Sections 1-6; CHAPTER 7, Sections 1-4; HN: CHAPTER 11, Section 1-5 Standard B-4: The student will demonstrate an understanding of the molecular
UNIT 8 BIOLOGY: Meiosis and Heredity Page 148 CP: CHAPTER 6, Sections 1-6; CHAPTER 7, Sections 1-4; HN: CHAPTER 11, Section 1-5 Standard B-4: The student will demonstrate an understanding of the molecular
Binomial Mixture Model-based Association Tests under Genetic Heterogeneity
 Binomial Mixture Model-based Association Tests under Genetic Heterogeneity Hui Zhou, Wei Pan Division of Biostatistics, School of Public Health, University of Minnesota, Minneapolis, MN 55455 April 30,
Binomial Mixture Model-based Association Tests under Genetic Heterogeneity Hui Zhou, Wei Pan Division of Biostatistics, School of Public Health, University of Minnesota, Minneapolis, MN 55455 April 30,
MATHEMATICAL MODELS - Vol. III - Mathematical Modeling and the Human Genome - Hilary S. Booth MATHEMATICAL MODELING AND THE HUMAN GENOME
 MATHEMATICAL MODELING AND THE HUMAN GENOME Hilary S. Booth Australian National University, Australia Keywords: Human genome, DNA, bioinformatics, sequence analysis, evolution. Contents 1. Introduction:
MATHEMATICAL MODELING AND THE HUMAN GENOME Hilary S. Booth Australian National University, Australia Keywords: Human genome, DNA, bioinformatics, sequence analysis, evolution. Contents 1. Introduction:
Bioinformatics. Genotype -> Phenotype DNA. Jason H. Moore, Ph.D. GECCO 2007 Tutorial / Bioinformatics.
 Bioinformatics Jason H. Moore, Ph.D. Frank Lane Research Scholar in Computational Genetics Associate Professor of Genetics Adjunct Associate Professor of Biological Sciences Adjunct Associate Professor
Bioinformatics Jason H. Moore, Ph.D. Frank Lane Research Scholar in Computational Genetics Associate Professor of Genetics Adjunct Associate Professor of Biological Sciences Adjunct Associate Professor
O 3 O 4 O 5. q 3. q 4. Transition
 Hidden Markov Models Hidden Markov models (HMM) were developed in the early part of the 1970 s and at that time mostly applied in the area of computerized speech recognition. They are first described in
Hidden Markov Models Hidden Markov models (HMM) were developed in the early part of the 1970 s and at that time mostly applied in the area of computerized speech recognition. They are first described in
GSBHSRSBRSRRk IZTI/^Q. LlML. I Iv^O IV I I I FROM GENES TO GENOMES ^^^H*" ^^^^J*^ ill! BQPIP. illt. goidbkc. itip31. li4»twlil FIFTH EDITION
 FIFTH EDITION IV I ^HHk ^ttm IZTI/^Q i I II MPHBBMWBBIHB '-llwmpbi^hbwm^^pfc ' GSBHSRSBRSRRk LlML I I \l 1MB ^HP'^^MMMP" jflp^^^^^^^^st I Iv^O FROM GENES TO GENOMES %^MiM^PM^^MWi99Mi$9i0^^ ^^^^^^^^^^^^^V^^^fii^^t^i^^^^^
FIFTH EDITION IV I ^HHk ^ttm IZTI/^Q i I II MPHBBMWBBIHB '-llwmpbi^hbwm^^pfc ' GSBHSRSBRSRRk LlML I I \l 1MB ^HP'^^MMMP" jflp^^^^^^^^st I Iv^O FROM GENES TO GENOMES %^MiM^PM^^MWi99Mi$9i0^^ ^^^^^^^^^^^^^V^^^fii^^t^i^^^^^
Calculation of IBD probabilities
 Calculation of IBD probabilities David Evans and Stacey Cherny University of Oxford Wellcome Trust Centre for Human Genetics This Session IBD vs IBS Why is IBD important? Calculating IBD probabilities
Calculation of IBD probabilities David Evans and Stacey Cherny University of Oxford Wellcome Trust Centre for Human Genetics This Session IBD vs IBS Why is IBD important? Calculating IBD probabilities
Model-Free Knockoffs: High-Dimensional Variable Selection that Controls the False Discovery Rate
 Model-Free Knockoffs: High-Dimensional Variable Selection that Controls the False Discovery Rate Lucas Janson, Stanford Department of Statistics WADAPT Workshop, NIPS, December 2016 Collaborators: Emmanuel
Model-Free Knockoffs: High-Dimensional Variable Selection that Controls the False Discovery Rate Lucas Janson, Stanford Department of Statistics WADAPT Workshop, NIPS, December 2016 Collaborators: Emmanuel
CISC 889 Bioinformatics (Spring 2004) Hidden Markov Models (II)
 Hidden Markov Models (II)") CISC 889 Bioinformatics (Spring 24) Hidden Markov Models (II) a. Likelihood: forward algorithm b. Decoding: Viterbi algorithm c. Model building: Baum-Welch algorithm Viterbi training Hidden Markov models
CISC 889 Bioinformatics (Spring 24) Hidden Markov Models (II) a. Likelihood: forward algorithm b. Decoding: Viterbi algorithm c. Model building: Baum-Welch algorithm Viterbi training Hidden Markov models
- mutations can occur at different levels from single nucleotide positions in DNA to entire genomes.
 February 8, 2005 Bio 107/207 Winter 2005 Lecture 11 Mutation and transposable elements - the term mutation has an interesting history. - as far back as the 17th century, it was used to describe any drastic
February 8, 2005 Bio 107/207 Winter 2005 Lecture 11 Mutation and transposable elements - the term mutation has an interesting history. - as far back as the 17th century, it was used to describe any drastic
Lecture 24. Ingo Ruczinski. November 24, Department of Biostatistics Johns Hopkins Bloomberg School of Public Health Johns Hopkins University
 Department of Biostatistics Johns Hopkins Bloomberg School of Public Health Johns Hopkins University November 24, 2015 1 2 3 4 5 1 Odds ratios for retrospective studies 2 Odds ratios approximating the
Department of Biostatistics Johns Hopkins Bloomberg School of Public Health Johns Hopkins University November 24, 2015 1 2 3 4 5 1 Odds ratios for retrospective studies 2 Odds ratios approximating the
Friday Harbor From Genetics to GWAS (Genome-wide Association Study) Sept David Fardo
 Sept David Fardo") Friday Harbor 2017 From Genetics to GWAS (Genome-wide Association Study) Sept 7 2017 David Fardo Purpose: prepare for tomorrow s tutorial Genetic Variants Quality Control Imputation Association Visualization
Friday Harbor 2017 From Genetics to GWAS (Genome-wide Association Study) Sept 7 2017 David Fardo Purpose: prepare for tomorrow s tutorial Genetic Variants Quality Control Imputation Association Visualization
Supporting Information
 Supporting Information Hammer et al. 10.1073/pnas.1109300108 SI Materials and Methods Two-Population Model. Estimating demographic parameters. For each pair of sub-saharan African populations we consider
Supporting Information Hammer et al. 10.1073/pnas.1109300108 SI Materials and Methods Two-Population Model. Estimating demographic parameters. For each pair of sub-saharan African populations we consider
Cloud-scale RNA-sequencing differential expression analysis with Myrna
 Cloud-scale RNA-sequencing differential expression analysis with Myrna Jeff Leek Johns Hopkins Bloomberg School of Public Health e: jleek@jhsph.edu t: http://www.twitter.com/leekgroup myrna: http://bowtie-bio.sourceforge.net/myrna/
Cloud-scale RNA-sequencing differential expression analysis with Myrna Jeff Leek Johns Hopkins Bloomberg School of Public Health e: jleek@jhsph.edu t: http://www.twitter.com/leekgroup myrna: http://bowtie-bio.sourceforge.net/myrna/
GS Analysis of Microarray Data
 GS01 0163 Analysis of Microarray Data Keith Baggerly and Kevin Coombes Section of Bioinformatics Department of Biostatistics and Applied Mathematics UT M. D. Anderson Cancer Center kabagg@mdanderson.org
GS01 0163 Analysis of Microarray Data Keith Baggerly and Kevin Coombes Section of Bioinformatics Department of Biostatistics and Applied Mathematics UT M. D. Anderson Cancer Center kabagg@mdanderson.org
GS Analysis of Microarray Data
 GS01 0163 Analysis of Microarray Data Keith Baggerly and Kevin Coombes Section of Bioinformatics Department of Biostatistics and Applied Mathematics UT M. D. Anderson Cancer Center kabagg@mdanderson.org
GS01 0163 Analysis of Microarray Data Keith Baggerly and Kevin Coombes Section of Bioinformatics Department of Biostatistics and Applied Mathematics UT M. D. Anderson Cancer Center kabagg@mdanderson.org
A sequential Monte Carlo framework for haplotype inference in CNV/SNP genotype data
 Iliadis et al. EURASIP Journal on Bioinformatics and Systems Biology 2014, 2014:7 RESEARCH Open Access A sequential Monte Carlo framework for haplotype inference in CNV/SNP genotype data Alexandros Iliadis,
Iliadis et al. EURASIP Journal on Bioinformatics and Systems Biology 2014, 2014:7 RESEARCH Open Access A sequential Monte Carlo framework for haplotype inference in CNV/SNP genotype data Alexandros Iliadis,
Bioinformatics 2 - Lecture 4
 Bioinformatics 2 - Lecture 4 Guido Sanguinetti School of Informatics University of Edinburgh February 14, 2011 Sequences Many data types are ordered, i.e. you can naturally say what is before and what
Bioinformatics 2 - Lecture 4 Guido Sanguinetti School of Informatics University of Edinburgh February 14, 2011 Sequences Many data types are ordered, i.e. you can naturally say what is before and what
Association Testing with Quantitative Traits: Common and Rare Variants. Summer Institute in Statistical Genetics 2014 Module 10 Lecture 5
 Association Testing with Quantitative Traits: Common and Rare Variants Timothy Thornton and Katie Kerr Summer Institute in Statistical Genetics 2014 Module 10 Lecture 5 1 / 41 Introduction to Quantitative
Association Testing with Quantitative Traits: Common and Rare Variants Timothy Thornton and Katie Kerr Summer Institute in Statistical Genetics 2014 Module 10 Lecture 5 1 / 41 Introduction to Quantitative
Lecture 12 April 25, 2018
 Stats 300C: Theory of Statistics Spring 2018 Lecture 12 April 25, 2018 Prof. Emmanuel Candes Scribe: Emmanuel Candes, Chenyang Zhong 1 Outline Agenda: The Knockoffs Framework 1. The Knockoffs Framework
Stats 300C: Theory of Statistics Spring 2018 Lecture 12 April 25, 2018 Prof. Emmanuel Candes Scribe: Emmanuel Candes, Chenyang Zhong 1 Outline Agenda: The Knockoffs Framework 1. The Knockoffs Framework
Some Statistical Models and Algorithms for Change-Point Problems in Genomics
 Some Statistical Models and Algorithms for Change-Point Problems in Genomics S. Robin UMR 518 AgroParisTech / INRA Applied MAth & Comput. Sc. Journées SMAI-MAIRCI Grenoble, September 2012 S. Robin (AgroParisTech
Some Statistical Models and Algorithms for Change-Point Problems in Genomics S. Robin UMR 518 AgroParisTech / INRA Applied MAth & Comput. Sc. Journées SMAI-MAIRCI Grenoble, September 2012 S. Robin (AgroParisTech
Comparative Gene Finding. BMI/CS 776 Spring 2015 Colin Dewey
 Comparative Gene Finding BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2015 Colin Dewey cdewey@biostat.wisc.edu Goals for Lecture the key concepts to understand are the following: using related genomes
Comparative Gene Finding BMI/CS 776 www.biostat.wisc.edu/bmi776/ Spring 2015 Colin Dewey cdewey@biostat.wisc.edu Goals for Lecture the key concepts to understand are the following: using related genomes
Hidden Markov Dirichlet Process: Modeling Genetic Recombination in Open Ancestral Space
 Hidden Markov Dirichlet Process: Modeling Genetic Recombination in Open Ancestral Space Kyung-Ah Sohn School of Computer Science Carnegie Mellon University Pittsburgh, PA 15213 ksohn@cs.cmu.edu Eric P.
Hidden Markov Dirichlet Process: Modeling Genetic Recombination in Open Ancestral Space Kyung-Ah Sohn School of Computer Science Carnegie Mellon University Pittsburgh, PA 15213 ksohn@cs.cmu.edu Eric P.
BIOINFORMATICS. Discrepancies in dbsnp confirmation rates and allele frequency distributions from varying genotyping error rates and patterns
 BIOINFORMATICS Vol. 2 no. 7 24, pages 122 132 DOI: 1.193/bioinformatics/bth34 Discrepancies in dbsnp confirmation rates and allele frequency distributions from varying genotyping error rates and patterns
BIOINFORMATICS Vol. 2 no. 7 24, pages 122 132 DOI: 1.193/bioinformatics/bth34 Discrepancies in dbsnp confirmation rates and allele frequency distributions from varying genotyping error rates and patterns
Multidimensional data analysis in biomedicine and epidemiology
 in biomedicine and epidemiology Katja Ickstadt and Leo N. Geppert Faculty of Statistics, TU Dortmund, Germany Stakeholder Workshop 12 13 December 2017, PTB Berlin Supported by Deutsche Forschungsgemeinschaft
in biomedicine and epidemiology Katja Ickstadt and Leo N. Geppert Faculty of Statistics, TU Dortmund, Germany Stakeholder Workshop 12 13 December 2017, PTB Berlin Supported by Deutsche Forschungsgemeinschaft
New imputation strategies optimized for crop plants: FILLIN (Fast, Inbred Line Library ImputatioN) FSFHap (Full Sib Family Haplotype)
 FSFHap (Full Sib Family Haplotype)") New imputation strategies optimized for crop plants: FILLIN (Fast, Inbred Line Library ImputatioN) FSFHap (Full Sib Family Haplotype) Kelly Swarts PAG Allele Mining 1/11/2014 Imputation is the projection
New imputation strategies optimized for crop plants: FILLIN (Fast, Inbred Line Library ImputatioN) FSFHap (Full Sib Family Haplotype) Kelly Swarts PAG Allele Mining 1/11/2014 Imputation is the projection
Complexity and Approximation of the Minimum Recombination Haplotype Configuration Problem
 Complexity and Approximation of the Minimum Recombination Haplotype Configuration Problem Lan Liu 1, Xi Chen 3, Jing Xiao 3, and Tao Jiang 1,2 1 Department of Computer Science and Engineering, University
Complexity and Approximation of the Minimum Recombination Haplotype Configuration Problem Lan Liu 1, Xi Chen 3, Jing Xiao 3, and Tao Jiang 1,2 1 Department of Computer Science and Engineering, University
Supplementary Information for Discovery and characterization of indel and point mutations
 Supplementary Information for Discovery and characterization of indel and point mutations using DeNovoGear Avinash Ramu 1 Michiel J. Noordam 1 Rachel S. Schwartz 2 Arthur Wuster 3 Matthew E. Hurles 3 Reed
Supplementary Information for Discovery and characterization of indel and point mutations using DeNovoGear Avinash Ramu 1 Michiel J. Noordam 1 Rachel S. Schwartz 2 Arthur Wuster 3 Matthew E. Hurles 3 Reed
Tutorial Session 2. MCMC for the analysis of genetic data on pedigrees:
 MCMC for the analysis of genetic data on pedigrees: Tutorial Session 2 Elizabeth Thompson University of Washington Genetic mapping and linkage lod scores Monte Carlo likelihood and likelihood ratio estimation
MCMC for the analysis of genetic data on pedigrees: Tutorial Session 2 Elizabeth Thompson University of Washington Genetic mapping and linkage lod scores Monte Carlo likelihood and likelihood ratio estimation
Chapter 2: Extensions to Mendel: Complexities in Relating Genotype to Phenotype.
 Chapter 2: Extensions to Mendel: Complexities in Relating Genotype to Phenotype. please read pages 38-47; 49-55;57-63. Slide 1 of Chapter 2 1 Extension sot Mendelian Behavior of Genes Single gene inheritance
Chapter 2: Extensions to Mendel: Complexities in Relating Genotype to Phenotype. please read pages 38-47; 49-55;57-63. Slide 1 of Chapter 2 1 Extension sot Mendelian Behavior of Genes Single gene inheritance
A segmentation-clustering problem for the analysis of array CGH data
 A segmentation-clustering problem for the analysis of array CGH data F. Picard, S. Robin, E. Lebarbier, J-J. Daudin UMR INA P-G / ENGREF / INRA MIA 518 APPLIED STOCHASTIC MODELS AND DATA ANALYSIS Brest
A segmentation-clustering problem for the analysis of array CGH data F. Picard, S. Robin, E. Lebarbier, J-J. Daudin UMR INA P-G / ENGREF / INRA MIA 518 APPLIED STOCHASTIC MODELS AND DATA ANALYSIS Brest
Page 1. References. Hidden Markov models and multiple sequence alignment. Markov chains. Probability review. Example. Markovian sequence
 Page Hidden Markov models and multiple sequence alignment Russ B Altman BMI 4 CS 74 Some slides borrowed from Scott C Schmidler (BMI graduate student) References Bioinformatics Classic: Krogh et al (994)
Page Hidden Markov models and multiple sequence alignment Russ B Altman BMI 4 CS 74 Some slides borrowed from Scott C Schmidler (BMI graduate student) References Bioinformatics Classic: Krogh et al (994)
NETWORK BIOLOGY AND COMPLEX DISEASES. Ahto Salumets
 NETWORK BIOLOGY AND COMPLEX DISEASES Ahto Salumets CENTRAL DOGMA OF BIOLOGY https://en.wikipedia.org/wiki/central_dogma_of_molecular_biology http://www.qaraqalpaq.com/genetics.html CHROMOSOMES SINGLE-NUCLEOTIDE
NETWORK BIOLOGY AND COMPLEX DISEASES Ahto Salumets CENTRAL DOGMA OF BIOLOGY https://en.wikipedia.org/wiki/central_dogma_of_molecular_biology http://www.qaraqalpaq.com/genetics.html CHROMOSOMES SINGLE-NUCLEOTIDE
Supplementary Materials for Integrated study of copy number states and genotype calls using high density SNP arrays
 Supplementary Materials for Integrated study of copy number states and genotype calls using high density SNP arrays A HapMap samples Originally, Illumina performed 73 CEU samples, 77 YRI samples, and 75
Supplementary Materials for Integrated study of copy number states and genotype calls using high density SNP arrays A HapMap samples Originally, Illumina performed 73 CEU samples, 77 YRI samples, and 75
BENCHMARK 1 STUDY GUIDE SPRING 2017
 BENCHMARK 1 STUDY GUIDE SPRING 2017 Name: There will be semester one content on this benchmark as well. Study your final exam review guide from last semester. New Semester Material: (Chapter 10 Cell Growth
BENCHMARK 1 STUDY GUIDE SPRING 2017 Name: There will be semester one content on this benchmark as well. Study your final exam review guide from last semester. New Semester Material: (Chapter 10 Cell Growth
Solutions to Problem Set 4
 Question 1 Solutions to 7.014 Problem Set 4 Because you have not read much scientific literature, you decide to study the genetics of garden peas. You have two pure breeding pea strains. One that is tall
Question 1 Solutions to 7.014 Problem Set 4 Because you have not read much scientific literature, you decide to study the genetics of garden peas. You have two pure breeding pea strains. One that is tall
SAT in Bioinformatics: Making the Case with Haplotype Inference
 SAT in Bioinformatics: Making the Case with Haplotype Inference Inês Lynce 1 and João Marques-Silva 2 1 IST/INESC-ID, Technical University of Lisbon, Portugal ines@sat.inesc-id.pt 2 School of Electronics
SAT in Bioinformatics: Making the Case with Haplotype Inference Inês Lynce 1 and João Marques-Silva 2 1 IST/INESC-ID, Technical University of Lisbon, Portugal ines@sat.inesc-id.pt 2 School of Electronics
Calculation of IBD probabilities
 Calculation of IBD probabilities David Evans University of Bristol This Session Identity by Descent (IBD) vs Identity by state (IBS) Why is IBD important? Calculating IBD probabilities Lander-Green Algorithm
Calculation of IBD probabilities David Evans University of Bristol This Session Identity by Descent (IBD) vs Identity by state (IBS) Why is IBD important? Calculating IBD probabilities Lander-Green Algorithm
Supporting Information
 Supporting Information Weghorn and Lässig 10.1073/pnas.1210887110 SI Text Null Distributions of Nucleosome Affinity and of Regulatory Site Content. Our inference of selection is based on a comparison of
Supporting Information Weghorn and Lässig 10.1073/pnas.1210887110 SI Text Null Distributions of Nucleosome Affinity and of Regulatory Site Content. Our inference of selection is based on a comparison of
Decision Theoretic Classification of Copy-Number-Variation in Cancer Genomes
 Decision Theoretic Classification of Copy-Number-Variation in Cancer Genomes Christopher Holmes (joint work with Chris Yau) Department of Statistics, & Wellcome Trust Centre for Human Genetics, University
Decision Theoretic Classification of Copy-Number-Variation in Cancer Genomes Christopher Holmes (joint work with Chris Yau) Department of Statistics, & Wellcome Trust Centre for Human Genetics, University
Learning Ancestral Genetic Processes using Nonparametric Bayesian Models
 Learning Ancestral Genetic Processes using Nonparametric Bayesian Models Kyung-Ah Sohn CMU-CS-11-136 November 2011 Computer Science Department School of Computer Science Carnegie Mellon University Pittsburgh,
Learning Ancestral Genetic Processes using Nonparametric Bayesian Models Kyung-Ah Sohn CMU-CS-11-136 November 2011 Computer Science Department School of Computer Science Carnegie Mellon University Pittsburgh,
Robust Detection and Identification of Sparse Segments in Ultra-High Dimensional Data Analysis
 Robust Detection and Identification of Sparse Segments in Ultra-High Dimensional Data Analysis Hongzhe Li hongzhe@upenn.edu, http://statgene.med.upenn.edu University of Pennsylvania Perelman School of
Robust Detection and Identification of Sparse Segments in Ultra-High Dimensional Data Analysis Hongzhe Li hongzhe@upenn.edu, http://statgene.med.upenn.edu University of Pennsylvania Perelman School of
Association studies and regression
 Association studies and regression CM226: Machine Learning for Bioinformatics. Fall 2016 Sriram Sankararaman Acknowledgments: Fei Sha, Ameet Talwalkar Association studies and regression 1 / 104 Administration
Association studies and regression CM226: Machine Learning for Bioinformatics. Fall 2016 Sriram Sankararaman Acknowledgments: Fei Sha, Ameet Talwalkar Association studies and regression 1 / 104 Administration
On coding genotypes for genetic markers with multiple alleles in genetic association study of quantitative traits
 Wang BMC Genetics 011, 1:8 http://www.biomedcentral.com/171-156/1/8 METHODOLOGY ARTICLE Open Access On coding genotypes for genetic markers with multiple alleles in genetic association study of quantitative
Wang BMC Genetics 011, 1:8 http://www.biomedcentral.com/171-156/1/8 METHODOLOGY ARTICLE Open Access On coding genotypes for genetic markers with multiple alleles in genetic association study of quantitative
Computational Methods for Learning Population History from Large Scale Genetic Variation Datasets
 Computational Methods for Learning Population History from Large Scale Genetic Variation Datasets Ming-Chi Tsai CMU-CB-13-102 July 2, 2013 School of Computer Science Carnegie Mellon University Pittsburgh,
Computational Methods for Learning Population History from Large Scale Genetic Variation Datasets Ming-Chi Tsai CMU-CB-13-102 July 2, 2013 School of Computer Science Carnegie Mellon University Pittsburgh,
Lecture 28. Ingo Ruczinski. December 3, Department of Biostatistics Johns Hopkins Bloomberg School of Public Health Johns Hopkins University
 Lecture 28 Department of Biostatistics Johns Hopkins Bloomberg School of Public Health Johns Hopkins University December 3, 2015 1 2 3 4 5 1 Familywise error rates 2 procedure 3 Performance of with multiple
Lecture 28 Department of Biostatistics Johns Hopkins Bloomberg School of Public Health Johns Hopkins University December 3, 2015 1 2 3 4 5 1 Familywise error rates 2 procedure 3 Performance of with multiple
Web-based Supplementary Materials for BM-Map: Bayesian Mapping of Multireads for Next-Generation Sequencing Data
 Web-based Supplementary Materials for BM-Map: Bayesian Mapping of Multireads for Next-Generation Sequencing Data Yuan Ji 1,, Yanxun Xu 2, Qiong Zhang 3, Kam-Wah Tsui 3, Yuan Yuan 4, Clift Norris 1, Shoudan
Web-based Supplementary Materials for BM-Map: Bayesian Mapping of Multireads for Next-Generation Sequencing Data Yuan Ji 1,, Yanxun Xu 2, Qiong Zhang 3, Kam-Wah Tsui 3, Yuan Yuan 4, Clift Norris 1, Shoudan
Introduction to population genetics & evolution
 Introduction to population genetics & evolution Course Organization Exam dates: Feb 19 March 1st Has everybody registered? Did you get the email with the exam schedule Summer seminar: Hot topics in Bioinformatics
Introduction to population genetics & evolution Course Organization Exam dates: Feb 19 March 1st Has everybody registered? Did you get the email with the exam schedule Summer seminar: Hot topics in Bioinformatics
Principles of QTL Mapping. M.Imtiaz
 Principles of QTL Mapping M.Imtiaz Introduction Definitions of terminology Reasons for QTL mapping Principles of QTL mapping Requirements For QTL Mapping Demonstration with experimental data Merit of QTL
Principles of QTL Mapping M.Imtiaz Introduction Definitions of terminology Reasons for QTL mapping Principles of QTL mapping Requirements For QTL Mapping Demonstration with experimental data Merit of QTL
Mixtures and Hidden Markov Models for analyzing genomic data
 Mixtures and Hidden Markov Models for analyzing genomic data Marie-Laure Martin-Magniette UMR AgroParisTech/INRA Mathématique et Informatique Appliquées, Paris UMR INRA/UEVE ERL CNRS Unité de Recherche
Mixtures and Hidden Markov Models for analyzing genomic data Marie-Laure Martin-Magniette UMR AgroParisTech/INRA Mathématique et Informatique Appliquées, Paris UMR INRA/UEVE ERL CNRS Unité de Recherche
Chapter 6 Linkage Disequilibrium & Gene Mapping (Recombination)
") 12/5/14 Chapter 6 Linkage Disequilibrium & Gene Mapping (Recombination) Linkage Disequilibrium Genealogical Interpretation of LD Association Mapping 1 Linkage and Recombination v linkage equilibrium ²
12/5/14 Chapter 6 Linkage Disequilibrium & Gene Mapping (Recombination) Linkage Disequilibrium Genealogical Interpretation of LD Association Mapping 1 Linkage and Recombination v linkage equilibrium ²
Expression QTLs and Mapping of Complex Trait Loci. Paul Schliekelman Statistics Department University of Georgia
 Expression QTLs and Mapping of Complex Trait Loci Paul Schliekelman Statistics Department University of Georgia Definitions: Genes, Loci and Alleles A gene codes for a protein. Proteins due everything.
Expression QTLs and Mapping of Complex Trait Loci Paul Schliekelman Statistics Department University of Georgia Definitions: Genes, Loci and Alleles A gene codes for a protein. Proteins due everything.
Classification of SNP genotypes by a Gaussian mixture model in competitive enzymatic assays
 Mathematical Statistics Stockholm University Classification of SNP genotypes by a Gaussian mixture model in competitive enzymatic assays Hedvig Norlén, Erik Pettersson, Afshin Ahmadian, Joakim Lundeberg
Mathematical Statistics Stockholm University Classification of SNP genotypes by a Gaussian mixture model in competitive enzymatic assays Hedvig Norlén, Erik Pettersson, Afshin Ahmadian, Joakim Lundeberg
Frequency Spectra and Inference in Population Genetics
 Frequency Spectra and Inference in Population Genetics Although coalescent models have come to play a central role in population genetics, there are some situations where genealogies may not lead to efficient
Frequency Spectra and Inference in Population Genetics Although coalescent models have come to play a central role in population genetics, there are some situations where genealogies may not lead to efficient
GS Analysis of Microarray Data
 GS01 0163 Analysis of Microarray Data Keith Baggerly and Bradley Broom Department of Bioinformatics and Computational Biology UT M. D. Anderson Cancer Center kabagg@mdanderson.org bmbroom@mdanderson.org
GS01 0163 Analysis of Microarray Data Keith Baggerly and Bradley Broom Department of Bioinformatics and Computational Biology UT M. D. Anderson Cancer Center kabagg@mdanderson.org bmbroom@mdanderson.org
Constructing a Pedigree
 Constructing a Pedigree Use the appropriate symbols: Unaffected Male Unaffected Female Affected Male Affected Female Male carrier of trait Mating of Offspring 2. Label each generation down the left hand
Constructing a Pedigree Use the appropriate symbols: Unaffected Male Unaffected Female Affected Male Affected Female Male carrier of trait Mating of Offspring 2. Label each generation down the left hand
Gene mapping, linkage analysis and computational challenges. Konstantin Strauch
 Gene mapping, linkage analysis an computational challenges Konstantin Strauch Institute for Meical Biometry, Informatics, an Epiemiology (IMBIE) University of Bonn E-mail: strauch@uni-bonn.e Genetics an
Gene mapping, linkage analysis an computational challenges Konstantin Strauch Institute for Meical Biometry, Informatics, an Epiemiology (IMBIE) University of Bonn E-mail: strauch@uni-bonn.e Genetics an
Alleles Notes. 3. In the above table, circle each symbol that represents part of a DNA molecule. Underline each word that is the name of a protein.
 Alleles Notes Different versions of the same gene are called alleles. Different alleles give the instructions for making different versions of a protein. This table shows examples for two human genes.
Alleles Notes Different versions of the same gene are called alleles. Different alleles give the instructions for making different versions of a protein. This table shows examples for two human genes.
Feature Selection via Block-Regularized Regression
 Feature Selection via Block-Regularized Regression Seyoung Kim School of Computer Science Carnegie Mellon University Pittsburgh, PA 3 Eric Xing School of Computer Science Carnegie Mellon University Pittsburgh,
Feature Selection via Block-Regularized Regression Seyoung Kim School of Computer Science Carnegie Mellon University Pittsburgh, PA 3 Eric Xing School of Computer Science Carnegie Mellon University Pittsburgh,
The genomes of recombinant inbred lines
 The genomes of recombinant inbred lines Karl W Broman Department of Biostatistics Johns Hopkins University http://www.biostat.jhsph.edu/~kbroman C57BL/6 2 1 Recombinant inbred lines (by sibling mating)
The genomes of recombinant inbred lines Karl W Broman Department of Biostatistics Johns Hopkins University http://www.biostat.jhsph.edu/~kbroman C57BL/6 2 1 Recombinant inbred lines (by sibling mating)
Dropping Your Genes. A Simulation of Meiosis and Fertilization and An Introduction to Probability
 Dropping Your Genes A Simulation of Meiosis and Fertilization and An Introduction to To fully understand Mendelian genetics (and, eventually, population genetics), you need to understand certain aspects
Dropping Your Genes A Simulation of Meiosis and Fertilization and An Introduction to To fully understand Mendelian genetics (and, eventually, population genetics), you need to understand certain aspects
Common Variants near MBNL1 and NKX2-5 are Associated with Infantile Hypertrophic Pyloric Stenosis
 Supplementary Information: Common Variants near MBNL1 and NKX2-5 are Associated with Infantile Hypertrophic Pyloric Stenosis Bjarke Feenstra 1*, Frank Geller 1*, Camilla Krogh 1, Mads V. Hollegaard 2,
Supplementary Information: Common Variants near MBNL1 and NKX2-5 are Associated with Infantile Hypertrophic Pyloric Stenosis Bjarke Feenstra 1*, Frank Geller 1*, Camilla Krogh 1, Mads V. Hollegaard 2,
A Statistical Framework for Expression Trait Loci (ETL) Mapping. Meng Chen
 Mapping. Meng Chen") A Statistical Framework for Expression Trait Loci (ETL) Mapping Meng Chen Prelim Paper in partial fulfillment of the requirements for the Ph.D. program in the Department of Statistics University of Wisconsin-Madison
A Statistical Framework for Expression Trait Loci (ETL) Mapping Meng Chen Prelim Paper in partial fulfillment of the requirements for the Ph.D. program in the Department of Statistics University of Wisconsin-Madison
Variation of Traits. genetic variation: the measure of the differences among individuals within a population
 Genetic variability is the measure of the differences among individuals within a population. Because some traits are more suited to certain environments, creating particular niches and fits, we know that
Genetic variability is the measure of the differences among individuals within a population. Because some traits are more suited to certain environments, creating particular niches and fits, we know that
Probability of Detecting Disease-Associated SNPs in Case-Control Genome-Wide Association Studies
 Probability of Detecting Disease-Associated SNPs in Case-Control Genome-Wide Association Studies Ruth Pfeiffer, Ph.D. Mitchell Gail Biostatistics Branch Division of Cancer Epidemiology&Genetics National
Probability of Detecting Disease-Associated SNPs in Case-Control Genome-Wide Association Studies Ruth Pfeiffer, Ph.D. Mitchell Gail Biostatistics Branch Division of Cancer Epidemiology&Genetics National
A DNA Sequence 2017/12/6 1
 A DNA Sequence ccgtacgtacgtagagtgctagtctagtcgtagcgccgtagtcgatcgtgtgg gtagtagctgatatgatgcgaggtaggggataggatagcaacagatgagc ggatgctgagtgcagtggcatgcgatgtcgatgatagcggtaggtagacttc gcgcataaagctgcgcgagatgattgcaaagragttagatgagctgatgcta
A DNA Sequence ccgtacgtacgtagagtgctagtctagtcgtagcgccgtagtcgatcgtgtgg gtagtagctgatatgatgcgaggtaggggataggatagcaacagatgagc ggatgctgagtgcagtggcatgcgatgtcgatgatagcggtaggtagacttc gcgcataaagctgcgcgagatgattgcaaagragttagatgagctgatgcta
Describe the process of cell division in prokaryotic cells. The Cell Cycle
 The Cell Cycle Objective # 1 In this topic we will examine the cell cycle, the series of changes that a cell goes through from one division to the next. We will pay particular attention to how the genetic
The Cell Cycle Objective # 1 In this topic we will examine the cell cycle, the series of changes that a cell goes through from one division to the next. We will pay particular attention to how the genetic
The Quantitative TDT
 The Quantitative TDT (Quantitative Transmission Disequilibrium Test) Warren J. Ewens NUS, Singapore 10 June, 2009 The initial aim of the (QUALITATIVE) TDT was to test for linkage between a marker locus
The Quantitative TDT (Quantitative Transmission Disequilibrium Test) Warren J. Ewens NUS, Singapore 10 June, 2009 The initial aim of the (QUALITATIVE) TDT was to test for linkage between a marker locus
25 : Graphical induced structured input/output models
 10-708: Probabilistic Graphical Models 10-708, Spring 2016 25 : Graphical induced structured input/output models Lecturer: Eric P. Xing Scribes: Raied Aljadaany, Shi Zong, Chenchen Zhu Disclaimer: A large
10-708: Probabilistic Graphical Models 10-708, Spring 2016 25 : Graphical induced structured input/output models Lecturer: Eric P. Xing Scribes: Raied Aljadaany, Shi Zong, Chenchen Zhu Disclaimer: A large
Meiosis and Fertilization Understanding How Genes Are Inherited 1
 Meiosis and Fertilization Understanding How Genes Are Inherited 1 Introduction In this activity, you will learn how you inherited two copies of each gene, one from your mother and one from your father.
Meiosis and Fertilization Understanding How Genes Are Inherited 1 Introduction In this activity, you will learn how you inherited two copies of each gene, one from your mother and one from your father.
Minimization of Boolean Expressions Using Matrix Algebra
 Minimization of Boolean Expressions Using Matrix Algebra Holger Schwender Collaborative Research Center SFB 475 University of Dortmund holger.schwender@udo.edu Abstract The more variables a logic expression
Minimization of Boolean Expressions Using Matrix Algebra Holger Schwender Collaborative Research Center SFB 475 University of Dortmund holger.schwender@udo.edu Abstract The more variables a logic expression
EM algorithm. Rather than jumping into the details of the particular EM algorithm, we ll look at a simpler example to get the idea of how it works
 EM algorithm The example in the book for doing the EM algorithm is rather difficult, and was not available in software at the time that the authors wrote the book, but they implemented a SAS macro to implement
EM algorithm The example in the book for doing the EM algorithm is rather difficult, and was not available in software at the time that the authors wrote the book, but they implemented a SAS macro to implement
Heterozygous BMN lines
 Optical density at 80 hours 0.8 0.6 0.4 0.2 0.8 0.6 0.4 0.2 0.8 0.6 0.4 0.2 0.8 0.6 0.4 0.2 a YPD b YPD + 1µM nystatin c YPD + 2µM nystatin d YPD + 4µM nystatin 1 3 5 6 9 13 16 20 21 22 23 25 28 29 30
Optical density at 80 hours 0.8 0.6 0.4 0.2 0.8 0.6 0.4 0.2 0.8 0.6 0.4 0.2 0.8 0.6 0.4 0.2 a YPD b YPD + 1µM nystatin c YPD + 2µM nystatin d YPD + 4µM nystatin 1 3 5 6 9 13 16 20 21 22 23 25 28 29 30
Evolutionary Models. Evolutionary Models
 Edit Operators In standard pairwise alignment, what are the allowed edit operators that transform one sequence into the other? Describe how each of these edit operations are represented on a sequence alignment
Edit Operators In standard pairwise alignment, what are the allowed edit operators that transform one sequence into the other? Describe how each of these edit operations are represented on a sequence alignment
Question: If mating occurs at random in the population, what will the frequencies of A 1 and A 2 be in the next generation?
 October 12, 2009 Bioe 109 Fall 2009 Lecture 8 Microevolution 1 - selection The Hardy-Weinberg-Castle Equilibrium - consider a single locus with two alleles A 1 and A 2. - three genotypes are thus possible:
October 12, 2009 Bioe 109 Fall 2009 Lecture 8 Microevolution 1 - selection The Hardy-Weinberg-Castle Equilibrium - consider a single locus with two alleles A 1 and A 2. - three genotypes are thus possible:
Drosophila melanogaster and D. simulans, two fruit fly species that are nearly
 Comparative Genomics: Human versus chimpanzee 1. Introduction The chimpanzee is the closest living relative to humans. The two species are nearly identical in DNA sequence (>98% identity), yet vastly different
Comparative Genomics: Human versus chimpanzee 1. Introduction The chimpanzee is the closest living relative to humans. The two species are nearly identical in DNA sequence (>98% identity), yet vastly different
Bioinformatics. Dept. of Computational Biology & Bioinformatics
 Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
Bioinformatics Dept. of Computational Biology & Bioinformatics 3 Bioinformatics - play with sequences & structures Dept. of Computational Biology & Bioinformatics 4 ORGANIZATION OF LIFE ROLE OF BIOINFORMATICS
ESTIMATION OF PARENT SPECIFIC DNA COPY NUMBER IN TUMORS USING HIGH-DENSITY GENOTYPING ARRAYS. Hao Chen Haipeng Xing Nancy Zhang
 ESTIMATION OF PARENT SPECIFIC DNA COPY NUMBER IN TUMORS USING HIGH-DENSITY GENOTYPING ARRAYS By Hao Chen Haipeng Xing Nancy Zhang Technical Report 251 March 2010 Division of Biostatistics STANFORD UNIVERSITY
ESTIMATION OF PARENT SPECIFIC DNA COPY NUMBER IN TUMORS USING HIGH-DENSITY GENOTYPING ARRAYS By Hao Chen Haipeng Xing Nancy Zhang Technical Report 251 March 2010 Division of Biostatistics STANFORD UNIVERSITY
7.014 Problem Set 6. Question 1. MIT Department of Biology Introductory Biology, Spring 2004
 MIT Department of Biology 7.014 Introductory Biology, Spring 2004 Name: 7.014 Problem Set 6 Please print out this problem set and record your answers on the printed copy. Problem sets will not be accepted
MIT Department of Biology 7.014 Introductory Biology, Spring 2004 Name: 7.014 Problem Set 6 Please print out this problem set and record your answers on the printed copy. Problem sets will not be accepted
Learning gene regulatory networks Statistical methods for haplotype inference Part I
 Learning gene regulatory networks Statistical methods for haplotype inference Part I Input: Measurement of mrn levels of all genes from microarray or rna sequencing Samples (e.g. 200 patients with lung
Learning gene regulatory networks Statistical methods for haplotype inference Part I Input: Measurement of mrn levels of all genes from microarray or rna sequencing Samples (e.g. 200 patients with lung