Ensemble refinement of protein crystal structures in PHENIX. Tom Burnley Piet Gros
|
|
|
- Olivia Perry
- 6 years ago
- Views:
Transcription
1 Ensemble refinement of protein crystal structures in PHENIX Tom Burnley Piet Gros
2 Incomplete modelling of disorder contributes to R factor gap Only ~5% of residues in the PDB are modelled with more than one conforma=on (x- ray structures) Mul=ple discrete models restricted due to increase in number of model parameters Vitkup et al., (2002) Lang et al., (2010)
3 Incomplete modelling of disorder contributes to R factor gap Only ~5% of residues in the PDB are modelled with more than one conforma=on (x- ray structures) Mul=ple discrete models restricted due to increase in number of model parameters Molecular dynamics simula=ons produce a Boltzmann- weighted popula=on of inter- related structures MD simula=ons can be restrained with x- ray data
 ) hkl 2 Simula,on temperature >2000K Trajectory resolves local minima Final model = end structure Brunger et al.")
4 Simulated Annealing/MD refinement Geometric restraints: E bond E angle E dih X- ray restraint: E = X ray whkl ( Fobs( hkl) k Fcalc( hkl) ) hkl 2 Simula,on temperature >2000K Trajectory resolves local minima Final model = end structure Brunger et al., 1987
 k F calc (hkl) ) 2 hkl Sampling F calc t = (1 e Δt/τ x )F t calc + e Δt/τ x F calc t Δt Gros et al.")
5 Time- averaged MD refinement Geometric restraints: E bond E angle E dih X- ray restraint: E X ray = w hkl ( F obs (hkl) k F calc (hkl) ) 2 hkl Sampling F calc t = (1 e Δt/τ x )F t calc + e Δt/τ x F calc t Δt Gros et al., 1990
F t calc + e Δt/τ x F calc t Δt")
6 Time- averaged MD refinement Geometric restraints: E bond E angle E dih X- ray restraint: E X ray = w hkl ( F obs (hkl) k F calc (hkl) ) 2 hkl Running average Simula,on temperature = 300K Final model = trajectory ensemble F calc t = (1 e Δt/τ x )F t calc + e Δt/τ x F calc t Δt
7 Ensemble refinement with TA restraints (1) (2) (1) Start: Tradi.onal structure (2) Fit TLS / remove alt. conf.
8 Ensemble refinement with TA restraints (1) (2) (3) (1) Start: Tradi.onal structure (2) Fit TLS / remove alt. conf. (3) TA restrained MD simula.on Collect structure / 0.04ps X- ray restraints accelerate sampling
9 Ensemble refinement with TA restraints (1) (2) (3) (1) Start: Tradi.onal structure (2) Fit TLS / remove alt. conf. (3) TA restrained MD simula.on (4) Final ensemble (4)


10 Molecular disorder / la@ce disorder Protein with local disorder Perfect crystal laoce
11 Molecular disorder / la@ce disorder Protein with local disorder Perfect crystal laoce 'Real' crystal laoce Averaged data

12 Molecular disorder / la@ce disorder Protein with local disorder Perfect crystal laoce 'Real' crystal laoce Averaged data Deconvolute: molecular disorder from laoce disorder
13 Rigid body disorder modelled with TLS LaOce distor.ons, domain mo.ons... Model rigid body mo=ons with TLS model
14 Rigid body disorder modelled with TLS Extract core rigid body mo=on by excluding atoms with large local fluctua=ons (defined as devia=ons from B TLS ).


15 Local disorder sampled within restrained MD LaOce distor.ons, domain mo.ons... Model rigid body mo=ons with TLS model 20 parameters per group 1 group / domain Fit TLS to star=ng structure B- factors Side- chains, loops... Local disorder sampled in MD MD simula=on restrained with X- ray data
16 Composite B- factor sum of TLS and atomic fluctuajons TLS Core TLS model RMSF Atomic fluctua=ons in ensemble TLS+RMSF TLS RMSF TLS+RMSF Start Total disorder in ensemble Start Input single structure ADPs
17 Dual explicit- bulk solvent model - Explicit solvent - Model with explicit atoms - Water picked every 250 steps standard rules: > 3 σ in difference map - < 3 Å distances B- factor from nearest TLS group

F t mask + e Δt/τ x F mask")
18 Dual explicit- bulk solvent model - Explicit solvent - Model with explicit atoms - Water picked every 250 steps standard rules: > 3 σ in difference map - < 3 Å distances B- factor from nearest TLS group - Bulk solvent - Model with density mask F mask t = (1 e Δt/τ x )F t mask + e Δt/τ x F mask t Δt
19 Development of ensemble refinement PHENIX Time- averaged x- ray restrained MD TLS fiong Explicit- and bulk- solvent model Maximum- likelihood target func.on


20 Development of ensemble refinement Tested with 20 datasets Resolu.on: 1-3 Å ASU size: residues CPU.me: hours models / ensemble
21 Ensemble refinement reduces Rfree Rfree: ensemble vs phenix.refine Rfree reduced in all cases - 4.9% (max) - 0.3% (min) - 1.8% (mean) Rf/Rw ra=o (mean): = 1.23 phenix.refine = 1.25 ensemble
 Rf/Rw ra=o (mean): = 1.23 phenix.refine = 1.25 ensemble Rfree: ensemble vs PDB_REDO - 5.1% (max) + 0.6% (min) - 2.")
22 Ensemble refinement reduces Rfree Rfree: ensemble vs phenix.refine Rfree reduced in all cases - 4.9% (max) - 0.3% (min) - 1.8% (mean) Rf/Rw ra=o (mean): = 1.23 phenix.refine = 1.25 ensemble Rfree: ensemble vs PDB_REDO - 5.1% (max) + 0.6% (min) - 2.1% (mean) Joosten et. al (2011)

23 B factor 5-20Ų 1uoy.pdb 188 ensemble 40ps acquisi.on.me 1 tls group

24 B factor 5-20Ų 1uoy.pdb 188 ensemble 40ps acquisi.on.me 1 tls group

25 1uoy.pdb phenix.refine 1 tls group mfo- DFc ±0.49 e/å³ (3.00 σ)

26 1uoy.pdb 188 ensemble 1 tls group mfo- DFc ±0.49 e/å³ (4.27 σ)


27 Improved real- space correla=on Burling et al. (1996): Excellent experimentally phased data for MBP: 1YTT (1.8- Å res.) Ensemble Phenix.refine B- factor
 Ensemble Diff.")
28 Disordered side chain in MBP (1YTT) Gln167 (A) Glu117 (A) Phenix.refine Diff. vector map (3σ) Ensemble Diff. vector map (3σ)

 Mul.- conformer (Rfree 20.")
29 Anisotropic side chain in MBP (1YTT) Burling et al. (1996) Phe121 (A) Experimental map (1.4σ) Mul.- conformer (Rfree 20.3%) Ensemble (Rfree 17.4%)
30 Diffuse solvent in MBP (1YTT) Burling et al. (1996) Published: Experimental map (1.4σ and 0.7σ)


31 Diffuse solvent in MBP (1YTT) Burling et al. (1996) Published: Experimental map (1.4σ and 0.7σ) Ensemble: Experimental map (1.4 σ) (0.7 σ)

")
32 Explicit waters in MBP (1YTT)
33 How similar are NCS copies? 1IEP (2.1- Å res.)
34 NCS copies show similar distribu=ons 1IEP (2.1- Å res.) - Global TLS model accounts for differences in packing of copies - Local fluctua=ons are similar between the two copies

35 1M52 (2.6- Å res.)


36 2XFA (2.1- Å res.)
 Fraser et al.")
37 Isomorphous crystals at 100 and 288 K 3K0N & 3K0M: Proline isomerase (Cyclophilin A) Fraser et al. (2009)
 288 K Phe113 Leu98 Ensemble refinement: 1.3% gain Rfree A:B = 2:1 Ser99 Phe113 Fraser et al. (2009), Eisenmesser et al.")
38 Mul=- conformers in ac=ve site 3K0M (1.3- Šres.) 100 K Leu98 Ser99 3K0N (1.4- Šres.) 288 K Phe113 Leu98 Ensemble refinement: 1.3% gain Rfree A:B = 2:1 Ser99 Phe113 Fraser et al. (2009), Eisenmesser et al. (2005) B- factor 5-25Ų 2.6% gain Rfree
39 Occupancies agree with NMR data NMR relaxa=on dispersion (283 K) L98, S99, F113 Minor popula=on ~10% S99 L98 3K0N & 3K0M: Proline isomerase Fraser et al. (2009), Eisenmesser et al. (2005) F113 Ensemble (288 K) B- factor 5-25Ų
40 Ima=nib- ABL Tyrosine Kinase (1IEP) Nagar et al. (2002)

41 Ima=nib- ABL Tyrosine Kinase (1IEP)
 Nagar et al (2002), Johnson")
42 Ima=nib- ABL Tyrosine Kinase (1IEP) Nagar et al (2002), Johnson (2009)
")



43 Inhibitor binding to HIV protease apo- protease (2PC0) complex with JE (1KZK) Compara=ve analysis of atomic flexibility: - Isomorphous crystals eliminate differences due to Xtal contacts - Non- isomorphous crystals allow evalua=on of Xtal contact effects

")


44 Conclusions Global disorder modeled by TLS and local disorder by MD Ensemble refinement improves Rfree and electron density maps Suitable for a broad resolu=on range (1Å 3Å) NCS copies show very similar fluctua=ons Clear representa=on of local disorder / uncertainty Distribu=on of atom posi=ons allows further structural analysis Resolve the finer details of protein structure(s)


45 Acknowledgements Piet Gros Gros Laboratory Pavel Afonine, Paul Adams PHENIX Developers Funding: Utrecht University / NWO
46 Number structures in ensemble vs resolujon

47 Number of structures in the ensemble Number of structures Averaging =me τ x Structures taken equidistant in =me to reproduce the Rwork within 0.1%
48 R- factor by resolu=on shell 1UOY


49 Par=al ligand/ion binding B- factor of Cd 2+ - ion more than twice its surrounding Simula=on run at Q=1 for Cd 2+ - ion (atoms coloured by kine=c energy) - High B- factor ligand/ion may indicate par=al occupancy


50 Par=al ligand/ion binding Cd 2+ : Q=1 Cd 2+ : Q=0.7 - Heat map (kine=c energy) is valida=on tool for Ensemble Refinement

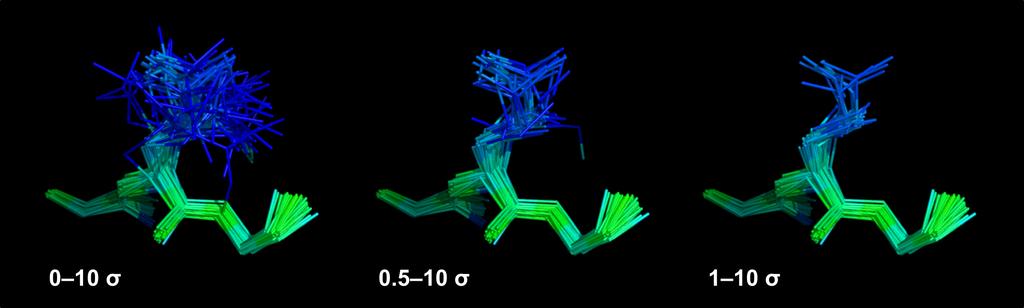
51 Ensemble Ensemble Single- structure

 2 +0.002 +0.30 +4.")
52 Geometric valida=on Bonds Angles Dih. angles Single structure Å Ens: (x ideal - x model ) Ens: (x ideal - x model ) (sta=s=cs for all 20 cases)
53 Ramachandran analysis 1UOY ( 1.5- Å res.) Outliers Allowed Favoured


54 Outliers occur more in flexible regions X- ray ensemble 1BV1 (2.0- Å res.) NMR ensemble
55 Ramachandran devia=ons by resolu=on Single structure Ensemble modal Ensemble mean Outliers Allowed - More outliers are observed at lower resolu=on - Geometric quality correlates with Rfree - Best run selected by Rfree
56 Ramachandran devia=ons by resolu=on Single structure Ensemble modal Ensemble mean Outliers Allowed - More outliers are observed at lower resolu=on - Geometric quality correlates with Rfree - Best run selected by Rfree
Summary of Experimental Protein Structure Determination. Key Elements
 Programme 8.00-8.20 Summary of last week s lecture and quiz 8.20-9.00 Structure validation 9.00-9.15 Break 9.15-11.00 Exercise: Structure validation tutorial 11.00-11.10 Break 11.10-11.40 Summary & discussion
Programme 8.00-8.20 Summary of last week s lecture and quiz 8.20-9.00 Structure validation 9.00-9.15 Break 9.15-11.00 Exercise: Structure validation tutorial 11.00-11.10 Break 11.10-11.40 Summary & discussion
X-ray Crystallography I. James Fraser Macromolecluar Interactions BP204
 X-ray Crystallography I James Fraser Macromolecluar Interactions BP204 Key take-aways 1. X-ray crystallography results from an ensemble of Billions and Billions of molecules in the crystal 2. Models in
X-ray Crystallography I James Fraser Macromolecluar Interactions BP204 Key take-aways 1. X-ray crystallography results from an ensemble of Billions and Billions of molecules in the crystal 2. Models in
The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C2-symmetric hexamer
 Volume 71 (2015) Supporting information for article: The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C2-symmetric hexamer Marina Vostrukhina, Alexander Popov, Elena Brunstein, Martin
Volume 71 (2015) Supporting information for article: The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C2-symmetric hexamer Marina Vostrukhina, Alexander Popov, Elena Brunstein, Martin
Q1 current best prac2ce
 Group- A Q1 current best prac2ce Star2ng from some common molecular representa2on with bond orders, configura2on on chiral centers (e.g. ChemDraw, SMILES) NEW! PDB should become resource for refinement
Group- A Q1 current best prac2ce Star2ng from some common molecular representa2on with bond orders, configura2on on chiral centers (e.g. ChemDraw, SMILES) NEW! PDB should become resource for refinement
RNA protects a nucleoprotein complex against radiation damage
 Supporting information Volume 72 (2016) Supporting information for article: RNA protects a nucleoprotein complex against radiation damage Charles S. Bury, John E. McGeehan, Alfred A. Antson, Ian Carmichael,
Supporting information Volume 72 (2016) Supporting information for article: RNA protects a nucleoprotein complex against radiation damage Charles S. Bury, John E. McGeehan, Alfred A. Antson, Ian Carmichael,
Table 1. Crystallographic data collection, phasing and refinement statistics. Native Hg soaked Mn soaked 1 Mn soaked 2
 Table 1. Crystallographic data collection, phasing and refinement statistics Native Hg soaked Mn soaked 1 Mn soaked 2 Data collection Space group P2 1 2 1 2 1 P2 1 2 1 2 1 P2 1 2 1 2 1 P2 1 2 1 2 1 Cell
Table 1. Crystallographic data collection, phasing and refinement statistics Native Hg soaked Mn soaked 1 Mn soaked 2 Data collection Space group P2 1 2 1 2 1 P2 1 2 1 2 1 P2 1 2 1 2 1 P2 1 2 1 2 1 Cell
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 10:24 pm GMT PDB ID : 1A30 Title : HIV-1 PROTEASE COMPLEXED WITH A TRIPEPTIDE INHIBITOR Authors : Louis, J.M.; Dyda, F.; Nashed, N.T.; Kimmel,
Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 10:24 pm GMT PDB ID : 1A30 Title : HIV-1 PROTEASE COMPLEXED WITH A TRIPEPTIDE INHIBITOR Authors : Louis, J.M.; Dyda, F.; Nashed, N.T.; Kimmel,
Electronic Supplementary Information (ESI) for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat
 for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat") Electronic Supplementary Information (ESI) for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat Jun Yi* and George B. Richter- Addo* Department of Chemistry
Electronic Supplementary Information (ESI) for Chem. Commun. Unveiling the three- dimensional structure of the green pigment of nitrite- cured meat Jun Yi* and George B. Richter- Addo* Department of Chemistry
Automated identification of functional dynamic contact networks from X-ray crystallography
 1 Automated identification of functional dynamic contact networks from X-ray crystallography Henry van den Bedem, Gira Bhabha, Kun Yang, Peter E. Wright and James S. Fraser Supplementary Figure 1 Supplementary
1 Automated identification of functional dynamic contact networks from X-ray crystallography Henry van den Bedem, Gira Bhabha, Kun Yang, Peter E. Wright and James S. Fraser Supplementary Figure 1 Supplementary
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 08:34 pm GMT PDB ID : 1RUT Title : Complex of LMO4 LIM domains 1 and 2 with the ldb1 LID domain Authors : Deane, J.E.; Ryan, D.P.; Maher, M.J.;
Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 08:34 pm GMT PDB ID : 1RUT Title : Complex of LMO4 LIM domains 1 and 2 with the ldb1 LID domain Authors : Deane, J.E.; Ryan, D.P.; Maher, M.J.;
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 06:13 pm GMT PDB ID : 5G5C Title : Structure of the Pyrococcus furiosus Esterase Pf2001 with space group C2221 Authors : Varejao, N.; Reverter,
Full wwpdb X-ray Structure Validation Report i Mar 8, 2018 06:13 pm GMT PDB ID : 5G5C Title : Structure of the Pyrococcus furiosus Esterase Pf2001 with space group C2221 Authors : Varejao, N.; Reverter,
Anisotropy in macromolecular crystal structures. Andrea Thorn July 19 th, 2012
 Anisotropy in macromolecular crystal structures Andrea Thorn July 19 th, 2012 Motivation Courtesy of M. Sawaya Motivation Crystal structures are inherently anisotropic. X-ray diffraction reflects this
Anisotropy in macromolecular crystal structures Andrea Thorn July 19 th, 2012 Motivation Courtesy of M. Sawaya Motivation Crystal structures are inherently anisotropic. X-ray diffraction reflects this
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 14, 2018 02:00 pm GMT PDB ID : 3RRQ Title : Crystal structure of the extracellular domain of human PD-1 Authors : Lazar-Molnar, E.; Ramagopal, U.A.; Nathenson,
Full wwpdb X-ray Structure Validation Report i Mar 14, 2018 02:00 pm GMT PDB ID : 3RRQ Title : Crystal structure of the extracellular domain of human PD-1 Authors : Lazar-Molnar, E.; Ramagopal, U.A.; Nathenson,
Computational engineering of cellulase Cel9A-68 functional motions through mutations in its linker region. WT 1TF4 (crystal) -90 ERRAT PROVE VERIFY3D
 -90 ERRAT PROVE VERIFY3D") Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 218 Supplementary Material: Computational engineering of cellulase Cel9-68 functional
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 218 Supplementary Material: Computational engineering of cellulase Cel9-68 functional
X-ray Crystallography
 2009/11/25 [ 1 ] X-ray Crystallography Andrew Torda, wintersemester 2009 / 2010 X-ray numerically most important more than 4/5 structures Goal a set of x, y, z coordinates different properties to NMR History
2009/11/25 [ 1 ] X-ray Crystallography Andrew Torda, wintersemester 2009 / 2010 X-ray numerically most important more than 4/5 structures Goal a set of x, y, z coordinates different properties to NMR History
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Jan 17, 2019 09:42 AM EST PDB ID : 6D3Z Title : Protease SFTI complex Authors : Law, R.H.P.; Wu, G. Deposited on : 2018-04-17 Resolution : 2.00 Å(reported)
Full wwpdb X-ray Structure Validation Report i Jan 17, 2019 09:42 AM EST PDB ID : 6D3Z Title : Protease SFTI complex Authors : Law, R.H.P.; Wu, G. Deposited on : 2018-04-17 Resolution : 2.00 Å(reported)
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Jan 14, 2019 11:10 AM EST PDB ID : 6GYW Title : Crystal structure of DacA from Staphylococcus aureus Authors : Tosi, T.; Freemont, P.S.; Grundling, A. Deposited
Full wwpdb X-ray Structure Validation Report i Jan 14, 2019 11:10 AM EST PDB ID : 6GYW Title : Crystal structure of DacA from Staphylococcus aureus Authors : Tosi, T.; Freemont, P.S.; Grundling, A. Deposited
Molecular Biology Course 2006 Protein Crystallography Part II
 Molecular Biology Course 2006 Protein Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry December 2006 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview
Molecular Biology Course 2006 Protein Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry December 2006 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview
Direct Method. Very few protein diffraction data meet the 2nd condition
 Direct Method Two conditions: -atoms in the structure are equal-weighted -resolution of data are higher than the distance between the atoms in the structure Very few protein diffraction data meet the 2nd
Direct Method Two conditions: -atoms in the structure are equal-weighted -resolution of data are higher than the distance between the atoms in the structure Very few protein diffraction data meet the 2nd
Macromolecular Crystallography Part II
 Molecular Biology Course 2009 Macromolecular Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry November 2009 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de From
Molecular Biology Course 2009 Macromolecular Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry November 2009 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de From
Model and data. An X-ray structure solution requires a model.
 Model and data An X-ray structure solution requires a model. This model has to be consistent with: The findings of Chemistry Reflection positions and intensities Structure refinement = Model fitting by
Model and data An X-ray structure solution requires a model. This model has to be consistent with: The findings of Chemistry Reflection positions and intensities Structure refinement = Model fitting by
Protein Crystallography Part II
 Molecular Biology Course 2007 Protein Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry November 2007 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview
Molecular Biology Course 2007 Protein Crystallography Part II Tim Grüne University of Göttingen Dept. of Structural Chemistry November 2007 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Overview
Crystal lattice Real Space. Reflections Reciprocal Space. I. Solving Phases II. Model Building for CHEM 645. Purified Protein. Build model.
 I. Solving Phases II. Model Building for CHEM 645 Purified Protein Solve Phase Build model and refine Crystal lattice Real Space Reflections Reciprocal Space ρ (x, y, z) pronounced rho F hkl 2 I F (h,
I. Solving Phases II. Model Building for CHEM 645 Purified Protein Solve Phase Build model and refine Crystal lattice Real Space Reflections Reciprocal Space ρ (x, y, z) pronounced rho F hkl 2 I F (h,
Plasmid Relevant features Source. W18N_D20N and TrXE-W18N_D20N-anti
 Table S1. E. coli plasmids Plasmid Relevant features Source pdg680 T. reesei XynII AA 2-190 with C-terminal His 6 tag optimized for E. coli expression in pjexpress401 Wan et al. (in press) psbn44d psbn44h
Table S1. E. coli plasmids Plasmid Relevant features Source pdg680 T. reesei XynII AA 2-190 with C-terminal His 6 tag optimized for E. coli expression in pjexpress401 Wan et al. (in press) psbn44d psbn44h
Molecular Replacement. Airlie McCoy
 Molecular Replacement Airlie McCoy Molecular Replacement Find orienta5on and posi5on where model overlies the target structure Borrow the phases Then it becomes a refinement problem the phases change known
Molecular Replacement Airlie McCoy Molecular Replacement Find orienta5on and posi5on where model overlies the target structure Borrow the phases Then it becomes a refinement problem the phases change known
Modelling against small angle scattering data. Al Kikhney EMBL Hamburg, Germany
 Modelling against small angle scattering data Al Kikhney EMBL Hamburg, Germany Validation of atomic models CRYSOL Rigid body modelling SASREF BUNCH CORAL Oligomeric mixtures OLIGOMER Flexible systems EOM
Modelling against small angle scattering data Al Kikhney EMBL Hamburg, Germany Validation of atomic models CRYSOL Rigid body modelling SASREF BUNCH CORAL Oligomeric mixtures OLIGOMER Flexible systems EOM
Data quality indicators. Kay Diederichs
 Data quality indicators Kay Diederichs Crystallography has been highly successful Now 105839 Could it be any better? 2 Confusion what do these mean? CC1/2 Rmerge Rsym Mn(I/sd) I/σ Rmeas CCanom Rpim Rcum
Data quality indicators Kay Diederichs Crystallography has been highly successful Now 105839 Could it be any better? 2 Confusion what do these mean? CC1/2 Rmerge Rsym Mn(I/sd) I/σ Rmeas CCanom Rpim Rcum
PHENIX Wizards and Tools
 PHENIX Wizards and Tools Tom Terwilliger Los Alamos National Laboratory terwilliger@lanl.gov l The PHENIX project Computational Crystallography Initiative (LBNL) Paul Adams, Ralf Grosse-Kunstleve, Peter
PHENIX Wizards and Tools Tom Terwilliger Los Alamos National Laboratory terwilliger@lanl.gov l The PHENIX project Computational Crystallography Initiative (LBNL) Paul Adams, Ralf Grosse-Kunstleve, Peter
Likelihood and SAD phasing in Phaser. R J Read, Department of Haematology Cambridge Institute for Medical Research
 Likelihood and SAD phasing in Phaser R J Read, Department of Haematology Cambridge Institute for Medical Research Concept of likelihood Likelihood with dice 4 6 8 10 Roll a seven. Which die?? p(4)=p(6)=0
Likelihood and SAD phasing in Phaser R J Read, Department of Haematology Cambridge Institute for Medical Research Concept of likelihood Likelihood with dice 4 6 8 10 Roll a seven. Which die?? p(4)=p(6)=0
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Jan 28, 2019 11:10 AM EST PDB ID : 6A5H Title : The structure of [4+2] and [6+4] cyclase in the biosynthetic pathway of unidentified natural product Authors
Full wwpdb X-ray Structure Validation Report i Jan 28, 2019 11:10 AM EST PDB ID : 6A5H Title : The structure of [4+2] and [6+4] cyclase in the biosynthetic pathway of unidentified natural product Authors
SUPPLEMENTARY INFORMATION
 doi:10.1038/nature11054 Supplementary Fig. 1 Sequence alignment of Na v Rh with NaChBac, Na v Ab, and eukaryotic Na v and Ca v homologs. Secondary structural elements of Na v Rh are indicated above the
doi:10.1038/nature11054 Supplementary Fig. 1 Sequence alignment of Na v Rh with NaChBac, Na v Ab, and eukaryotic Na v and Ca v homologs. Secondary structural elements of Na v Rh are indicated above the
Introduction to Comparative Protein Modeling. Chapter 4 Part I
 Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Introduction to Comparative Protein Modeling Chapter 4 Part I 1 Information on Proteins Each modeling study depends on the quality of the known experimental data. Basis of the model Search in the literature
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 10, 2018 01:44 am GMT PDB ID : 1MWP Title : N-TERMINAL DOMAIN OF THE AMYLOID PRECURSOR PROTEIN Authors : Rossjohn, J.; Cappai, R.; Feil, S.C.; Henry,
Full wwpdb X-ray Structure Validation Report i Mar 10, 2018 01:44 am GMT PDB ID : 1MWP Title : N-TERMINAL DOMAIN OF THE AMYLOID PRECURSOR PROTEIN Authors : Rossjohn, J.; Cappai, R.; Feil, S.C.; Henry,
Structure Investigation of Fam20C, a Golgi Casein Kinase
 Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Structure Investigation of Fam20C, a Golgi Casein Kinase Sharon Grubner National Taiwan University, Dr. Jung-Hsin Lin University of California San Diego, Dr. Rommie Amaro Abstract This research project
Supplementary Figures
 1 Supplementary Figures Supplementary Figure 1 Type I FGFR1 inhibitors (a) Chemical structures of a pyrazolylaminopyrimidine inhibitor (henceforth referred to as PAPI; PDB-code of the FGFR1-PAPI complex:
1 Supplementary Figures Supplementary Figure 1 Type I FGFR1 inhibitors (a) Chemical structures of a pyrazolylaminopyrimidine inhibitor (henceforth referred to as PAPI; PDB-code of the FGFR1-PAPI complex:
HOMOLOGY MODELING. The sequence alignment and template structure are then used to produce a structural model of the target.
 HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
HOMOLOGY MODELING Homology modeling, also known as comparative modeling of protein refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental
Homology Modeling (Comparative Structure Modeling) GBCB 5874: Problem Solving in GBCB
 GBCB 5874: Problem Solving in GBCB") Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Homology Modeling (Comparative Structure Modeling) Aims of Structural Genomics High-throughput 3D structure determination and analysis To determine or predict the 3D structures of all the proteins encoded
Macromolecular X-ray Crystallography
 Protein Structural Models for CHEM 641 Fall 07 Brian Bahnson Department of Chemistry & Biochemistry University of Delaware Macromolecular X-ray Crystallography Purified Protein X-ray Diffraction Data collection
Protein Structural Models for CHEM 641 Fall 07 Brian Bahnson Department of Chemistry & Biochemistry University of Delaware Macromolecular X-ray Crystallography Purified Protein X-ray Diffraction Data collection
TLS and all that. Ethan A Merritt. CCP4 Summer School 2011 (Argonne, IL) Abstract
 Abstract") TLS and all that Ethan A Merritt CCP4 Summer School 2011 (Argonne, IL) Abstract We can never know the position of every atom in a crystal structure perfectly. Each atom has an associated positional uncertainty.
TLS and all that Ethan A Merritt CCP4 Summer School 2011 (Argonne, IL) Abstract We can never know the position of every atom in a crystal structure perfectly. Each atom has an associated positional uncertainty.
Supporting Information
 Supporting Information Structural Analysis of the Binding of Type I, I 1/2, and II Inhibitors to Eph Tyrosine Kinases Jing Dong, *1 Hongtao Zhao, 1 Ting Zhou, 1 Dimitrios Spiliotopoulos, 1 Chitra Rajendran,
Supporting Information Structural Analysis of the Binding of Type I, I 1/2, and II Inhibitors to Eph Tyrosine Kinases Jing Dong, *1 Hongtao Zhao, 1 Ting Zhou, 1 Dimitrios Spiliotopoulos, 1 Chitra Rajendran,
8. Strategies for Macromolecular Refinement
 8. Strategies for Macromolecular Refinement SHELXL is designed to be easy to use and general for all space groups and uses a conventional structure-factor calculation rather than a FFT summation; the latter
8. Strategies for Macromolecular Refinement SHELXL is designed to be easy to use and general for all space groups and uses a conventional structure-factor calculation rather than a FFT summation; the latter
Linking data and model quality in macromolecular crystallography. Kay Diederichs
 Linking data and model quality in macromolecular crystallography Kay Diederichs Crystallography is highly successful Can we do better? Error in experimental data Error = random + systematic Multiplicity
Linking data and model quality in macromolecular crystallography Kay Diederichs Crystallography is highly successful Can we do better? Error in experimental data Error = random + systematic Multiplicity
SUPPLEMENTARY INFORMATION. doi: /nature07461
 Figure S1 Electrophysiology. a ph-activation of. Two-electrode voltage clamp recordings of Xenopus oocytes expressing in comparison to waterinjected oocytes. Currents were recorded at 40 mv. The ph of
Figure S1 Electrophysiology. a ph-activation of. Two-electrode voltage clamp recordings of Xenopus oocytes expressing in comparison to waterinjected oocytes. Currents were recorded at 40 mv. The ph of
Understanding protein motions by computational modeling and statistical approaches
 Retrospective Theses and Dissertations Iowa State University Capstones, Theses and Dissertations 28 Understanding protein motions by computational modeling and statistical approaches Lei Yang Iowa State
Retrospective Theses and Dissertations Iowa State University Capstones, Theses and Dissertations 28 Understanding protein motions by computational modeling and statistical approaches Lei Yang Iowa State
7.91 Amy Keating. Solving structures using X-ray crystallography & NMR spectroscopy
 7.91 Amy Keating Solving structures using X-ray crystallography & NMR spectroscopy How are X-ray crystal structures determined? 1. Grow crystals - structure determination by X-ray crystallography relies
7.91 Amy Keating Solving structures using X-ray crystallography & NMR spectroscopy How are X-ray crystal structures determined? 1. Grow crystals - structure determination by X-ray crystallography relies
Overview & Applications. T. Lezon Hands-on Workshop in Computational Biophysics Pittsburgh Supercomputing Center 04 June, 2015
 Overview & Applications T. Lezon Hands-on Workshop in Computational Biophysics Pittsburgh Supercomputing Center 4 June, 215 Simulations still take time Bakan et al. Bioinformatics 211. Coarse-grained Elastic
Overview & Applications T. Lezon Hands-on Workshop in Computational Biophysics Pittsburgh Supercomputing Center 4 June, 215 Simulations still take time Bakan et al. Bioinformatics 211. Coarse-grained Elastic
Molecular Dynamics. Molecules in motion
 Molecular Dynamics Molecules in motion 1 Molecules in mo1on Molecules are not sta1c, but move all the 1me Source: h9p://en.wikipedia.org/wiki/kine1c_theory 2 Gasses, liquids and solids Gasses, liquids
Molecular Dynamics Molecules in motion 1 Molecules in mo1on Molecules are not sta1c, but move all the 1me Source: h9p://en.wikipedia.org/wiki/kine1c_theory 2 Gasses, liquids and solids Gasses, liquids
Macromolecular Crystallography Part II
 Molecular Biology Course 2010 Macromolecular Crystallography Part II University of Göttingen Dept. of Structural Chemistry November 2010 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Crystallography
Molecular Biology Course 2010 Macromolecular Crystallography Part II University of Göttingen Dept. of Structural Chemistry November 2010 http://shelx.uni-ac.gwdg.de tg@shelx.uni-ac.gwdg.de Crystallography
Ensemble of Climate Models
 Ensemble of Climate Models Claudia Tebaldi Climate Central and Department of Sta7s7cs, UBC Reto Knu>, Reinhard Furrer, Richard Smith, Bruno Sanso Outline Mul7 model ensembles (MMEs) a descrip7on at face
Ensemble of Climate Models Claudia Tebaldi Climate Central and Department of Sta7s7cs, UBC Reto Knu>, Reinhard Furrer, Richard Smith, Bruno Sanso Outline Mul7 model ensembles (MMEs) a descrip7on at face
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Feb 17, 2018 01:16 am GMT PDB ID : 1IFT Title : RICIN A-CHAIN (RECOMBINANT) Authors : Weston, S.A.; Tucker, A.D.; Thatcher, D.R.; Derbyshire, D.J.; Pauptit,
Full wwpdb X-ray Structure Validation Report i Feb 17, 2018 01:16 am GMT PDB ID : 1IFT Title : RICIN A-CHAIN (RECOMBINANT) Authors : Weston, S.A.; Tucker, A.D.; Thatcher, D.R.; Derbyshire, D.J.; Pauptit,
Pipelining Ligands in PHENIX: elbow and REEL
 Pipelining Ligands in PHENIX: elbow and REEL Nigel W. Moriarty Lawrence Berkeley National Laboratory Physical Biosciences Division Ligands in Crystallography Drug design Biological function studies Generate
Pipelining Ligands in PHENIX: elbow and REEL Nigel W. Moriarty Lawrence Berkeley National Laboratory Physical Biosciences Division Ligands in Crystallography Drug design Biological function studies Generate
Proteins. Central Dogma : DNA RNA protein Amino acid polymers - defined composition & order. Perform nearly all cellular functions Drug Targets
 Proteins Central Dogma : DNA RNA protein Amino acid polymers - defined composition & order Perform nearly all cellular functions Drug Targets Fold into discrete shapes. Proteins - cont. Specific shapes
Proteins Central Dogma : DNA RNA protein Amino acid polymers - defined composition & order Perform nearly all cellular functions Drug Targets Fold into discrete shapes. Proteins - cont. Specific shapes
Details of Protein Structure
 Details of Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Anne Mølgaard, Kemisk Institut, Københavns Universitet Learning Objectives
Details of Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Anne Mølgaard, Kemisk Institut, Københavns Universitet Learning Objectives
Protein crystallography. Garry Taylor
 Protein crystallography Garry Taylor X-ray Crystallography - the Basics Grow crystals Collect X-ray data Determine phases Calculate ρ-map Interpret map Refine coordinates Do the biology. Nitrogen at -180
Protein crystallography Garry Taylor X-ray Crystallography - the Basics Grow crystals Collect X-ray data Determine phases Calculate ρ-map Interpret map Refine coordinates Do the biology. Nitrogen at -180
Phase problem: Determining an initial phase angle α hkl for each recorded reflection. 1 ρ(x,y,z) = F hkl cos 2π (hx+ky+ lz - α hkl ) V h k l
 = F hkl cos 2π (hx+ky+ lz - α hkl ) V h k l") Phase problem: Determining an initial phase angle α hkl for each recorded reflection 1 ρ(x,y,z) = F hkl cos 2π (hx+ky+ lz - α hkl ) V h k l Methods: Heavy atom methods (isomorphous replacement Hg, Pt)
Phase problem: Determining an initial phase angle α hkl for each recorded reflection 1 ρ(x,y,z) = F hkl cos 2π (hx+ky+ lz - α hkl ) V h k l Methods: Heavy atom methods (isomorphous replacement Hg, Pt)
Full wwpdb X-ray Structure Validation Report i
 Full wwpdb X-ray Structure Validation Report i Mar 13, 2018 04:03 pm GMT PDB ID : 5NMJ Title : Chicken GRIFIN (crystallisation ph: 6.5) Authors : Ruiz, F.M.; Romero, A. Deposited on : 2017-04-06 Resolution
Full wwpdb X-ray Structure Validation Report i Mar 13, 2018 04:03 pm GMT PDB ID : 5NMJ Title : Chicken GRIFIN (crystallisation ph: 6.5) Authors : Ruiz, F.M.; Romero, A. Deposited on : 2017-04-06 Resolution
wwpdb X-ray Structure Validation Summary Report
 wwpdb X-ray Structure Validation Summary Report io Jan 31, 2016 06:45 PM GMT PDB ID : 1CBS Title : CRYSTAL STRUCTURE OF CELLULAR RETINOIC-ACID-BINDING PROTEINS I AND II IN COMPLEX WITH ALL-TRANS-RETINOIC
wwpdb X-ray Structure Validation Summary Report io Jan 31, 2016 06:45 PM GMT PDB ID : 1CBS Title : CRYSTAL STRUCTURE OF CELLULAR RETINOIC-ACID-BINDING PROTEINS I AND II IN COMPLEX WITH ALL-TRANS-RETINOIC
BIOINF 4371 Drug Design 1 Oliver Kohlbacher & Jens Krüger
 BIOINF 4371 Drug Design 1 Oliver Kohlbacher & Jens Krüger Winter 2013/2014 11. Docking Part IV: Receptor Flexibility Overview Receptor flexibility Types of flexibility Implica5ons for docking Examples
BIOINF 4371 Drug Design 1 Oliver Kohlbacher & Jens Krüger Winter 2013/2014 11. Docking Part IV: Receptor Flexibility Overview Receptor flexibility Types of flexibility Implica5ons for docking Examples
NMR, X-ray Diffraction, Protein Structure, and RasMol
 NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
NMR, X-ray Diffraction, Protein Structure, and RasMol Introduction So far we have been mostly concerned with the proteins themselves. The techniques (NMR or X-ray diffraction) used to determine a structure
*Corresponding Author *K. F.: *T. H.:
 Theoretical Analysis of Activity Cliffs among Benzofuranone Class Pim1 Inhibitors Using the Fragment Molecular Orbital Method with Molecular Mechanics Poisson-Boltzmann Surface Area (FMO+MM-PBSA) Approach
Theoretical Analysis of Activity Cliffs among Benzofuranone Class Pim1 Inhibitors Using the Fragment Molecular Orbital Method with Molecular Mechanics Poisson-Boltzmann Surface Area (FMO+MM-PBSA) Approach
Refinement of Disorder with SHELXL
 Refinement of Disorder with SHELXL Peter Müller MIT pmueller@mit.edu Single Crystal Structure Determination A crystal is a potentially endless, three-dimensional, periodic discontinuum built up by atoms,
Refinement of Disorder with SHELXL Peter Müller MIT pmueller@mit.edu Single Crystal Structure Determination A crystal is a potentially endless, three-dimensional, periodic discontinuum built up by atoms,
Molecular replacement. New structures from old
 Molecular replacement New structures from old The Phase Problem phase amplitude Phasing by molecular replacement Phases can be calculated from atomic model Rotate and translate related structure Models
Molecular replacement New structures from old The Phase Problem phase amplitude Phasing by molecular replacement Phases can be calculated from atomic model Rotate and translate related structure Models
Introduction to" Protein Structure
 Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
Introduction to" Protein Structure Function, evolution & experimental methods Thomas Blicher, Center for Biological Sequence Analysis Learning Objectives Outline the basic levels of protein structure.
research papers Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias 1.
 Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias Thomas C. Terwilliger,
Acta Crystallographica Section D Biological Crystallography ISSN 0907-4449 Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias Thomas C. Terwilliger,
Native State of Complement Protein C3d Analyzed via Hydrogen Exchange and Conformational Sampling
 Devaurs et al. Native State of Complement Protein C3d Analyzed via Hydrogen Exchange and Conformational Sampling Didier Devaurs 1, Malvina Papanastasiou 2, Dinler A Antunes 1, Jayvee R Abella 1, Mark Moll
Devaurs et al. Native State of Complement Protein C3d Analyzed via Hydrogen Exchange and Conformational Sampling Didier Devaurs 1, Malvina Papanastasiou 2, Dinler A Antunes 1, Jayvee R Abella 1, Mark Moll
GC376 (compound 28). Compound 23 (GC373) (0.50 g, 1.24 mmol), sodium bisulfite (0.119 g,
. Compound 23 (GC373) (0.50 g, 1.24 mmol), sodium bisulfite (0.119 g,") Supplemental Material Synthesis of GC376 GC376 (compound 28). Compound 23 (GC373) (0.50 g, 1.24 mmol), sodium bisulfite (0.119 g, 1.12 mmol), ethyl acetate (2 ml), ethanol (1 ml) and water (0.40 ml) were
Supplemental Material Synthesis of GC376 GC376 (compound 28). Compound 23 (GC373) (0.50 g, 1.24 mmol), sodium bisulfite (0.119 g, 1.12 mmol), ethyl acetate (2 ml), ethanol (1 ml) and water (0.40 ml) were
Structural and mechanistic insight into the substrate. binding from the conformational dynamics in apo. and substrate-bound DapE enzyme
 Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 215 Structural and mechanistic insight into the substrate binding from the conformational
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is the Owner Societies 215 Structural and mechanistic insight into the substrate binding from the conformational
Protein Structure Determination Using NMR Restraints BCMB/CHEM 8190
 Protein Structure Determination Using NMR Restraints BCMB/CHEM 8190 Programs for NMR Based Structure Determination CNS - Brünger, A. T.; Adams, P. D.; Clore, G. M.; DeLano, W. L.; Gros, P.; Grosse-Kunstleve,
Protein Structure Determination Using NMR Restraints BCMB/CHEM 8190 Programs for NMR Based Structure Determination CNS - Brünger, A. T.; Adams, P. D.; Clore, G. M.; DeLano, W. L.; Gros, P.; Grosse-Kunstleve,
Structure factors again
 Structure factors again Remember 1D, structure factor for order h F h = F h exp[iα h ] = I 01 ρ(x)exp[2πihx]dx Where x is fractional position along unit cell distance (repeating distance, origin arbitrary)
Structure factors again Remember 1D, structure factor for order h F h = F h exp[iα h ] = I 01 ρ(x)exp[2πihx]dx Where x is fractional position along unit cell distance (repeating distance, origin arbitrary)
SUPPLEMENTARY INFORMATION
 Supplementary Table S1 Kinetic Analyses of the AMSH-LP mutants AMSH-LP K M (μm) k cat x 10-3 (s -1 ) WT 71.8 ± 6.3 860 ± 65.4 T353A 76.8 ± 11.7 46.3 ± 3.7 F355A 58.9 ± 10.4 5.33 ± 0.30 proximal S358A 75.1
Supplementary Table S1 Kinetic Analyses of the AMSH-LP mutants AMSH-LP K M (μm) k cat x 10-3 (s -1 ) WT 71.8 ± 6.3 860 ± 65.4 T353A 76.8 ± 11.7 46.3 ± 3.7 F355A 58.9 ± 10.4 5.33 ± 0.30 proximal S358A 75.1
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows
 MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
MD Simulation in Pose Refinement and Scoring Using AMBER Workflows Yuan Hu (On behalf of Merck D3R Team) D3R Grand Challenge 2 Webinar Department of Chemistry, Modeling & Informatics Merck Research Laboratories,
APPENDIX E. Crystallographic Data for TBA Eu(DO2A)(DPA) Temperature Dependence
(DPA) Temperature Dependence") APPENDIX E Crystallographic Data for TBA Eu(DO2A)(DPA) Temperature Dependence Temperature Designation CCDC Page 100 K MLC18 761599 E2 200 K MLC17 762705 E17 300 K MLC19 763335 E31 E2 CALIFORNIA INSTITUTE
APPENDIX E Crystallographic Data for TBA Eu(DO2A)(DPA) Temperature Dependence Temperature Designation CCDC Page 100 K MLC18 761599 E2 200 K MLC17 762705 E17 300 K MLC19 763335 E31 E2 CALIFORNIA INSTITUTE
SUPPLEMENTARY INFORMATION
 Supplementary Table 1: Amplitudes of three current levels. Level 0 (pa) Level 1 (pa) Level 2 (pa) TrkA- TrkH WT 200 K 0.01 ± 0.01 9.5 ± 0.01 18.7 ± 0.03 200 Na * 0.001 ± 0.01 3.9 ± 0.01 12.5 ± 0.03 200
Supplementary Table 1: Amplitudes of three current levels. Level 0 (pa) Level 1 (pa) Level 2 (pa) TrkA- TrkH WT 200 K 0.01 ± 0.01 9.5 ± 0.01 18.7 ± 0.03 200 Na * 0.001 ± 0.01 3.9 ± 0.01 12.5 ± 0.03 200
SOCS3 binds specific receptor JAK complexes to control cytokine signaling by direct kinase inhibition SUPPLEMENTARY INFORMATION
 SOCS3 binds specific receptor JAK complexes to control cytokine signaling by direct kinase inhibition Nadia J. Kershaw 1,2, James M. Murphy 1,2, Nicholas P.D. Liau 1,2, Leila N. Varghese 1,2, Artem Laktyushin
SOCS3 binds specific receptor JAK complexes to control cytokine signaling by direct kinase inhibition Nadia J. Kershaw 1,2, James M. Murphy 1,2, Nicholas P.D. Liau 1,2, Leila N. Varghese 1,2, Artem Laktyushin
Acta Cryst. (2017). D73, doi: /s
. D73, doi: /s") Acta Cryst. (2017). D73, doi:10.1107/s2059798317010932 Supporting information Volume 73 (2017) Supporting information for article: Designing better diffracting crystals of biotin carboxyl carrier protein
Acta Cryst. (2017). D73, doi:10.1107/s2059798317010932 Supporting information Volume 73 (2017) Supporting information for article: Designing better diffracting crystals of biotin carboxyl carrier protein
SUPPLEMENTARY INFORMATION
 doi:1.138/nature1737 Supplementary Table 1 variant Description FSEC - 2B12 a FSEC - 6A1 a K d (leucine) c Leucine uptake e K (wild-type like) K (Y18F) K (TS) K (TSY) K288A mutant, lipid facing side chain
doi:1.138/nature1737 Supplementary Table 1 variant Description FSEC - 2B12 a FSEC - 6A1 a K d (leucine) c Leucine uptake e K (wild-type like) K (Y18F) K (TS) K (TSY) K288A mutant, lipid facing side chain
ABSTRACT. Chong Zhang
 ABSTRACT Improved Biomolecular Crystallography at Low Resolution with the Deformable Complex Network Approach by Chong Zhang It is often a challenge to atomically determine the structure of large macromolecular
ABSTRACT Improved Biomolecular Crystallography at Low Resolution with the Deformable Complex Network Approach by Chong Zhang It is often a challenge to atomically determine the structure of large macromolecular
Improving Protein Function Prediction with Molecular Dynamics Simulations. Dariya Glazer Russ Altman
 Improving Protein Function Prediction with Molecular Dynamics Simulations Dariya Glazer Russ Altman Motivation Sometimes the 3D structure doesn t score well for a known function. The experimental structure
Improving Protein Function Prediction with Molecular Dynamics Simulations Dariya Glazer Russ Altman Motivation Sometimes the 3D structure doesn t score well for a known function. The experimental structure
Stabilizing the CH2 domain of an Antibody by Engineering in an Enhanced Aromatic Sequon
 Stabilizing the CH2 domain of an Antibody by Engineering in an Enhanced Aromatic Sequon Wentao Chen,, Leopold Kong, Stephen Connelly, Julia M. Dendle,, Yu Liu,, Ian A. Wilson,#, Evan T. Powers, *, Jeffery
Stabilizing the CH2 domain of an Antibody by Engineering in an Enhanced Aromatic Sequon Wentao Chen,, Leopold Kong, Stephen Connelly, Julia M. Dendle,, Yu Liu,, Ian A. Wilson,#, Evan T. Powers, *, Jeffery
Supporting Information
 Supporting Information Allosteric communication disrupted by small molecule binding to the Imidazole glycerol phosphate synthase protein-protein interface. Ivan Rivalta*,#, George P. Lisi #, Ning-Shiuan
Supporting Information Allosteric communication disrupted by small molecule binding to the Imidazole glycerol phosphate synthase protein-protein interface. Ivan Rivalta*,#, George P. Lisi #, Ning-Shiuan
X-ray Diffraction. Diffraction. X-ray Generation. X-ray Generation. X-ray Generation. X-ray Spectrum from Tube
 X-ray Diffraction Mineral identification Mode analysis Structure Studies X-ray Generation X-ray tube (sealed) Pure metal target (Cu) Electrons remover inner-shell electrons from target. Other electrons
X-ray Diffraction Mineral identification Mode analysis Structure Studies X-ray Generation X-ray tube (sealed) Pure metal target (Cu) Electrons remover inner-shell electrons from target. Other electrons
Structural basis for catalytically restrictive dynamics of a high-energy enzyme state
 Supplementary Material Structural basis for catalytically restrictive dynamics of a high-energy enzyme state Michael Kovermann, Jörgen Ådén, Christin Grundström, A. Elisabeth Sauer-Eriksson, Uwe H. Sauer
Supplementary Material Structural basis for catalytically restrictive dynamics of a high-energy enzyme state Michael Kovermann, Jörgen Ådén, Christin Grundström, A. Elisabeth Sauer-Eriksson, Uwe H. Sauer
SUPPLEMENTARY INFORMATION
 www.nature.com/nature 1 Figure S1 Sequence alignment. a Structure based alignment of the plgic of E. chrysanthemi (ELIC), the acetylcholine binding protein from the snail Lymnea stagnalis (AchBP, PDB code
www.nature.com/nature 1 Figure S1 Sequence alignment. a Structure based alignment of the plgic of E. chrysanthemi (ELIC), the acetylcholine binding protein from the snail Lymnea stagnalis (AchBP, PDB code
Biophysical Journal Volume 96 June
 Biophysical Journal Volume 96 June 2009 4449 4463 4449 A Rapid Coarse Residue-Based Computational Method for X-Ray Solution Scattering Characterization of Protein Folds and Multiple Conformational States
Biophysical Journal Volume 96 June 2009 4449 4463 4449 A Rapid Coarse Residue-Based Computational Method for X-Ray Solution Scattering Characterization of Protein Folds and Multiple Conformational States
ENZYME MECHANISMS, PROTEASES, STRUCTURAL BIOLOGY
 Supplementary Information SUBJECT AREAS: ENZYME MECHANISMS, PROTEASES, STRUCTURAL BIOLOGY Correspondence and requests for materials should be addressed to N.T. (ntanaka@pharm.showa-u.ac.jp) or W.O. (owataru@vos.nagaokaut.ac.jp)
Supplementary Information SUBJECT AREAS: ENZYME MECHANISMS, PROTEASES, STRUCTURAL BIOLOGY Correspondence and requests for materials should be addressed to N.T. (ntanaka@pharm.showa-u.ac.jp) or W.O. (owataru@vos.nagaokaut.ac.jp)
Determining Protein Structure BIBC 100
 Determining Protein Structure BIBC 100 Determining Protein Structure X-Ray Diffraction Interactions of x-rays with electrons in molecules in a crystal NMR- Nuclear Magnetic Resonance Interactions of magnetic
Determining Protein Structure BIBC 100 Determining Protein Structure X-Ray Diffraction Interactions of x-rays with electrons in molecules in a crystal NMR- Nuclear Magnetic Resonance Interactions of magnetic
Viewing and Analyzing Proteins, Ligands and their Complexes 2
 2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
2 Viewing and Analyzing Proteins, Ligands and their Complexes 2 Overview Viewing the accessible surface Analyzing the properties of proteins containing thousands of atoms is best accomplished by representing
Physiochemical Properties of Residues
 Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Physiochemical Properties of Residues Various Sources C N Cα R Slide 1 Conformational Propensities Conformational Propensity is the frequency in which a residue adopts a given conformation (in a polypeptide)
Joana Pereira Lamzin Group EMBL Hamburg, Germany. Small molecules How to identify and build them (with ARP/wARP)
") Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Joana Pereira Lamzin Group EMBL Hamburg, Germany Small molecules How to identify and build them (with ARP/wARP) The task at hand To find ligand density and build it! Fitting a ligand We have: electron
Determination of the structure and dynamics of proteins using NMR chemical shifts (CS) and CS enhanced protein data bank (CS-PDB)
 and CS enhanced protein data bank (CS-PDB)") Determination of the structure and dynamics of proteins using NMR chemical shifts (CS) and CS enhanced protein data bank (CS-PDB) Biao Fu The Vendruscolo group, Uni. Of Cambridge Intensive Training Course,
Determination of the structure and dynamics of proteins using NMR chemical shifts (CS) and CS enhanced protein data bank (CS-PDB) Biao Fu The Vendruscolo group, Uni. Of Cambridge Intensive Training Course,
SHELXC/D/E. Andrea Thorn
 SHELXC/D/E Andrea Thorn What is experimental phasing? Experimental phasing is what you do if MR doesn t work. What is experimental phasing? Experimental phasing methods depend on intensity differences.
SHELXC/D/E Andrea Thorn What is experimental phasing? Experimental phasing is what you do if MR doesn t work. What is experimental phasing? Experimental phasing methods depend on intensity differences.
Experimental Techniques in Protein Structure Determination
 Experimental Techniques in Protein Structure Determination Homayoun Valafar Department of Computer Science and Engineering, USC Two Main Experimental Methods X-Ray crystallography Nuclear Magnetic Resonance
Experimental Techniques in Protein Structure Determination Homayoun Valafar Department of Computer Science and Engineering, USC Two Main Experimental Methods X-Ray crystallography Nuclear Magnetic Resonance
Experimental and Computational Mutagenesis to Investigate the. Positioning of a General Base within an Enzyme Active Site
 Experimental and Computational Mutagenesis to Investigate the Positioning of a General Base within an Enzyme Active Site Jason P. Schwans, Philip Hanoian, Benjamin J. Lengerich, Fanny Sunden, Ana Gonzalez
Experimental and Computational Mutagenesis to Investigate the Positioning of a General Base within an Enzyme Active Site Jason P. Schwans, Philip Hanoian, Benjamin J. Lengerich, Fanny Sunden, Ana Gonzalez
Imaging and monitoring with industrial seismic noise.
 Imaging and monitoring with industrial seismic noise. M. Campillo also : Boston, May 2016 Passive imaging: Long range correla@ons ()*&#"'!"#"$%"&'!!!!!!"! #! Source in A the signal recorded in B characterizes
Imaging and monitoring with industrial seismic noise. M. Campillo also : Boston, May 2016 Passive imaging: Long range correla@ons ()*&#"'!"#"$%"&'!!!!!!"! #! Source in A the signal recorded in B characterizes
Supporting Information
 Supporting Information Structural Basis of the Antiproliferative Activity of Largazole, a Depsipeptide Inhibitor of the Histone Deacetylases Kathryn E. Cole 1, Daniel P. Dowling 1,2, Matthew A. Boone 3,
Supporting Information Structural Basis of the Antiproliferative Activity of Largazole, a Depsipeptide Inhibitor of the Histone Deacetylases Kathryn E. Cole 1, Daniel P. Dowling 1,2, Matthew A. Boone 3,
Supplementary Figure 1. Proposed mechanism for AusE, PrhA, and these mutants. (a) 5 is desaturated
 5 is desaturated") S1 Supplementary Figure 1. Proposed mechanism for AusE, PrhA, and these mutants. (a) 5 is desaturated to form 6 through hydrogen atom abstraction at C-2 followed by the second hydrogen atom abstraction
S1 Supplementary Figure 1. Proposed mechanism for AusE, PrhA, and these mutants. (a) 5 is desaturated to form 6 through hydrogen atom abstraction at C-2 followed by the second hydrogen atom abstraction
Unfolding CspB by means of biased molecular dynamics
 Chapter 4 Unfolding CspB by means of biased molecular dynamics 4.1 Introduction Understanding the mechanism of protein folding has been a major challenge for the last twenty years, as pointed out in the
Chapter 4 Unfolding CspB by means of biased molecular dynamics 4.1 Introduction Understanding the mechanism of protein folding has been a major challenge for the last twenty years, as pointed out in the
IgE binds asymmetrically to its B cell receptor CD23
 Supplementary Information IgE binds asymmetrically to its B cell receptor CD23 Balvinder Dhaliwal 1*, Marie O. Y. Pang 2, Anthony H. Keeble 2,3, Louisa K. James 2,4, Hannah J. Gould 2, James M. McDonnell
Supplementary Information IgE binds asymmetrically to its B cell receptor CD23 Balvinder Dhaliwal 1*, Marie O. Y. Pang 2, Anthony H. Keeble 2,3, Louisa K. James 2,4, Hannah J. Gould 2, James M. McDonnell
Protein Simulations in Confined Environments
 Critical Review Lecture Protein Simulations in Confined Environments Murat Cetinkaya 1, Jorge Sofo 2, Melik C. Demirel 1 1. 2. College of Engineering, Pennsylvania State University, University Park, 16802,
Critical Review Lecture Protein Simulations in Confined Environments Murat Cetinkaya 1, Jorge Sofo 2, Melik C. Demirel 1 1. 2. College of Engineering, Pennsylvania State University, University Park, 16802,
SUPPLEMENTARY INFORMATION
 5 N 4 8 20 22 24 2 28 4 8 20 22 24 2 28 a b 0 9 8 7 H c (kda) 95 0 57 4 28 2 5.5 Precipitate before NMR expt. Supernatant before NMR expt. Precipitate after hrs NMR expt. Supernatant after hrs NMR expt.
5 N 4 8 20 22 24 2 28 4 8 20 22 24 2 28 a b 0 9 8 7 H c (kda) 95 0 57 4 28 2 5.5 Precipitate before NMR expt. Supernatant before NMR expt. Precipitate after hrs NMR expt. Supernatant after hrs NMR expt.